
Tissue-specific and mosaic imprinting defects underlie opposite congenital growth disorders in mice
A humanized mouse line carrying a mutation of the H19/IGF2 imprinting control region demonstrates how tissue-specific and mosaic imprinting alterations result in growth disorders with opposite clinical pictures and asymmetric growth of bilateral organs.
Published in the journal:
. PLoS Genet 14(2): e32767. doi:10.1371/journal.pgen.1007243
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1007243
Summary
A humanized mouse line carrying a mutation of the H19/IGF2 imprinting control region demonstrates how tissue-specific and mosaic imprinting alterations result in growth disorders with opposite clinical pictures and asymmetric growth of bilateral organs.
Introduction
Imprinted genes show monoallelic and parent-of-origin-dependent expression and play key roles in the control of growth and development. In humans, altered expression of imprinted genes is associated with Imprinting Disorders (IDs) that are characterized by growth, metabolic and behavioural disturbances [1–2]. Most imprinted genes are organized in clusters, in which their parental-specific expression is dependent on Imprinting Control Regions (ICRs). ICRs correspond to 2–5 kb-long sequences with differential DNA methylation on their maternal and paternal alleles. Parental-specific ICR methylation is acquired during gametogenesis and maintained in the zygote and somatic cells throughout development despite extensive demethylation occurring in the embryo before implantation and de novo methylation after implantation [3]. An evolutionary conserved cluster of imprinted genes of about 1 Mbp is located on human chromosome 11p15.5 and mouse distal chromosome 7. The cluster is organized in two functionally independent domains, each with its own ICR. In the telomeric domain, the H19/IGF2 intergenic differentially methylated region (also known as and herein termed Imprinting Center 1, IC1) controls the reciprocal imprinting of the maternally expressed H19 and paternally expressed Insulin like Growth Factor 2 (IGF2) genes [3]. IGF2 is required for normal foetal growth [4]. The liver is the main endocrine source of IGF2 in post-natal life, but autocrine/paracrine activity is found in most embryonic tissues, particularly in placenta, where it is needed for correct allocation of maternal resources to fetal growth [5, 6]. H19 is a long non-coding RNA with inhibitory activity on foetal growth [7]. Both IGF2/Igf2 and H19 are down-regulated after birth in both humans and mice but their deficiencies have long-lasting effects on somatic growth [4, 7–11].
In mouse embryos, H19 and Igf2 are co-expressed in endoderm - and mesoderm-derived tissues, and their expression depends on the same downstream enhancers on the maternal and paternal chromosomes, respectively [12–15]. IC1 is structurally different in humans and mice—human IC1 (hIC1) is ~ 5kb-long and contains seven CCCTC-binding factor (CTCF) target sites (CTS), whereas mouse IC1 (mIC1) is ~ 2kb-long and contains four CTS (Fig 1A). CTCF binding to IC1 is required for the formation of an insulator with enhancer blocking activity in both species [16–17]. Because IC1 is methylated on the paternal allele and CTCF binding is inhibited by DNA methylation, the insulator is formed only on the maternal chromosome where it prevents the activation of IGF2 but allows activation of H19 by the enhancers. The opposite happens on the paternal chromosome, where IGF2 is activated and H19 silenced.

Opposite hIC1 methylation and imprinting defects are associated with the Beckwith-Wiedemann syndrome (BWS, MIM #130650) and the Silver Russell syndrome (SRS, MIM #180860), two IDs characterised by congenital overgrowth and congenital undergrowth, respectively [1]. In particular, hIC1 gain of methylation (GOM) resulting in IGF2 activation and H19 repression on the maternal chromosome is found in 5–10% of BWS cases. Conversely, hIC1 loss of methylation (LOM) leading to IGF2 repression and H19 activation on the paternal chromosome occurs in about 50% of SRS patients. In a number of BWS cases with hIC1 GOM, small deletions and single nucleotide variations within hIC1 co-segregate with the clinical phenotype and abnormal methylation upon maternal transmission but lead to normal phenotype on paternal transmission [11, 18–25]. Different paternally inherited hIC1 deletions have been recently described in a few cases of SRS with IC1 LOM [26]. Although the extent of these deletions is similar to those found in BWS families, the sequence and the CTSs involved are different.
In the last twenty years, several mutations have been introduced into the endogenous mIC1 locus [27–35]. This work has been instrumental to demonstrate the fundamental role that H19/IGF2 imprinting has in the aetiology of congenital overgrowth and undergrowth associated with imprinting defects. However, due to its structural differences with the orthologous mouse locus, the mechanism by which hIC1 mutations affect epigenotype and phenotype in both BWS and SRS is still obscure.
To clarify the role of hIC1 mutations in the origin of imprinting defects and in the pathogenesis of BWS and SRS, we generated a knock-in (KI) mouse line in which the endogenous mIC1 was replaced by the orthologous hIC1 allele carrying a mutation (hIC1Δ2.2) that is associated with BWS on maternal transmission [36]. We compared the H19hIC1Δ2.2 line with wildtype mice and the previously described line carrying a humanized H19 allele with the wildtype human ICR (H19hIC1) [37]. The results demonstrate growth and molecular abnormalities of the mice with maternal and paternal transmission of the mutant KI that resemble those of BWS and SRS, respectively, including asymmetric organ growth. Importantly, tissue-specific and mosaic dysregulation of H19/Igf2 imprinting indicates new pathogenetic mechanisms of congenital growth disorders and lateralized/regional over/under-growth associated with imprinting defects.
Results
Generation of the H19hIC1Δ2.2 and H19hIC1 lines
In order to study the relationship between genotype, epigenotype and phenotype of IDs in the mouse, we replaced the endogenous mIC1 with a mutant hIC1 allele (H19hIC1Δ2.2) previously found in BWS [19, 36], by homologous recombination in mouse embryonic stem (ES) cells (Fig 1A). Chimeras were obtained and germ line transmission was confirmed by Southern blotting (Fig 1B). The transgenic line was then bred to pCX-NLS - Cre transgenic line to remove the NeoR cassette and its excision was confirmed by PCR (Fig 1C. See also Materials and methods).
We crossed KI mice on a C56BL/6 (B6) background to Balb/C and used polymorphisms present between these two strains to distinguish parental alleles. To compare the behaviour of the mutant with that of the wildtype hIC1 allele (H19hIC1) in the same genetic background, the previously described H19hIC1 line [37] was generated anew by targeting ES cells and breeding the mice using similar procedures to what done with the H19hIC1Δ2.2 line. Subsequent experiments were performed on both humanized KI lines. The wildtype +/+ littermates were also assayed as control.
Maternal transmission of the H19hIC1Δ2.2 allele results in overgrowth and tissue-specific Igf2 activation
Phenotypic analysis
To determine the consequence of maternal transmission of the H19hIC1Δ2.2 allele on somatic growth, we measured the body weight of the F1 progeny of H19hIC1Δ2.2/+ X H19+/+ crosses from birth to 14 weeks of age. Compared to their H19+/+ littermates, H19hIC1Δ2.2/+ mice were significantly heavier, longer and larger at birth and overgrowth persisted postnatally until adult age (Fig 2A and 2B). In addition, kidney and tongue but not liver were significantly heavier in adult H19hIC1Δ2.2/+ mice, if normalized to the weight of the whole animal, indicating the presence of nephromegaly and macroglossia (Fig 2C). Consistent with previously published data [37], no growth difference was observed between H19hIC1/+ and H19+/+ mice (S1 Fig). Thus, in contrast to H19hIC1, maternal transmission of H19hIC1Δ2.2 results in congenital macrosomia and organomegaly that persist in the adulthood.

Molecular analysis
To examine expression and methylation of the H19/Igf2 locus upon maternal transmission of the KI alleles, RNA and DNA were extracted from liver, kidney and tongue of newborn (P1) mice. When measured by quantitative real-time PCR (RT-qPCR), H19 RNA levels were significantly lower in H19hIC1Δ2.2/+ mice compared with their H19+/+ littermates in all analyzed tissues, while levels of Igf2 RNA were higher in tongue, but similar or not significantly increased in liver and kidney (Fig 3A). Allele-specific RNA analysis revealed that, while the imprinted H19 expression (paternal allele repressed) was normally maintained in all tissues of H19hIC1Δ2.2/+ mice as well as H19+/+ littermates, the maternal Igf2 allele was derepressed in tongue (44% mat/mat+pat expression) and kidney (31%) of H19hIC1Δ2.2/+ mice (Fig 3B). In contrast, the relative expression (8%) of maternal Igf2 in liver of H19hIC1Δ2.2/+ and H19+/+ mice was comparable. Consistent with previous data [37], total expression levels and imprinting of both H19 and Igf2 in H19hIC1/+ and H19+/+ mice were comparable (S2A and S2B Fig).

In BWS, maternal transmission of IC1 mutations is associated with IC1 GOM [36]. To investigate if this phenotype was reproduced in the mutant KI mice, we assayed the methylation of two different IC1 regions, CTS1 and CTS6, in H19hIC1Δ2.2/+ and H19hIC1/+ mice, by pyrosequencing of bisulfite-converted DNA. The results demonstrated that in contrast with H19hIC1 that maintained very low (1–4%) methylation levels (Fig 4A and Ref. 37), H19hIC1Δ2.2 was aberrantly methylated at similar levels (62–75% in CTS1 and 55–58% in CTS6) in liver, kidney and tongue. As expected, the endogenous paternal mIC1 was fully methylated and the maternal mIC1 was unmethylated. Hypermethylation of H19hIC1Δ2.2 was confirmed by bisulfite sequencing. (Fig 4B). This assay shows complete methylation of 7 out of 15 molecules indicating epigenetic mosaicism. Methylation analysis was repeated in three consecutive generations of H19hIC1Δ2.2/+ mice with consistent results (S3 Fig).

In summary, H19hIC1Δ2.2 exhibits aberrant hIC1 methylation upon maternal transmission and is associated with abnormal H19 repression and tissue-specific Igf2 activation.
Paternal transmission of the H19hIC1Δ2.2 allele results in growth restriction and tissue-specific Igf2 repression
Phenotypic analysis
We next assessed the effect of paternal transmission of the H19hIC1Δ2.2 allele. Paternal transmission of H19hIC1 resulted in embryonic lethality, confirming our previous data [37]. In contrast, the F1 progeny of heterozygous H19hIC1Δ2.2 males bred to H19+/+ females presented H19+/+ and KI pups in 1 : 1 ratio. However, compared with their H19+/+ littermates, H19+/hIC1Δ2.2 mice weighed less and were smaller at birth and maintained growth restriction postnatally until adult age (Fig 5A and 5B). H19+/hIC1Δ2.2 mice also appeared very lean and the weight of their liver and tongue, normalized to that of the whole animal, was significantly lower than that of H19+/+ mice (Fig 5C).

To compare the effects of paternal transmission of H19hIC1 and H19hIC1Δ2.2 on somatic growth, we collected the embryos at a stage (E15.5) preceding the death of H19+/hIC1 mice in utero. Body and placenta of embryos from both KI strains were smaller than H19+/+ littermates. However, H19+/hIC1 embryos were even smaller (30% lighter than H19+/+ littermates) than H19+/hIC1Δ2.2 embryos (13% lighter than H19+/+ littermates) (Fig 5D). A more dramatic difference was found in the placental weights (Fig 5E, H19+/hIC1 line ~30% lighter than H19+/+ littermates; H19+/hIC1Δ2.2 line ~8% lighter than H19+/+ littermates).
Thus, although paternal transmission of both H19hIC1 and H19hIC1Δ2.2 results in growth restriction, the phenotype of H19+/hIC1Δ2.2 is less severe than that of H19+/hIC1 embryos and is compatible with life.
Molecular analysis
Gene expression and DNA methylation at the H19/Igf2 locus were measured in liver, kidney and tongue of P1 H19+/hIC1Δ2.2 and H19+/+ littermates. Total H19 expression was significantly increased in tongue, while Igf2 levels were severely decreased in liver and more moderately in kidney of H19+/hIC1Δ2.2 mice (Fig 6A). Analysis of allele-specific expression revealed that in H19+/hIC1Δ2.2 mice the paternal H19 allele was strongly derepressed in liver (44% pat/mat+pat expression) and more moderately in tongue (15%, see Fig 6B). In kidney, the relative expression of the paternal H19 allele (8%) was comparable to that of H19+/+ mice. The imprinted Igf2 expression (maternal allele repressed) was properly maintained in all three tissues in H19+/hIC1Δ2.2 as well as their H19+/+ littermates (Fig 6B).

To compare H19/Igf2 expression of H19+/hIC1Δ2.2 and H19+/hIC1, E15.5 conceptuses were also analysed. Total H19 expression was increased about 2-fold in embryo body and placenta of H19+/hIC1, but H19 activation was less evident and did not reach statistical significance in H19+/hIC1Δ2.2. In contrast, Igf2 expression was downregulated in embryo and placenta of both KI lines, although repression in embryos was less dramatic in H19+/hIC1Δ2.2 than in H19+/hIC1 mice (S4A and S4B Fig). Allele-specific RNA analysis demonstrated that the paternal H19 allele was strongly activated (close to 50% of total expression) in both embryo body and placenta of E15.5 H19+/hIC1, but only to a lower extent (25% in embryo body and 35% in placenta) in H19+/hIC1Δ2.2. The imprinted Igf2 expression was properly maintained in both H19+/hIC1 and H19+/hIC1Δ2.2 (S4C and S4D Fig and Ref. 37).
We then measured IC1 methylation in H19+/hIC1Δ2.2 and H19+/hIC1 mice at E15.5 by pyrosequencing. In contrast with the extremely low (2–3%) methylation of H19hIC1, the H19hIC1Δ2.2 allele was found partially (35–43%) methylated in both embryo body and placenta (Fig 7A). Bisulfite-sequencing confirmed the differential methylation of the H19hIC1Δ2.2 and H19hIC1 alleles (Fig 7B). The analysis of liver, kidney and tongue of P1 mice demonstrated that methylation of the H19hIC1Δ2.2 allele was maintained postnatally. Methylation levels (64–77%) were similar in these three adult tissues. They were higher than in embryonic tissues, but still lower than the endogenous paternal mIC1 allele that was fully methylated (S5A and S5B Fig). Methylation analysis was repeated in three consecutive generations of KI mice derived from breeding of H19+/hIC1Δ2.2 males with +/+ females with consistent results (S6A Fig). Also, methylation of the hIC1Δ2.2 allele did not change after the passage from the female to male germline, as demonstrated by the methylation values of mice of two consecutive generations derived from reciprocal breeding (S6B Fig).

In summary, paternal transmission of H19hIC1Δ2.2 results in IC1 hypomethylation that is less severe than that of H19hIC1 and is associated with an imprinting defect on the paternal chromosome causing moderate H19 activation accompanied by Igf2 repression that is particularly prominent in liver and placenta.
Differential Igf2 expression and imprinting is associated with kidney asymmetry in mutant KI mice
To investigate the presence of kidney asymmetry, a clinical sign often found in BWS [2], we measured the weight of left and right kidneys of the mice carrying the mutant KI. Maternal transmission was first assessed. A significant difference between the two kidneys (with no bias toward the left or right organ) was found in adult and newborn H19hIC1Δ2.2/+ mice, but not in H19+/+ littermates (Fig 8A and 8B). In contrast, no difference was observed in H19hIC1/+ mice (S7 Fig). hIC1 methylation and H19/Igf2 expression were then assessed in the organs of the neonates carrying the mutant KI. While DNA methylation and H19 expression were comparable (Fig 8C and 8D), significant differences of total and allele-specific expression of Igf2 were found between the heavier and lighter kidneys of H19hIC1Δ2.2/+ (with higher expression in the larger organ) mice (Fig 8E and 8F). Next, we investigated the presence of kidney asymmetry in the mice with paternal transmission of the KI. As for maternal transmission, weight differences between the two kidneys in H19+/hIC1Δ2.2 mice were significantly higher than in their H19+/+ littermates, both at neonatal and adult stages (Fig 8G and 8H). Also, comparable hIC1 methylation and global H19 RNA levels were found in left and right kidneys of all tested animals (Fig 8I and 8J). In contrast, paternal H19 expression was relatively more up-regulated in the lighter kidneys of the H19+/hIC1Δ2.2 mice (Fig 8K). Although not statistically significant (P<0.1), a trend toward stronger Igf2 repression was observed in the smaller with respect to the larger kidney (Fig 8I).

Low methylation of H19hIC1Δ2.2 in germ cells
Having demonstrated that H19hIC1Δ2.2 is methylated in somatic cells upon both maternal and paternal transmission, we asked if methylation was already present in germ cells. For this purpose, we measured DNA methylation levels in oocytes and sperm by pyrosequencing. As previously demonstrated, H19hIC1 remains properly hypomethylated in female gametes but methylation is inefficiently established on H19hIC1 in male gametes (Fig 9A and Ref. 37). Similarly, H19hIC1Δ2.2 methylation was close to 0% and comparable with the endogenous mIC1 in oocytes (Fig 9A). mIC2 was also analysed as control and to rule out contamination of somatic cells. The expected methylation value close to 100% was found in H19hIC1Δ2.2/+, H19hIC1/+ and H19+/+ oocytes.
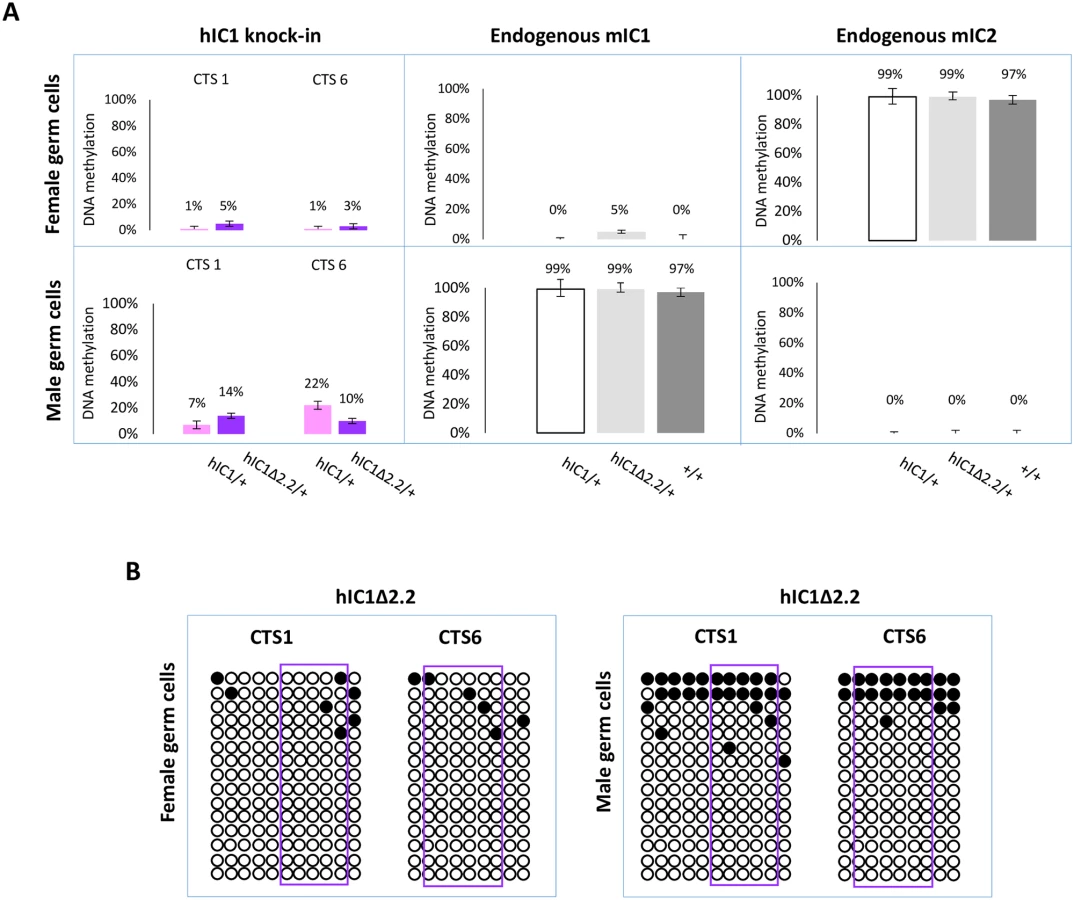
In sperm, methylation levels were relatively low (10–14%) on H19hIC1Δ2.2 as well as H19hIC1 (7–22%), while the endogenous mIC1 was almost 100% methylated (Fig 9A). The endogenous mIC2, analysed as control, was correctly unmethylated in all three lines (H19hIC1Δ2.2/+, H19hIC1/+ and H19+/+). The low methylation status of H19hIC1Δ2.2 was confirmed by bisulphite sequencing, in both oocytes and sperm (Fig 9B).
Overall, these results demonstrate that while DNA methylation of mutant hIC1 is normally absent in oocytes, methylation is not efficiently established on the mutant KI as well as the wildtype KI in male germ cells, indicating that paternal hypomethylation of the H19hIC1Δ2.2 allele is seemingly acquired as early as germline development and persists into embryo development, while maternal hypermethylation of hIC1Δ2.2 alleles does not occur until after fertilization.
Discussion
Gain and loss of IC1 methylation result in H19/IGF2 imprinting defects that are characteristic of BWS and SRS, respectively. In some patients, genetic mutations have been found associated with DNA methylation abnormalities in cis. However, the mechanism by which the genotype affects the epigenotype and phenotype in these cases is unknown. By employing a KI mouse line, this study demonstrates that a human genetic IC1 mutation reproduces several molecular and phenotypic abnormalities of BWS and SRS. The analysis of this mouse model provides mechanistic insights into the origin of prenatal overgrowth and undergrowth associated with H19/Igf2 imprinting defects, which are useful for understanding the aetiopathogenesis of BWS and SRS.
We have previously demonstrated that maternal transmission of hIC1 can functionally replace mIC1 in the mouse by properly regulating IC1 methylation and H19/Igf2 imprinting [37]. We now observe that mice maternally inheriting the mutant KI allele acquire methylation on hIC1Δ2.2 after fertilization, exhibit H19 repression and biallelic Igf2 activation, and pre - and post-natal overgrowth. Upon paternal transmission, hIC1 lacks methylation resulting in complete Igf2 silencing, H19 activation, severe growth restriction particularly in placenta and embryonic lethality [37]. In contrast, paternal hIC1Δ2.2 is partially methylated and results in a more moderate imprinting defect and pre/post-natal undergrowth which is compatible with life.
Methylation is established only in a minority of male germ cells on both hIC1Δ2.2 and hIC1 and appears unstable indicating evolutionarily divergent mechanisms of imprinting establishment between human and mouse [37]. However, while hIC1 is completely unmethylated, hIC1Δ2.2 shows partial methylation on both paternal and maternal chromosomes in embryo and placenta. Thus, methylation is likely acquired de novo and in mosaic form on hIC1Δ2.2 in somatic cells, during development. The functional difference between hIC1Δ2.2 and hIC1 is likely due to the lack of three CTSs in the mutant allele, which shows lower affinity for CTCF in human cells [36]. The results are consistent with hIC1 having an intrinsic propensity to acquire methylation that is inhibited by CTCF binding [32]. The different behaviour of hIC1Δ2.2 and mIC1, which share a similar number of CTSs (4) and ZFP57 binding sites (6), suggests that the CTSs alone are not sufficient to maintain insulator function and that CTS spacing or other transcription factor binding sites contribute to IC1 function. It is possible that one or more of these elements are reduced or missing and this exposes the mutant hIC1 allele to the action of de novo DNA methyl-transferases in pre - and/or post-implantation embryos.
The mechanisms by which hIC1 methylation is altered in BWS and SRS are unknown. Our mouse model indicates that the maternal hIC1Δ2.2 methylation is acquired post-zygotically in BWS, but does not allow to distinguish if the partial IC1 methylation of SRS is due to a primary germ cell imprint establishment defect, or a post-zygotic maintenance defect, or both. However, the observation that the hIC1 methylation status can drastically change from gametes to somatic cells, suggests that maintenance mechanisms have a critical role in the origin of imprinting defects on both maternal and paternal chromosomes.
Molecular analyses show that, although hIC1Δ2.2 is similarly methylated in neonatal liver, kidney and tongue, allele-specific H19/Igf2 expression is differently altered, suggesting that hIC1 enhancer blocking function is regulated in a tissue-specific manner (Fig 10), consistent with what was previously shown for mIC1 [35]. In particular, the insulator activity of hIC1Δ2.2 appears to be robust in liver and placenta, as demonstrated by weak expression of the Igf2 allele in cis with the KI. In contrast, the relatively high Igf2 expression indicates that the insulator activity of hIC1Δ2.2 is weak in tongue. The kidney shows intermediate and mosaic insulator activity resulting in differential Igf2 expression between left and right organs (see below). Tissue-specific differences in insulator activity may result from different post-translational modifications of CTCF [35]. Importantly, the observation that Igf2 expression is properly regulated in specific tissue contexts in the presence of abnormal IC1 methylation paves the way to new exciting avenues for BWS and SRS therapy. Concerning H19, this gene is significantly down-regulated in all three tissues of H19hIC1Δ2.2/+ mice and up-regulated in liver and tongue of H19+/hIC1Δ2.2 mice, with respect to wildtype littermates, consistent with the partial methylation of hIC1Δ2.2 and the demonstrated repressor activity of methylated IC1 [32]. In liver of H19+/hIC1Δ2.2 neonates, a strong derepression of the imprinted allele was not accompanied by a global (mat + pat) increase of the H19 RNA, possibly because of other physiological perturbations limiting its expression.

Overall, gene expression results are consistent with the hypothesis that the organomegaly of H19hIC1Δ2.2/+ mice is associated with autocrine/paracrine effects of IGF2, while defective hepatic and placental IGF2 expression underlie the growth restriction of H19+/hIC1Δ2.2 mice. Opposite deregulation of the growth inhibitory H19 transcript is likely playing an additional role in the growth abnormalities of the mice with maternal and paternal transmission of the mutant KI.
Although in humans this mutation is associated only with BWS, the hIC1Δ2.2 mice display several features of BWS and SRS, on maternal and paternal transmission of the KI, respectively. Growth abnormalities originate prenatally and persist in adulthood. In particular, H19+/hIC1Δ2.2 mice do not catch-up growth during development, as seen in the majority of children with SRS [38]. In addition, nephromegaly and macroglossia that are observed in H19hIC1Δ2.2/+ mice are also distinctive clinical signs of BWS with IC1 molecular defects [39]. Finally, the tissue-specific differences of hIC1 function we observed in the mouse may explain some of the aetiopathogenetic mechanisms of BWS and SRS. The model we propose predicts that maternal IC1 GOM in BWS causes IGF2 activation primarily in peripheral tissues, such as tongue and kidney resulting in macroglossia and organomegaly, while paternal IC1 LOM in SRS causes IGF2 repression primarily in liver and placenta leading to deficient growth stimulation through defective endocrine secretion and placenta function.
Both H19hIC1Δ2.2/+ and H19+/hIC1Δ2.2 mice show differential growth of the kidneys indicating that asymmetric growth of bilateral organs occurs in these animals, as in many cases of BWS with IC1 defects [40]. Derepression of the maternal Igf2 allele and repression of the paternal Igf2 allele are incomplete in the kidney of P1 H19hIC1Δ2.2/+ and P1 H19+/hIC1Δ2.2 mice, respectively (see Figs 3 and 6), indicating mosaic expression of H19/Igf2 in these tissues. Further analyses showed that, although IC1 methylation and H19 RNA levels were similar, Igf2 expression was higher in the larger than in the smaller kidney due to imprinting defects, indicating that Igf2 is mostly responsible for the asymmetric kidney growth. These results raise the hypothesis that mosaic IGF2 expression may also cause lateralized somatic overgrowth in humans. Our results are consistent with the findings of Ginart et al [41] demonstrating that the incomplete derepression of the paternal H19 allele in mutant mice can result from an epigenetic mosaicism at a single cell level.
Overall, our findings show that a mutant human IC1 sequence can reproduce the opposite growth and molecular phenotypes of BWS and SRS in mouse, when introduced at the orthologous locus. Several mouse lines carrying mutations in the H19/Igf2 locus have been described so far. Some of these mice, including a 1.3 kb deletion of mIC1 (Δ2,3) that results in tissue-specific loss of Igf2 imprinting, show similarities with our H19hIC1Δ2.2 model [28, 34–35]. However, the methylation defects, growth abnormalities and H19/Igf2 dysregulation of H19hIC1Δ2.2 more closely reproduce the phenotypic features and contribute better in understanding the molecular pathogenesis of BWS and SRS. Such humanized mouse models will be useful for more accurately unravelling pathogenetic mechanisms and for developing new therapeutic strategies in these rare congenital growth disorders.
Materials and methods
Targeting of the mIC1 locus and generation of the KI mice
To generate the H19hIC Δ2.2 mouse line, we performed gene targeting by homologous recombination in E14 embryonic stem (ES) cells [42] to target the endogenous mIC1 with a plasmid containing the H19hIC1Δ2.2 allele and neomycin resistance cassette (NeoR; Fig 1A). Briefly, a PciI–MluI restriction fragment of 620 bp spanning the break-point of the 2.2 kb deletion found in a BWS family [18] was extracted from the EΔ2.2 (B5/b1)pL vector [36] and subcloned in the Δ3.8kb-5’ pre-targeting vector containing the wildtype hIC1 region [28]. Sanger sequencing of the fragment was performed and no variant was found in respect to the reference human genome hg19. The subsequent steps to obtain the targeting vector were performed as previously described [37]. To compare H19hIC1 and H19hIC1Δ2.2 in the same strain background and avoid animal transfer from US to Italy, the hIC1 KI line was generated anew by performing gene targeting in E14 ES cells with the original vector used in the previous study [37].
Injection into B6 blastocysts of the H19hIC1-neo and H19hIC1Δ2.2-neo KI ES clones and generation of chimeras were performed by Cogentech Facility S.c.a.r.l. (Milan, Italy). Chimeras were crossed to B6 mice and germline transmission of the KI was confirmed in the agouti progeny by PCR-genotyping using primers flanking the deletion break point (hDMDB5SeqF: 5’–GGTAGTGAGGGATAGAACAC – 3’; hDMDB1RepR: 5’–GAGTGTCCTATTCCCAGATGAC – 3’) (Fig 1B). The NeoR cassette was excised by crossing heterozygous KI with pCX-NLS - Cre transgenic mice [43] on a B6 background. Excision was tested by PCR using primers flanking the NeoR cassette (NeoEXL3 : 5’–ACAGAATCGGTTGTGGCTGT – 3’ H19SeqR1 : 5’–CCACAGAGTCAGCATCCAC – 3’) (Fig 1C). KI mice without the NeoR cassette were crossed with B6 mice and only the progeny carrying the KI and lacking the Cre gene were selected to expand and assay the KI lines. All animal experimentation was conducted in accordance with the guidelines of the Animal Care and Use Committee of Campania University “Luigi Vanvitelli” (Naples, Italy) and was authorized by the Italian Ministry of Health.
Extraction of nucleic acids
Liver, kidneys and tongue were collected from mice at birth and at 14 weeks of age. Placenta and whole body excluding the head were recovered from conceptuses of 15.5 days post coitum (E15.5). Genomic DNA was isolated from tissues following the standard protocol of proteinase K digestion and phenol-chloroform extraction. Total RNA was extracted using TRI Reagent (Sigma-Aldrich Italia, Milan) and following the manufacturer’s protocol. Concentrations of nucleic acids were determined with Nanodrop spectrophotometer. Sperm was isolated from adult mice and DNA was extracted as described previously [44]. Unfertilized oocytes were collected from 4–5 superovulated females of 8 weeks and resuspended in 0.03% SDS, 10 mg glycogen, 10 mg proteinase K and 1 x PBS to a final volume of 20μl. Suspension was incubated at 37°C for 90 min followed by 15 min at 95°C before sodium bisulphite treatment for DNA methylation analysis (see below).
Gene expression analysis
About 1 μg of total RNA was treated with RNase-free DNase, and first-strand cDNA was synthesized using the QuantiTech Reverse Transcription Kit (Qiagen Italy, Milan), according to the manufacturer’s protocol. Total expression of H19 and Igf2 was measured by SYBR Green quantitative real-time RT-PCR (Applied Biosystems Italy, Milan). Reactions were set up in triplicate and run on ABI PRISM 7500 using the default cycling conditions. Relative expression was determined using the ΔΔCT method, and the gene expression values were normalized to the expression of the Gapdh, Arpp0 and beta-actin reference genes. Primer sequences are available on request. Allele-specific expression analysis was performed by typing for the polymorphisms present in the F1 progeny between the C57BL/6 (B6) and Balb/C mouse strains. The MspI Restriction Fragment Length Polymorphism (RFLP) of H19 (GRCm38/mm10 chr 7 : 142,577,609; sequenced region: chr7 : 142,577,530–142,577,732) and the (CA)n repeat of Igf2 (GRCm38/mm10 chr 7 : 142,652,936–142,652,973; sequenced region: chr7 : 142,652,821–142,653,091) were analysed as described by Pedone et al, [45]. The forward primer of Igf2 was labelled with FAM and the PCR products were run on the ABI 3130XL fluorescent capillary system (Applied Biosystems Italy, Milan).
DNA methylation analysis
The methylation status of cytosines in gDNA was determined by bisulphite treatment followed by pyrosequencing or cloning and sequencing. About 1 μg of genomic DNA was treated by sodium bisulphite using the Epitech Kit (Qiagen Italy, Milan), according to the manufacturer’s instructions. For the pyrosequencing, converted DNA was amplified with primers of which the reverse primer was biotinylated. The PCR products were run on the PyroMark Q24 platform, using PyromMark Gold Q96 Reagent [Qiagen Italy, Milan]. For bisulphite cloning and sequencing, converted DNA was amplified, the PCR products were cloned in Topo pCR2.1 vector (Topo-TA cloning kit, Termo Fisher Scientific Italy, Milan) and the clones were sequenced by Sanger method at Microtech Sequencing Core (Naples, Italy). Primers and PCR conditions are reported in S1 Table.
Statistical analyses
Unless otherwise indicated, data are expressed as the mean ± standard error of the mean (SEM). The significance of the difference between two groups (KI and wildtype) was determined with a two-tailed Student’s t-test with two-sample unequal variance. The number of samples, animals, biological or technical replicates are indicated in the respective figure legends. Differences with P-values ≤ 0.05 were considered significant.
Supporting Information
Zdroje
1. Eggermann T, Perez de Nanclares G, Maher ER, Temple IK, Tümer Z, Monk D, Mackay DJ, Grønskov K, Riccio A, Linglart A, Netchine I (2015) Imprinting disorders: a group of congenital disorders with overlapping patterns of molecular changes affecting imprinted loci. Clin Epigenetics 7 : 123 doi: 10.1186/s13148-015-0143-8 26583054
2. Soellner L, Begemann M, Mackay DJ, Grønskov K, Tümer Z, Maher ER, Temple IK, Monk D, Riccio A, Linglart A, Netchine I, Eggermann T (2017) Recent Advances in Imprinting Disorders. Clin Genet 91 : 3–13 doi: 10.1111/cge.12827 27363536
3. Barlow DP, Bartolomei MS (2014) Genomic imprinting in mammals. Cold Spring Harb Perspect Biol 1 : 6–7
4. DeChiara TM, Efstratiadis A, Robertson EJ (1990) A growth deficiency phenotype in heterozygous mice carrying an insulin-like growth factor II gene disrupted by targeting. Nature 345 : 78–80 doi: 10.1038/345078a0 2330056
5. O'Dell SD, Day IN (1998) Insulin-like growth factor II (IGF-II). Int J Biochem Cell Biol 30 : 767–771 9722981
6. Sferruzzi-Perri AN, Sandovici I, Constancia M, Fowden AL (2017) Placental phenotype and the insulin-like growth factors: resource allocation to fetal growth. J Physiol 595 : 5057–5093 doi: 10.1113/JP273330 28337745
7. Gabory A, Jammes H, Dandolo L (2010) The H19 locus: role of an imprinted non-coding RNA in growth and development. Bioessays 32 : 473–480 doi: 10.1002/bies.200900170 20486133
8. Lui JC, Baron J (2013) Evidence that Igf2 down-regulation in postnatal tissues and up-regulation in malignancies is driven by transcription factor E2f3. Proc Natl Acad Sci USA 110 : 6181–6186 doi: 10.1073/pnas.1219079110 23530192
9. Gicquel C, Rossignol S, Cabrol S, Houang M, Steunou V, Barbu V, Danton F, Thibaud N, Le Merrer M, Burglen L, Bertrand AM, Netchine I, Le Bouc Y (2005) Epimutation of the telomeric imprinting center region on chromosome 11p15 in Silver-Russell syndrome. Nat Genet 37 : 1003–1007 doi: 10.1038/ng1629 16086014
10. Begemann M, Zirn B, Santen G, Wirthgen E, Soellner L, Büttel HM, Schweizer R, van Workum W, Binder G, Eggermann T (2015) Paternally Inherited IGF2 Mutation and Growth Restriction. N Engl J Med 373 : 349–356 doi: 10.1056/NEJMoa1415227 26154720
11. Sparago A., Cerrato F., Vernucci M., Ferrero G.B., Cirillo-Silengo M. and Riccio A (2004) Microdeletions in the human H19 DMR result in loss of IGF2 imprinting and Beckwith-Wiedemann syndrome. Nat Genet 36 : 958–960 doi: 10.1038/ng1410 15314640
12. Leighton PA, Saam JR, Ingram RS, Stewart CL, Tilghman SM (1995) An enhancer deletion affects both H19 and Igf2 expression. Genes Dev 9 : 2079–2089 7544754
13. Ainscough JF, Koide T, Tada M, Barton S, Surani MA (1997) Imprinting of Igf2 and H19 from a 130 kb YAC transgene. Development 124 : 3621–3632 9342054
14. Kaffer CR, Grinberg A, Pfeifer K (2001) Regulatory mechanisms at the mouse Igf2/H19 locus. Mol Cell Biol 21 : 8189–8196 doi: 10.1128/MCB.21.23.8189-8196.2001 11689707
15. Davies K, Bowden L, Smith P, Dean W, Hill D, Furuumi H, Sasaki H, Cattanach B, Reik W (2002) Disruption of mesodermal enhancers for Igf2 in the minute mutant. Development 129 : 1657–1668 11923202
16. Hark AT, Schoenherr CJ, Katz DJ, Ingram RS, Levorse JM, Tilghman SM (2000) CTCF mediates methylation-sensitive enhancer-blocking activity at the H19/Igf2 locus. Nature 6785 : 486–489
17. Bell AC, Felsenfeld G (2000) Methylation of a CTCF-dependent boundary controls imprinted expression of the Igf2 gene. Nature 6785 : 482–485
18. Cerrato F, Sparago A, Di Matteo I, Zou X, Dean W, Sasaki H, Smith P, Genesio R, Bruggemann M, Reik W and Riccio A (2005) The two-domain hypothesis in Beckwith-Wiedemann Syndrome: Autonomous imprinting of the telomeric domain of the distal chromosome 7 cluster. Hum Mol Genet. 14 : 503–511 doi: 10.1093/hmg/ddi047 15640248
19. Prawitt D, Enklaar T, Gartner-Rupprecht B, Spangenberg C, Oswald M, Lausch E, Schmidtke P, Reutzel D, Fees S, Lucito R, Korzon M, Brozek I, Limon J, Housman DE, Pelletier J and Zabel B (2005) Microdeletion of target sites for insulator protein CTCF in a chromosome 11p15 imprinting center in Beckwith-Wiedemann syndrome and Wilms' tumor. Proc Natl Acad Sci USA 102 : 4085–4090 doi: 10.1073/pnas.0500037102 15743916
20. Sparago A., Russo S., Cerrato F., Ferraiuolo S., Castorina P., Selicorni A., Schwienbacher C., Negrini M., Ferrero G.B., Silengo M.C., Anichini C., Larizza L., Riccio A (2007) Mechanisms causing imprinting defects in familial Beckwith–Wiedemann syndrome with Wilms' tumour. Hum Mol Genet 16 : 254–264 doi: 10.1093/hmg/ddl448 17158821
21. Demars J, Shmela ME, Rossignol S, Okabe J, Netchine I, Azzi S, Cabrol S, Le Caignec C, David A, Le Bouc Y, El-Osta A, Gicquel C (2010) Analysis of the IGF2/H19 imprinting control region uncovers new genetic defects, including mutations of OCT-binding sequences, in patients with 11p15 fetal growth disorders. Hum Mol Genet 19 : 803–814 doi: 10.1093/hmg/ddp549 20007505
22. De Crescenzo A, Coppola F, Falco P, Bernardo I, Ausanio G, Cerrato F, Falco L, Riccio A (2011) A novel microdeletion in the IGF2/H19 imprinting centre region defines a recurrent mutation mechanism in familial Beckwith-Wiedemann syndrome. Eur J Med Genet 54 : 451–454
23. Poole RL, Leith DJ, Docherty LE, Shmela ME, Gicquel C, Splitt M, Temple IK, Mackay DJ (2012) Beckwith-Wiedemann syndrome caused by maternally inherited mutation of an OCT-binding motif in the IGF2/H19-imprinting control region, ICR1. Eur J Hum Genet 20 : 240–243 doi: 10.1038/ejhg.2011.166 21863054
24. Berland S, Appelbäck M, Bruland O, Beygo J, Buiting K, Mackay DJ, Karen Temple I, Houge G (2013) Evidence for anticipation in Beckwith-Wiedemann syndrome. Eur J Hum Genet 21 : 1344–1348 doi: 10.1038/ejhg.2013.71 23572028
25. Abi Habib W, Azzi S, Brioude F, Steunou V, Thibaud N, Das Neves C, Le Jule M, Chantot-Bastaraud S, Keren B, Lyonnet S, Michot C, Rossi M, Pasquier L, Gicquel C, Rossignol S, Le Bouc Y, Netchine I (2014) Extensive investigation of the IGF2/H19 imprinting control region reveals novel OCT4/SOX2 binding site defects associated with specific methylation patterns in Beckwith-Wiedemann syndrome. Hum Mol Genet 23 : 5763–5773 doi: 10.1093/hmg/ddu290 24916376
26. Abi Habib W, Brioude F, Azzi S, Salem J, Das Neves C, Personnier C, Chantot-Bastaraud S, Keren B, Le Bouc Y, Harbison MD, Netchine I (2017) 11p15 ICR1 Partial Deletions Associated with IGF2/H19 DMR Hypomethylation and Silver-Russell Syndrome. Hum Mutat 38 : 105–111 doi: 10.1002/humu.23131 27701793
27. Drewell RA, Brenton JD, Ainscough JF, Barton SC, Hilton KJ, Arney KL, Dandolo L, Surani MA (2000) Deletion of a silencer element disrupts H19 imprinting independently of a DNA methylation epigenetic switch. Development 127 : 3419–3428 10903168
28. Thorvaldsen JL, Mann MR, Nwoko O, Duran KL, Bartolomei MS (2002) Analysis of sequence upstream of the endogenous H19 gene reveals elements both essential and dispensable for imprinting. Mol Cell Biol 22 : 2450–2462. doi: 10.1128/MCB.22.8.2450-2462.2002 11909940
29. Schoenherr CJ, Levorse JM, Tilghman SM (2003) CTCF maintains differential methylation at the Igf2/H19 locus. Nat Genet 33 : 66–69 doi: 10.1038/ng1057 12461525
30. Pant V, Mariano P, Kanduri C, Mattsson A, Lobanenkov V, Heuchel R, Ohlsson R (2003) The nucleotides responsible for the direct physical contact between the chromatin insulator protein CTCF and the H19 imprinting control region manifest parent of origin-specific long-distance insulation and methylation-free domains. Genes Dev 17 : 586–590 doi: 10.1101/gad.254903 12629040
31. Pant V, Kurukuti S, Pugacheva E, Shamsuddin S, Mariano P, Renkawitz R, Klenova E, Lobanenkov V, Ohlsson R (2004) Mutation of a single CTCF target site within the H19 imprinting control region leads to loss of Igf2 imprinting and complex patterns of de novo methylation upon maternal inheritance. Mol Cell Biol 24 : 3497–3504 doi: 10.1128/MCB.24.8.3497-3504.2004 15060168
32. Engel N, West AG, Felsenfeld G, Bartolomei M (2004) Antagonist between DNA hypermethylation and enhancer-blocking activity at the H19 DMD is uncovered by CpGg mutations. Nat Genet 36 : 883–888 doi: 10.1038/ng1399 15273688
33. Engel N, Thorvaldsen JL, Bartolomei MS (2006) CTCF binding sites promote transcription initiation and prevent DNA methylation on the maternal allele at the imprinted H19/Igf2 locus. Hum Mol Genet 15 : 2945–2954 doi: 10.1093/hmg/ddl237 16928784
34. Thorvaldsen JL, Fedoriw AM, Nguyen S, Bartolomei MS (2006) Developmental profile of H19 differentially methylated domain (DMD) deletion alleles reveals multiple roles of the DMD in regulating allelic expression and DNA methylation at the imprinted H19/Igf2 locus. Mol Cell Biol. 26 : 1245–1258 doi: 10.1128/MCB.26.4.1245-1258.2006 16449639
35. Ideraabdullah FY, Thorvaldsen JL, Myers JA, Bartolomei MS (2014) Tissue-specific insulator function at H19/Igf2 revealed by deletions at the imprinting control region. Hum Mol Genet 23 : 6246–6259 doi: 10.1093/hmg/ddu344 24990148
36. Beygo J, Citro V, Sparago A, De Crescenzo A, Cerrato F, Heitmann M, Rademacher K, Guala A, Enklaar T, Anichini C, Cirillo Silengo M, Graf N, Prawitt D, Vittoria Cubellis M, Horsthemke B, Buiting K, Riccio A (2013) The molecular function and clinical phenotype of partial deletions of the IGF2/H19 imprinting control region depends on the spatial arrangement of the remaining CTCF-binding sites. Hum Mol Genet 22 : 544–57 doi: 10.1093/hmg/dds465 23118352
37. Hur SK, Freschi A, Ideraabdullah F, Thorvaldsen JL, Luense LJ, Weller AH, Berger SL, Cerrato F, Riccio A, Bartolomei MS (2016) Humanized H19/Igf2 locus reveals diverged imprinting mechanism between mouse and human and reflects Silver-Russell syndrome phenotypes. Proc Natl Acad Sci USA 113 : 10938–10943 doi: 10.1073/pnas.1603066113 27621468
38. Wakeling EL, Brioude F, Lokulo-Sodipe O, O'Connell SM, Salem J, Bliek J, Canton AP, Chrzanowska KH, Davies JH, Dias RP, Dubern B, Elbracht M, Giabicani E, Grimberg A, Grønskov K, Hokken-Koelega AC, Jorge AA, Kagami M, Linglart A et al, (2017) Diagnosis and management of Silver-Russell syndrome: first international consensus statement. Nat Rev Endocrinol 13 : 105–124 doi: 10.1038/nrendo.2016.138 27585961
39. Mussa A, Russo S, Larizza L, Riccio A, Ferrero GB (2016) (Epi)genotype-phenotype correlations in Beckwith-Wiedemann syndrome: a paradigm for genomic medicine. Clin Genet 89 : 403–415
40. Mussa A, Peruzzi L, Chiesa N, De Crescenzo A, Russo S, Melis D, Tarani L, Baldassarre G, Larizza L, Riccio A, Silengo M, Ferrero GB (2012) Nephrological findings and genotype-phenotype correlation in Beckwith-Wiedemann syndrome. Pediatr Nephrol 27 : 397–406 doi: 10.1007/s00467-011-2009-4 22015620
41. Ginart P, Kalish JM, Jiang CL, Yu AC, Bartolomei MS, Raj A (2016) isualizing allele-specific expression in single cells reveals epigenetic mosaicism in an H19 loss-of-imprinting mutant. Genes Dev 30 : 567–578 doi: 10.1101/gad.275958.115 26944681
42. Kühn R, Rajewsky K, Müller W (1991) Generation and analysis of interleukin-4 deficient mice. Science 254 : 707–710 1948049
43. Nagy A (2000) Cre recombinase: the universal reagent for genome tailoring. Genesis 26 : 99–109 10686599
44. Weyrich A (2012) Preparation of genomic DNA from mammalian sperm. Curr Protoc Mol Biol Chapter 2: Unit 2.13.1–3
45. Pedone PV, Pikaart MJ, Cerrato F, Vernucci M, Ungaro P, Bruni CB, Riccio A (1999) Role of histone acetylation and DNA methylation in the maintenance of the imprinted expression of the H19 and Igf2 genes. FEBS Lett 458 : 45–50 10518931
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2018 Číslo 2
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Tissue-specific and mosaic imprinting defects underlie opposite congenital growth disorders in mice
- Sex: Not all that it’s cracked up to be?
- Fish mutant, where is thy phenotype?
- Nuclear re-localization of Dicer in primary mouse embryonic fibroblast nuclei following DNA damage
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy