
A Microhomology-Mediated Break-Induced Replication Model for the Origin of Human Copy Number Variation
Chromosome structural changes with nonrecurrent endpoints associated with genomic disorders offer windows into the mechanism of origin of copy number variation (CNV). A recent report of nonrecurrent duplications associated with Pelizaeus-Merzbacher disease identified three distinctive characteristics. First, the majority of events can be seen to be complex, showing discontinuous duplications mixed with deletions, inverted duplications, and triplications. Second, junctions at endpoints show microhomology of 2–5 base pairs (bp). Third, endpoints occur near pre-existing low copy repeats (LCRs). Using these observations and evidence from DNA repair in other organisms, we derive a model of microhomology-mediated break-induced replication (MMBIR) for the origin of CNV and, ultimately, of LCRs. We propose that breakage of replication forks in stressed cells that are deficient in homologous recombination induces an aberrant repair process with features of break-induced replication (BIR). Under these circumstances, single-strand 3′ tails from broken replication forks will anneal with microhomology on any single-stranded DNA nearby, priming low-processivity polymerization with multiple template switches generating complex rearrangements, and eventual re-establishment of processive replication.
Published in the journal:
. PLoS Genet 5(1): e32767. doi:10.1371/journal.pgen.1000327
Category:
Review
doi:
https://doi.org/10.1371/journal.pgen.1000327
Summary
Chromosome structural changes with nonrecurrent endpoints associated with genomic disorders offer windows into the mechanism of origin of copy number variation (CNV). A recent report of nonrecurrent duplications associated with Pelizaeus-Merzbacher disease identified three distinctive characteristics. First, the majority of events can be seen to be complex, showing discontinuous duplications mixed with deletions, inverted duplications, and triplications. Second, junctions at endpoints show microhomology of 2–5 base pairs (bp). Third, endpoints occur near pre-existing low copy repeats (LCRs). Using these observations and evidence from DNA repair in other organisms, we derive a model of microhomology-mediated break-induced replication (MMBIR) for the origin of CNV and, ultimately, of LCRs. We propose that breakage of replication forks in stressed cells that are deficient in homologous recombination induces an aberrant repair process with features of break-induced replication (BIR). Under these circumstances, single-strand 3′ tails from broken replication forks will anneal with microhomology on any single-stranded DNA nearby, priming low-processivity polymerization with multiple template switches generating complex rearrangements, and eventual re-establishment of processive replication.
Introduction
In the past few years, we have learnt that a major component of the differences between individuals is variation in the number of copies of segments of the genome, and of genes included in these segments (copy number variation or CNV) (for definition of abbreviations, see Table 1). A considerable portion of the genome is involved in CNV [1]–[11]—with estimates of up to 12% [4]—which can arise meiotically and also somatically as shown by the finding that identical twins can differ in CNV [12]. CNV has been a significant component of primate evolution [13]–[16]. Here we draw on evidence on the mechanism of DNA transactions in Escherichia coli, yeast, Drosophila, mammals, and human cancer to derive a model for the origin of CNV based on the mechanism of BIR occurring at sites of microhomology (microhomology-mediated BIR or MMBIR).

Genomic Disorders
Although we can see that considerable variation in copy number is tolerated or is advantageous to its carrier, some genes are dosage-sensitive, and duplication or deletion involving these genes gives rise to human clinical phenotypes collectively referred to as genomic disorders [17]. This has allowed the ascertainment of structural changes and thus the study of the origin of CNV. For recurrent rearrangements, much CNV stems from homologous recombination between segments that already occur as two or more copies. When this happens, sequences that lie between the repeats that recombine will be either duplicated or deleted, thus changing the copy number. This process is referred to as nonallelic homologous recombination, or NAHR [18]. The repeated sequences that recombine might occasionally be highly repetitive sequences that occur widely in the human genome [19] but are usually sequences that occur only twice or a few times (i.e., low-copy repeats, LCRs, or segmental duplications, SDs). The LCRs tend to occur in clusters in highly complex regions of the genome. These repeated segments might be short (about 10 kilobases (kb)), or up to several hundreds of kb in length, and they occur in either orientation. Some examples of genomic complex regions are shown in Figure 1.

The endpoints of CNVs that arose by NAHR occur in a few positions where there is sufficient homology for homologous recombination. Although many genomic disorders arise by NAHR [20], some rearrangements have endpoints in many different positions. These CNVs arose de novo by rearrangements at sites that lack extensive homology. Recent evidence on the distribution of nonpathological CNVs in two individuals suggests that most differences in copy number from the reference sequence arose by nonrecurrent events [2]. Thus nonrecurrent chromosomal changes arise quite frequently [21]. Because the nonrecurrent events presumably reflect the origin of most genome complexity, the study of them is important to the understanding of genomic disorders, genetic variability due to CNV, and human evolution.
Pelizaeus-Merzbacher disease (PMD; Online Mendelian Inheritance in Man (OMIM) accession code 312080; http://www.ncbi.nlm.nih.gov/omim/) is a recessive X-linked genomic disorder affecting the central nervous system that arises by nonrecurrent chromosomal changes. The changes involve duplication, triplication, or deletion of the PLP1 gene. The clinical phenotype allows identification of individuals showing nonrecurrent chromosomal changes in the PLP region. In a study of the structural variation in the genomes of patients with PMD, Lee et al. [22] describe some aspects of the fine structure of newly arising CNVs with nonrecurrent endpoints and report three striking properties of their structure that help us to understand the origin of CNVs. First, the authors report that the novel junctions form at sites of microhomology, i.e., lengths of homology 2 to 5 nucleotides long that are too short to support homologous recombination. Such junctions have been reported previously in cases of nonrecurrent endpoints of deletions and duplications [19],[23],[24]. Second, they observed that the new structures are complex, showing duplication and deletion interspersed with nonduplicated or with triplicated lengths, and showing duplicated segments in either orientation. These characteristics were reported previously [25]–[31]. Third, although these events did not arise by NAHR, the novel junctions tend to occur in close proximity to LCRs [32]–[34]. Figures 2 and 3 illustrate examples of these complex non-recurrent events. Nonrecurrent rearrangements had previously been attributed to a mechanism of nonhomologous end-joining (NHEJ) [19],[20],[24],[33]. However, the characteristics of microhomology junctions and structural complexity in these new structures, as revealed by nucleotide sequencing and high-resolution array comparative genomic hybridization, led Lee et al. [22] to propose that the rearrangements arose through a replication-based mechanism termed FoSTeS (fork stalling and template switching), a mechanism proposed previously for amplification in E. coli [35]. Replication-based models have also been proposed to explain the origin of gross chromosomal rearrangements seen in a low proportion of patients with cystic fibrosis and hemophilia A. Analysis of deletions of the genes involved reveals complex structures similar to those described for PLP1 [28],[29],[31].
![Complex rearrangements involving <i>PLP1</i> detected by junction analysis (A) and oligonucleotide array comparative genomic hybridization analysis (B) <em class="ref">[22]</em>.](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image/fa97ce188b2f3761459896c44c110c7d.png)

Genome Rearrangements in Cancer
The amount of structural variation in cancer cells is sometimes so extreme [36] that it is not possible to determine which changes occurred within the same event. However, it can be seen that duplications are often discontinuous, and junction regions include insertions of nearby, unlinked, and unknown sequences, and deletions and inversions [37], showing that rearrangement events in cancer cells are complex. Many studies report microhomology at junctions of a large proportion of the structural variation (e. g., [37]–[39]). Studies of translocation endpoints in leukemia and other cancers find that many junctions have microhomology and are associated with insertions and deletions of various lengths [40]–[42]. These observations are compatible with at least some of the genomic instability seen in tumor formation and progression having stemmed from the same underlying mechanism as the formation of nonrecurrent duplications in genomic disorders.
Involvement of Replication in Chromosomal Structural Change
In the Lac assay system in E. coli [43], amplification of the lac operon to 20–100 copies occurs in response to the stress of starvation [44],[45]. The novel junctions of the amplified segments (amplicons) show that endpoints occurred at sites of microhomology of 2–15 bp [35],[46]. Some of the amplicons are complex, containing both direct and inverted repeats. Many others cannot be identified by outward-facing polymerase chain reaction (an observation also encountered frequently for PLP1 duplication junction analysis [22]), which would reveal the junctions of simple tandem repeats, and so are presumed to be complex, rather than simple tandem repeats [35],[46],[47]. By these criteria, about 25% of amplicons are complex. Thus, with respect to microhomology and complexity, the chromosomal structural changes in this system resemble those found in nonrecurrent events in human genomic disorders.
Homologous recombination requires RecA protein (Rad51 in eukaryotes) (reviewed in [48]). Microhomology-mediated deletion formation in E. coli (less than 25 nucleotides of homology) has long been known to be RecA-independent [49]–[52]. RecA-independent short homology-mediated deletions (25–50 nucleotides) have previously been attributed to template switching within a replication fork during DNA replication (reviewed in [53]). The evidence for this is, first, that mutations in genes encoding replication functions affect the formation of these events; second, that mutations affecting post-replicational mismatch repair affect them, placing the event very near to the replication fork; third, that mutation of 3′ exonucleases has an effect that is consistent with the ends being used to prime DNA synthesis; and fourth, that it is very difficult to obtain mutations affecting the process by transposon mutagenesis, suggesting essential functions.
In the E. coli Lac system, study of genetic requirements of stress-induced amplification has revealed some details of the mechanism. First, the events involve 3′ DNA ends. This is seen by an increase in amplification when a 3′ exonuclease gene (xonA) is deleted, and a decrease when the 3′ exonuclease is over-expressed. Similar manipulation of 5′-exonuclease has no effect [35]. This suggests that amplification results from free 3′ ends in the cell most of which are normally removed by exonuclease. As above, the involvement of 3′ ends but not 5′ ends is consistent with priming of DNA synthesis.
Second, lagging-strand processing at replication forks is implicated by a requirement for the 5′ exonuclease domain of DNA polymerase I (Pol I) [35],[45]. Pol I is involved in lagging-strand replication, base excision repair, and nucleotide excision repair, but these excision repair processes are not involved in amplification [35], so lagging strands at replication forks are implicated in amplification.
Third, there is a requirement for the proteins of double-strand break (DSB) repair by homologous recombination [35] (the RecBC system, reviewed in [48]). That this is actually a requirement for DSB repair (not just the proteins) is shown by the discovery that in vivo double-strand cleavage of DNA near lac enhances amplification rates [54].
Taken together, these observations suggest a model for amplification in the Lac system in E. coli in which replication is restarted at sites of repair of DNA double-strand ends [35]. The hypothesis proposed was that template switching occurs during replication restart at stalled replication forks. Because the distances involved exceed the lengths that are expected to be exposed as single-stranded at a single replication fork, it was proposed that the switches occurred between different replication forks [35].
The idea that chromosomal structural changes originate from DNA replication has received support from a study of microhomology-mediated SD formation in yeast [55]. These authors support the idea that the mechanism of SD formation involves replication by showing that its frequency is enhanced by treatment with camptothecin and is dependent on Pol32, a component of Polδ (discussed below). Camptothecin is a topoisomerase I inhibitor that leaves nicks in DNA. These nicks are believed to become collapsed forks when a replication fork reaches them. Thus, increasing the frequency of fork collapse increases the frequency of duplication formation. These authors also report that situations that lead to fork stalling rather than collapse have little effect on the frequency of duplication formation [55]. Thus, it appears that the substrate for duplication is a single double-strand end at a collapsed replication fork.
This long-distance template-switch model was also used by Lee et al. [22] to explain the observations of nonrecurrent chromosomal changes seen in Pelizaeus-Merzbacher disease discussed above and the juxtaposition of multiple genomic sequences normally separated by large genomic distances [22],[56]. Experiments on the integration of nonhomologous DNA into mammalian cells revealed microhomology junctions and insertion of sequence from other parts of the genome at the junctions. These observations were interpreted in terms of a similar model of repeated copying and switching to another template [57].
Break-Induced Replication
A more specific model for restarting replication at collapsed (broken) replication forks, BIR [58], has been developed for yeast, and a similar mechanism was proposed to explain telomere maintenance in yeast and human cell lines that have lost telomerase activity (reviewed in [59]). Recent evidence [60],[61] suggests that the BIR mechanism can be modified to explain the complexity of chromosomal structural changes described above for human and E. coli. Figure 4 illustrates the mechanism of BIR. When the replicative helicase encounters a nick on the template strand (Figure 4A), one arm of a replication fork breaks off (Figure 4B). There is no second end to be involved in the mechanisms of DSB repair that are available at a DSB consisting of two double-strand ends: homologous recombination or nonhomologous end-joining. The 5′ end of the broken arm is resected by an exonuclease to leave a 3′ overhang (Figure 4C). This 3′ tail invades a homologous sequence, normally the sister chromatid from which it came. This invasion is mediated by RecA/Rad51 protein (Figure 4D). The 3′ end primes DNA synthesis and establishes a replication fork consisting of both leading and lagging strand synthesis [61] (Figure 4E). This replication is of low processivity, and the extended arm is separated from the sister chromatid (Figure 4E). Such separation might be achieved by migration of the Holliday junction shown in Figure 4D and 4E. The 3′ end reinvades and the process is repeated (Figure 4G and 4H). After a few cycles of invasion, extension, and separation, the replication fork becomes more processive, and replication continues to the end of the chromosome arm or to the end of the replicon. The change from low processivity to highly processive replication can be attributed to a switch in the DNA polymerases involved [61]. Initial extension from a double-strand end was shown to require the primase complex and Polδ, notably the nonessential Pol32 subunit, whereas the more processive Polε was required for the 30-kb extension to the telomere. Figure 4I shows the completed pair of chromatids with the new material segregating conservatively as suggested for E. coli [62]. This would result if the Holliday junction followed the replication fork. Another possibility is that the Holliday junction is resolved so that there will be semi-conservative segregation of old and new DNA strands [60], (reviewed in [63]). Evidence for conservative segregation of new DNA strands in BIR, suggesting that the Holliday junction was not resolved, was reported for E. coli [62].
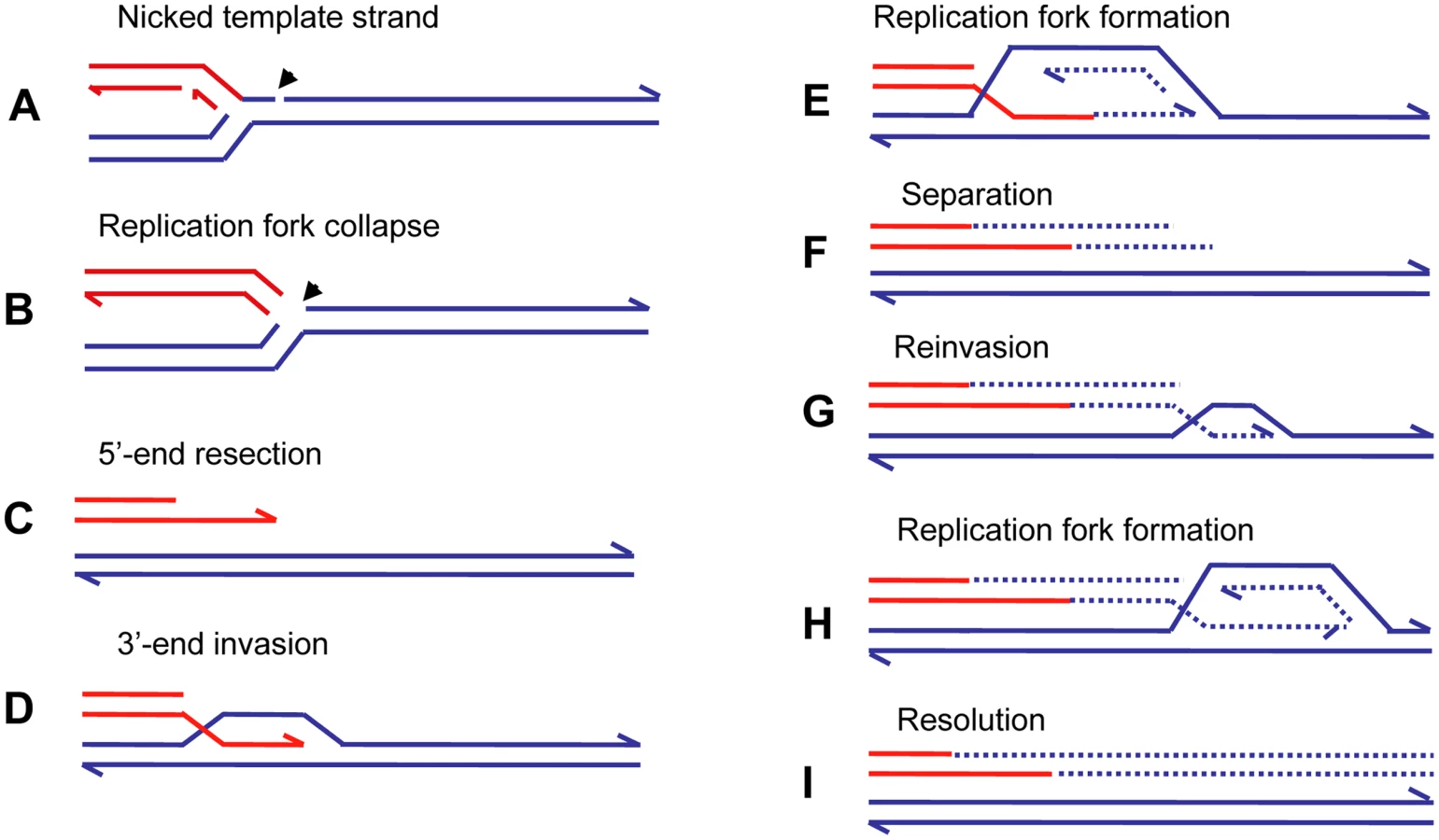
The repeated extension and separation have been interpreted as repeated attempts to find the other side of a break consisting of two double-strand ends. When, eventually, none is found because this is a collapsed fork rather than a two ended DSB, the remainder of the chromosome is replaced by replication [60],[63]. The pattern of repeated rounds of template switching followed by a long length of replication is supported by observations of BIR in yeast. BIR can be induced experimentally by transforming a chromosomal fragment into a yeast cell [64]. Using such a system, Smith et al. [60] placed a chromosomal fragment with a centromere and one telomere-forming sequence into a diploid yeast cell. The fragment had homology to both homologues of chromosome III. These homologues were differentially marked. Selection for a marker on the fragment selected for cells in which the fragment had acquired a second telomere. These authors found that most fragments had completed the replication of 50 kb to the end of the chromosome to which the fragment had homology. The striking result was that many of the chromosomes recovered had switched from one homologue to the other. In some cases, more than one switch was seen. The switches were confined to the first 10 kb, after which a single homologue was copied. In a few percent of cases, the switch was to a different chromosome at sites of repeated homology consisting of the long terminal repeat of a retrotransposon. Thus, BIR was demonstrated to produce complexity of the sorts reported above for E. coli amplification and for nonrecurrent end-points in human genomic disorders.
BIR has been suggested as the mechanism that underlies SD and other structural changes in yeast, e.g., [55],[65],[66], and human, e.g., [31],[67]. As discussed below, BIR is strongly RecA/Rad51-dependent and homology-dependent, and so cannot account for the observations of microhomology associated with complex rearrangements without substantial change.
Microhomology-Mediated BIR (MMBIR)
BIR, as described above, is usually an accurate process, because the repeated invasions are RecA/Rad51-mediated and involve long lengths of homology between DNA sequences. Invasion catalyzed by RecA/Rad51 requires extensive homology of about 50 bp in E. coli [68] and more in eukaryotes [69],[70]. This does not fit with the microhomology junctions described above. We therefore suggest that in these systems, replication forks are reestablished in a RecA/Rad51-independent manner. Rad51-independent BIR occurs in yeast at a much lower efficiency than the Rad51-dependent BIR [71],[72], though its frequency is very much enhanced, at the expense of fidelity, by the presence of unusual structures such as an inverted repeat [71]. However, telomere recombination in the absence of telomerase is proficient in the absence of Rad51 and is mediated by very short homologies [73],[74] (reviewed in [59]). The fact that telomere recombination occurs by BIR is supported by the finding that it requires the same set of enzymes as BIR that is initiated in the middle of a chromosome [61]. Absence or shortage of RecA/Rad51 might arise because the cells are stressed, as described below. That microhomology-mediated SD formation occurs in yeast by a BIR mechanism is supported by the finding that, like homology-mediated BIR [61], it requires Pol32 [55].
In mammalian cells, there is a surprisingly efficient microhomology-mediated DSB repair pathway. Most, if not all, experimental research on microhomology-mediated DSB repair has been performed with nuclease-induced breaks. This recently described pathway was characterized in recombination events induced by I-SceI or RAG1/RAG2 nucleases in cells deficient in classical NHEJ and in cancer cells [75],[76]. Nucleases generate two-ended breaks at random with respect to ongoing replication forks. However, BIR acts under circumstances when DSB repair, including NHEJ, is not an option, because after replication fork breakage, there is only a single end with no second end to which the one end can be annealed or ligated. Spontaneous damage to DNA occurs predominantly during replication [77]–[79], so that mechanisms that repair single DNA ends are more appropriately invoked for spontaneous damage than are mechanisms that act on two-ended DSBs. We suggest that a novel pathway, microhomology-mediated BIR (MMBIR), is used to repair single double-strand ends when stretches of single-stranded DNA are available and share microhomology with the 3′ single-strand end from the collapsed fork.
Single-stranded DNA might be expected to occur in replication forks, from stalled transcription complexes, at excision repair tracts, or at secondary structures in DNA such as cruciforms or hairpins caused by complex genomic architecture, and possibly in other situations such as in promoter regions and replication origins. The dimensions of most of the template switches discussed here (tens to hundreds of kb distant, i.e., the length of a duplication or deletion) preclude mechanisms of replication slippage within a single replication fork. An ability of any single-stranded DNA region that shares microhomology with the single-stranded 3′ end to take part in the events would explain why MMBIR is inexact and liable to lead to chromosomal structural changes. Very short homology should not be a barrier to replication fork restart because polymerase eta, used in DSB repair in vertebrates [80],[81], is efficient in initiating new DNA synthesis from mismatched primers, and even primers as short as 2–3 bp [82].
The presence of inverted repeats could generate hairpin loops that expose single-stranded sequence [22],[32]. In addition, hairpin structures might increase the likelihood of replication fork stalling, which might then initiate BIR. Such major roles for secondary DNA structures in the generation of chromosomal structural changes offers an explanation for the clustering of structural changes, producing complex chromosomal regions such as that illustrated in Figure 1. The model of MMBIR is presented in Figure 5.
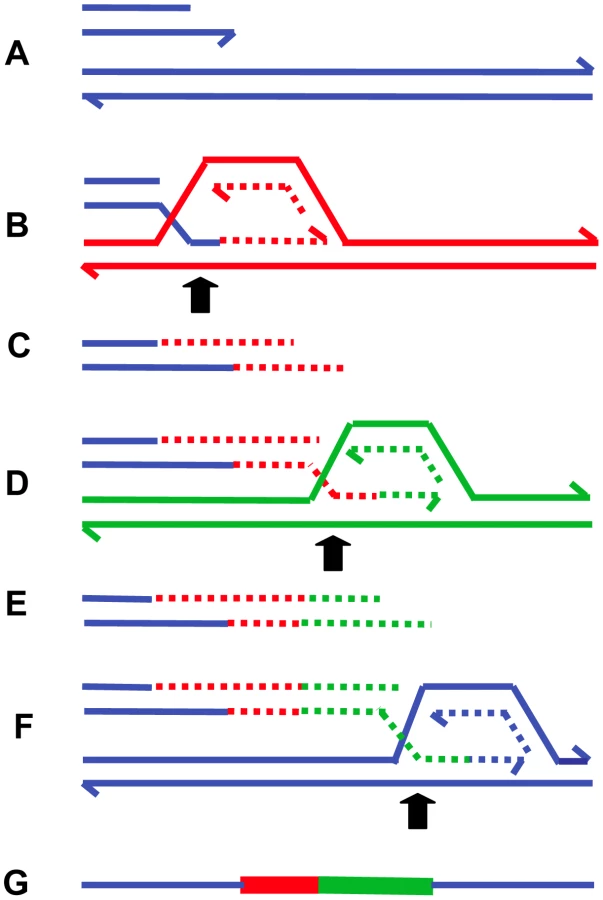
The clear distinction between NHEJ and BIR mediated by microhomology is that, in the second instance, microhomology junctions are followed by shorter or longer stretches of DNA sequence derived from elsewhere. Ten to 20% of nonhomologous junctions in mammalian cells have sequence inserted at the junction [83]. Some events that had previously been interpreted as occurring by an NHEJ mechanism might have occurred by MMBIR with a single template switch. In addition, events that appeared to be simple end-joining events might have had complexity that was not revealed by the techniques in use.
Control of BIR and MMBIR
A major question remains—why do cells use microhomology - and not homology-driven repair? The likely answer is that Rad51 is not available or is in short supply. This might be caused by stress responses. Evidence supporting this comes from cancer research. Hypoxia in the tumor microenvironment is correlated with genetic instability [84],[85] (reviewed in [86]). It has been shown that hypoxia leads to repression of RAD51 and BRCA1 [87],[88] and to reduced homologous recombination [87],[89] (reviewed in [89],[90]). This has been interpreted as a switch from high-fidelity homologous recombination to lower fidelity NHEJ caused by stress [87],[88]. At collapsed replication forks, where NHEJ is not possible, we suggest that down-regulation of RAD51 prevents BIR from following the Rad51 - homology-dependent BIR route but still allows a Rad51-independent BIR route that requires very much less homology, as observed in telomere recombination in budding yeast [73],[74]. If Rad51 is down-regulated but not absent, a condition might prevail in which some homologous invasion is allowed, but not enough to prevent some illegitimate events occurring, as was witnessed in Drosophila with reduced gene dosage of Rad51 [91]. We do not know whether the error-prone nature of this repair is aided by down-regulation of mismatch repair, which has also been reported for stressed cancer cells [92],[93]. There might be other changes in gene expression under stress that promote genomic instability (e.g., [94]).
A similar switch from high fidelity to low-fidelity DSB repair is seen in E. coli in response to the stress of starvation [54]. Similarly the microhomology-mediated amplification seen in the Lac system in E. coli discussed above is induced by stress, as evidenced by the observation that the event occurred after the beginning of starvation [44], and by the finding that adaptive amplification in this system requires the starvation and general stress response transcriptional activator RpoS [95].
The mechanism of MMBIR, as described above, features annealing of single-stranded DNA with minimal homology. Hence the enzyme responsible for this has a central role in the proposed mechanism. We suggest that annealing is catalyzed by Rad52. Rad52 is essential for the single-strand annealing reaction that deletes sequence between direct repeats [96], and it anneals single strands in vitro [97]. Chromosomal rearrangements in yeast that have microhomology at the junctions have been seen to occur in the absence of Rad51, but they require Rad52 [42],[66],[98]. In one of these cases, frequent switches were associated with microhomology junctions in a Rad51-independent, Rad52-dependent process that produced translocations and inversions at sites of highly diverged genes [66]. These authors proposed that these events occurred by template switching during BIR [66]. In vitro, Rad51 inhibits the single-strand annealing activity of Rad52 [99], suggesting that the absence of Rad51 might exercise tight control of the switch from strand invasion to annealing of single strands. However, the formation of microhomology-mediated Rad51-independent SDs in yeast was found to be Rad52-independent [55]. Rad52 is also not required for microhomology-mediated end-joining [100]. These observations show that microhomology junction formation can be mediated by a different protein in yeast, as well as by Rad52.
In summary, we are suggesting that, because stress induces a reduction in the amount of Rad51 available, while leaving Rad52 unchanged, the amount of homologous interaction that is used for repair is reduced, leaving annealing of single DNA strands as the main mechanism available for the repair of collapsed replication forks. Thus, classical BIR will be reduced, and MMBIR will be substituted.
Long-Range Discontinuities in Duplications
The idea that there is a cell-wide physiological condition that favors nonhomologous interactions has further implications. If a condition prevails that allows one such event, it is possible that further nonhomologous events will occur in the same cell. The possibility of multiple rounds of events was suggested for a yeast system to correct for an inversion that would produce a dicentric chromosome [66]. We also note that, in human duplications, there are discontinuities (short regions that are not duplicated) and triplicated regions within duplications on a scale of hundreds of kb or Mb apart (Figures 2 and 3). These long-distance interruptions are not readily explained by template switching during the early stages of a single BIR event, where switching occurs after one template is copied for hundreds of bp to a few kb (Figure 2 and [60]), but rather suggest that more than one BIR event occurred along the same chromosome. MMBIR requires, in addition to a cell-wide stress response, a specific DNA structure: a single double-strand end. To explain why single double-strand ends should occur serially along the same chromosome, we propose that the Holliday junction formed during BIR follows the replication fork, as we have suggested above as the mechanism of separation of the extended broken end. If the replication fork formed by BIR stalls for any reason, the Holliday junction might then process through the fork, separating the newly synthesized DNA from its template, and so generating a collapsed fork anew (as in Figure 4E and 4F) and leading to the long range discontinuities seen in duplicated segments, as illustrated in Figure 3.
Chromosome Structural Consequences of MMBIR
The ways in which MMBIR would lead to the various chromosomal structural changes are summarized in Table 2. Translocations would be formed by a switch to a different chromosome. Duplication would occur when the switch was to either the sister or the homologue behind the position at which the fork collapsed (with respect to the direction of movement of the fork). Deletion happens when there is a switch to a position ahead of the fork collapse. A switch to a sequence that has already been duplicated, behind the end of the duplicated sequence, would produce a triplication. Switching to the same molecule behind the position of fork collapse has the potential to initiate rolling-circle replication and consequent amplification. Switching to either the sister molecule or the homologue in inverted orientation would give an inverted chromosomal segment. If long-distance replication follows, this might form a dicentric chromosome, so that this would have to be followed by a second inversion to allow a cell to be viable. This need for a second switch has led to the idea that there might be more than one round of switching events involved in the formation of some structural changes [66] as discussed above. Alternatively, a second inverted template switch within a single series of switches would restore a viable chromosomal structure.

Implications of the Model
We suggest that the replicative mechanism described here contributes to genomic disorders that show nonrecurrent endpoints, contributes to much of the chromosomal structural instability that occurs somatically in cancer formation and tumor progression and also to the origin of the genomic constitutional structural complexity that underlies NAHR genomic disorders, and is a driving force in evolution. We offer evidence from diverse organisms that such a mechanism exists, and suggest that the model offers directions for future research that will further elucidate the molecular details.
The mechanism of MMBIR affects human biology at many levels. First, at the cellular level, the mechanism might apply to the events underlying much cancer formation and progression. Second, at the organismal level, we propose that MMBIR acting in the germline will give rise to CNV, and the accompanying genomic disorders and chromosomal syndromes. At the same time MMBIR could create LCRs that provides the homology required for NAHR, leading to genomic disorders in future generations. Third, at the species level, we suggest that complex genomic regions generate secondary structures that increase the likelihood of MMBIR, so that complex architecture becomes more complex on an evolutionary timescale, as has been documented for primate evolution [13],[16]. We suggest that MMBIR might underlie genomic rearrangements and CNV associated with the emergence of primate-specific traits [10],[13],[101]. Furthermore, MMBIR provides material on which natural selection and evolution operate: variation in copy number might change the expression levels of included genes and also provide redundant copies of genes that could then be mutated and changed to encode new functions [102]–[104]. Further, the formation of nonhomologous junctions might shuffle exons of different genes to attain new functions (F. Zhang and J. Lupski, unpublished observations). Indeed, these regions of complex genomic architecture have been referred to as gene nurseries, i.e., regions in which new genes are formed [13],[14].
The MMBIR model predicts that complex genomic rearrangements will often be accompanied by extensive loss of heterozygosity and, in some cases, by loss of imprinting, because the chromosome that is copied might be either the sister or the homologue. Such loss of heterozygosity could lead to regional uniparental disomy [105] as a potential novel mechanism for disease. We also predict that the events described here will be seen in model systems under conditions where the cells are stressed, and study of DNA repair activities in stressed cells might be a fertile field for investigation.
Zdroje
1. IafrateAJ
FeukL
RiveraMN
ListewnikML
DonahoePK
2004 Detection of large-scale variation in the human genome. Nat Genet 36 949 951
2. KorbelJO
UrbanAE
AffourtitJP
GodwinB
GrubertF
2007 Paired-end mapping reveals extensive structural variation in the human genome. Science 318 420 426
3. SebatJ
LakshmiB
TrogeJ
AlexanderJ
YoungJ
2004 Large-scale copy number polymorphism in the human genome. Science 305 525 528
4. RedonR
IshikawaS
FitchKR
FeukL
PerryGH
2006 Global variation in copy number in the human genome. Nature 444 444 454
5. WongKK
deLeeuwRJ
DosanjhNS
KimmLR
ChengZ
2007 A comprehensive analysis of common copy-number variations in the human genome. Am J Hum Genet 80 91 104
6. KhajaR
ZhangJ
MacDonaldJR
HeY
Joseph-GeorgeAM
2006 Genome assembly comparison identifies structural variants in the human genome. Nat Genet 38 1413 1418
7. NewmanTL
TuzunE
MorrisonVA
HaydenKE
VenturaM
2005 A genome-wide survey of structural variation between human and chimpanzee. Genome Res 15 1344 1356
8. FieglerH
RedonR
AndrewsD
ScottC
AndrewsR
2006 Accurate and reliable high-throughput detection of copy number variation in the human genome. Genome Res 16 1566 1574
9. KomuraD
ShenF
IshikawaS
FitchKR
ChenW
2006 Genome-wide detection of human copy number variations using high-density DNA oligonucleotide arrays. Genome Res 16 1575 1584
10. LupskiJR
2007 Structural variation in the human genome. N Engl J Med 356 1169 1171
11. TuzunE
SharpAJ
BaileyJA
KaulR
MorrisonVA
2005 Fine-scale structural variation of the human genome. Nat Genet 37 727 732
12. BruderCEG
PoitrowskiA
GijsbersAACJ
AnderssonR
EricksonS
2008 Phenotypically concordant and discordant monozygotic twins display different DNA copy-number-variation profiles. Am J Hum Genet 82 1 9
13. DumasL
KimYH
Karimpour-FardA
CoxM
HopkinsJ
2007 Gene copy number variation spanning 60 million years of human and primate evolution. Genome Res 17 1266 1277
14. NahonJL
2003 Birth of ‘human-specific’ genes during primate evolution. Genetica 118 193 208
15. BaileyJA
EichlerEE
2006 Primate segmental duplications: crucibles of evolution, diversity and disease. Nat Rev Genet 7 552 564
16. StankiewiczP
ShawCJ
WithersM
InoueK
LupskiJR
2004 Serial segmental duplications during primate evolution result in complex human genome architecture. Genome Res 14 2209 2220
17. LupskiJR
1998 Genomic disorders: structural features of the genome can lead to DNA rearrangement and human disease traits. Trends Genet 14 417 422
18. StankiewiczP
LupskiJR
2002 Genome architecture, rearrangements and genomic disorders. Trends Genet 18 74 82
19. ShawCJ
LupskiJR
2005 Non-recurrent 17p11.2 deletions are generated by homologous and non-homologous mechanisms. Hum Genet 116 1 7
20. LupskiJR
StankiewiczP
2005 Genomic disorders: molecular mechanisms for rearrangements and conveyed phenotypes. PLoS Genet 1 e49 doi:10.1371/journal.pgen.0010049
21. LupskiJR
2006 Genome structural variation and sporadic disease traits. Nat Genet 38 974 976
22. LeeJA
CarvalhoCM
LupskiJR
2007 A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell 131 1235 1247
23. NobileCT
RizziL
SimionatiF
NigroB
CardazzoV
PatarnelloB
ValleT
DanieliG, GA
2002 Analysis of 22 deletion breakpoints in dystrophin intron 49. Hum Genet 110 418 421
24. InoueK
OsakaH
ThurstonVC
ClarkeJT
YoneyamaA
2002 Genomic rearrangements resulting in PLP1 deletion occur by nonhomologous end joining and cause different dysmyelinating phenotypes in males and females. Am J Hum Genet 71 838 853
25. LeeJA
2006 Molecular analysis of the non-recurrent genomic duplications causing Pelizaeus-Merzbacher disease and its allelic disorder paraplegia type 2. 371 [PhD thesis] Houston (Texas): Department of Molecular and Human Genetics, Baylor College of Medicine
26. PotockiLB, W
Treadwell-DeeringD
CarvalhoCM
EifertA
FriedmanEM
2007 Characterization of Potocki-Lupski syndrome (dup(17)(p11.2p11.2)) and delineation of a dosage-sensitive critical interval that can convey an autism phenotype. Am J Hum Genet 80 633 649
27. VissersLE
StankiewiczP
YatsenkoSA
CrawfordE
CreswickH
2007 Complex chromosome 17p rearrangements associated with low-copy repeats in two patients with congenital anomalies. Hum Genet 121 697 709
28. ChenJM
ChuzhanovaN
StensonPD
FérecC
CooperDN
2005 Intrachromosomal serial replication slippage in trans gives rise to diverse genomic rearrangements involving inversions. Hum Mutat 26 362 373
29. FérecC
CasalsT
ChuzhanovaN
MacekMJ
BienvenuT
2006 Gross genomic rearrangements involving deletions in the CFTR gene: characterization of six new events from a large cohort of hitherto unidentified cystic fibrosis chromosomes and meta-analysis of the underlying mechanisms. Eur J Hum Genet 14 562 567
30. del GaudioD
FangP
ScagliaF
WardPA
CraigenWJ
2006 Increased MECP2 gene copy number as the result of genomic duplication in neurodevelopmentally delayed males. Genet Med 8 784 792
31. SheenCR
JewellUR
MorrisCM
BrennanSO
FérecC
2007 Double complex mutations involving F8 and FUNDC2 caused by distinct break-induced replication. Hum Mutat 28 1198 2006
32. StankiewiczP
ShawCJ
DapperJD
WakuiK
ShafferLG
2003 Genome architecture catalyzes nonrecurrent chromosomal rearrangements. Am J Hum Genet 72 1101 1116
33. LeeJA
InoueK
CheungSW
ShawCA
StankiewiczP
2006 Role of genomic architecture in PLP1 duplication causing Pelizaeus-Merzbacher disease. Hum Mol Genet 15 2250 2265
34. LeeJA
MadridRE
SperleK
RittersonCM
HobsonGM
2006 Spastic paraplegia type 2 associated with axonal neuropathy and apparent PLP1 position effect. Ann Neurol 59 398 403
35. SlackA
ThorntonPC
MagnerDB
RosenbergSM
HastingsPJ
2006 On the mechanism of gene amplification induced under stress in Escherichia coli. PLoS Genet 2 e48 doi:10.1371/journal.pgen.0020048
36. VolikS
RaphaelBJ
HuangG
StrattonMR
BignelG
2006 Decoding the fine-scale structure of a breast cancer genome and transcriptome. Genome Res 16 394 404
37. BignellGR
SantariusT
PoleJC
ButlerAP
PerryJ
2007 Architectures of somatic genomic rearrangement in human cancer amplicons at sequence-level resolution. Genome Res 17 1296 1303
38. CanningS
DryjaTP
1989 Short, direct repeats at the breakpoints of deletions of the retinoblastoma gene. Proc Natl Acad Sci U S A 86 5044 5048
39. KohnoT
YokataJ
2006 Molecular processes of chromosome 9p21 deletions causing inactivation of the p16 tumor suppressor gene in human cancer: deduction from structural analysis of breakpoints for deletions. DNA Repair (Amst) 5 1273 1281
40. ZhangY
Zeleznik-LeN
EmmanuelN
JayathilakaN
ChenJ
2004 Characterization of genomic breakpoints in MLL and CBP in leukemia patients with t(11;16). Genes Chromosomes Cancer 41 257 265
41. ZhangY
StrisselP
StrickR
ChenJ
NuciforaG
2004 Genomic DNA breakpoints in AML1/RUNX1 and ETO cluster with topoisomerase II DNA cleavage and DNase I hypersensitive sites in t(8;21) leukemia. Proc Natl Acad Sci U S A 99 3070 3075
42. ChenC
UmezuK
KolodnerRD
1998 Chromosomal rearrangements occur in S. cerevisiae rfa1 mutator mutants due to mutagenic lesions processed by double-strand-break repair. Mol Cell 2 9 22
43. CairnsJ
FosterPL
1991 Adaptive reversion of a frameshift mutation in Escherichia coli. Genetics 128 695 701
44. HastingsPJ
BullHJ
KlumpJR
RosenbergSM
2000 Adaptive amplification: an inducible chromosomal instability mechanism. Cell 103 723 731
45. HastingsPJ
SlackA
PetrosinoJF
RosenbergSM
2004 Adaptive amplification and point mutation are independent mechanisms: Evidence for various stress-inducible mutation mechanisms. PLoS Biol 2 e399 doi:10.1371/journal.pbio.0020399
46. KugelbergE
KofoidE
ReamsAB
AnderssonDI
RothJR
2006 Multiple pathways of selected gene amplification during adaptive mutation. Proc Natl Acad Sci U S A 103 17319 17324
47. HastingsPJ
2007 Adaptive amplification. Critical Rev Biochem Mol Biol 42 1 13
48. FriedbergEC
WalkerGC
SiedeW
WoodRD
SchultzRA
2005 DNA Repair and Mutagenesis Washington (DC) ASM Press 1164
49. IkedaH
ShimizuH
UkitaT
KumagaiM
1995 A novel assay for illegitimate recombination in Escherichia coli: stimulation of lambda bio transducing phage formation by ultra-violet light and its independence from RecA function. Adv Biophys 31 197 208
50. AlbertiniAM
HoferM
CalosMP
MillerJH
1982 On the formation of spontaneous deletions: the importance of short sequence homologies in the generation of large deletions. Cell 29 319 328
51. FarabaughPJ
SchmeissnerU
HoferM
MillerJH
1978 Genetic studies of the lac repressor. VII. On the molecular nature of spontaneous hotspots in the lacI gene of Escherichia coli. J Mol Biol 126 847 857
52. ShimizuH
YamaguchiH
AshizawaY
KohnoY
AsamiM
1997 Short-homology-independent illegitimate recombination in Escherichia coli: distinct mechanism from short-homology-dependent illegitimate recombination. J Mol Biol 266 297 305
53. BzymekM
LovettST
2001 Instability of repetitive DNA sequences: the role of replication in multiple mechanisms. Proc Natl Acad Sci U S A 98 8319 8325
54. PonderRG
FonvilleNC
RosenbergSM
2005 A switch from high-fidelity to error-prone DNA double-strand break repair underlies stress-induced mutation. Mol Cell 19 791 804
55. PayenC
KoszulR
DujonB
FischerG
2008 Segmental duplications arise from Pol32-dependent repair of broken forks through two alternative replication-based mechanisms. PLoS Genet 4 e1000175 doi:10.1371/journal.pgen.1000175
56. BranzeiD
FoianiM
2007 Template Switching: From Replication Fork Repair to Genome Rearrangements. Cell 131 1228 1230
57. MerrihewRV
MarburgerK
PenningtonSL
RothDB
WilsonJH
1996 High-frequency illegitimate integration of transfected DNA at preintegrated target sites in a mammalian genome. Mol Cell Biol 16 10 18
58. MorrowDM
ConnellyC
HieterP
1997 “Break-copy” duplication: a model for chromosome fragment formation in Saccharomyces cerevisiae. Genetics 147 371 382
59. McEachernMJ
HaberJE
2006 Break-Induced Replication and Recombinational Telomere Elongation in Yeast. Annu Rev Biochem 75 111 135
60. SmithCE
LlorenteB
SymingtonLS
2007 Template switching during break-induced replication. Nature 447 102 105
61. LydeardJR
JainS
YamaguchiM
HaberJE
2007 Break-induced replication and telomerase-independent telomere maintenance require Pol32. Nature 448 820 823
62. MotamediM
SzigetySK
RosenbergSM
1999 Double-strand-break repair in Escherichia coli: physical evidence for a DNA replication mechanism in vivo. Genes Dev 13 2889 2903
63. LlorenteB
SmithCE
SymingtonLS
2008 Break-induced replication: what is it and what is it for? Cell Cycle 7 859 864
64. HeiterP
MannC
SnyderM
DavisRW
1985 Mitotic stability of yeast chromosomes: A colony color assay that measures nondisjunction and chromosome loss. Cell 40 381 392
65. DeemA
BarkerK
VanhulleK
DowningB
VaylA
2008 Defective break-induced replication leads to half-crossovers in Saccharomyces cerevisiae. Genetics 179 1845 1860
66. SchmidtKH
WuJ
KolodnerRD
2006 Control of translocations between highly diverged genes by Sgs1, the Saccharomyces cerevisiae homolog of the Bloom's syndrome protein. Mol Cell Biol 26 5406 5420
67. BautersM
Van EschH
FriezMJ
Boespflug-TanguyO
ZenkerM
2008 Nonrecurrent MECP2 duplications mediated by genomic architecture-driven DNA breaks and break-induced replication repair. Genome Res 18 847 858
68. LovettST
HurleyRL
SuteraVAJr
AubuchonRH
LebedevaMA
2002 Crossing over between regions of limited homology in Escherichia coli. RecA-dependent and RecA-independent pathways. Genetics 160 851 859
69. LiskayRM
LetsouA
StachelekJL
1987 Homology requirement for efficient gene conversion between duplicated chromosomal sequences in mammalian cells. Genetics 115 161 167
70. ReiterLT
HastingsPJ
NelisE
De JongheP
Van BroeckhovenC
1998 Human meiotic recombination products revealed by sequencing a hotspot for homologous strand exchange in multiple HNPP deletion patients. Am J Hum Genet 62 1023 1033
71. VanHulleK
LemoineFJ
NarayananV
DowningB
HullK
2007 Inverted DNA repeats channel repair of distant double-strand breaks into chromatid fusions and chromosomal rearrangements. Mol Cell Biol 27 2601 2614
72. DavisAP
SymingtonLS
2004 RAD51-dependent break-induced replication in yeast. Mol Cell Biol 24 2344 2351
73. LeS
MooreJK
HaberJE
GreiderCW
1999 RAD50 and RAD51 define two pathways that collaborate to maintain telomeres in the absence of telomerase. Genetics 152 143 152
74. TengSC
ZakianVA
1999 Telomere-telomere recombination is an efficient bypass pathway for telomere maintenance in Saccharomyces cerevisiae. Mol Cell Biol 19 8083 8093
75. BentleyJ
DiggleCP
HarndenP
KnowlesMA
KiltieAE
2004 DNA double strand break repair in human bladder cancer is error prone and involves microhomology-associated end-joining. Nucleic Acids Res 32 5249 5259
76. CorneoB
WendlandRL
DerianoL
CuiX
KleinIA
2007 Rag mutations reveal robust alternative end joining. Nature 449 483 486
77. LisbyM
BarlowJH
BurgessRC
RothsteinR
2004 Choreography of the DNA damage response: spatiotemporal relationships among checkpoint and repair proteins. Cell 118 699 713
78. PenningtonJM
RosenbergSM
2007 Spontaneous DNA breakage in single living cells of Escherichia coli. Nat Gen 39 797 802
79. Saleh-GohariN
BryantHE
SchultzN
ParkerKM
CasselTN
2005 Spontaneous homologous recombination is induced by collapsed replication forks that are caused by endogenous DNA single-strand breaks. Mol Cell Biol 25 7158 7169
80. McIlwraithMJ
VaismanA
LiuY
FanningE
WoodgateR
2005 Human DNA polymerase eta promotes DNA synthesis from strand invasion intermediates of homologous recombination. Mol Cell 20 783 792
81. KawamotoT
ArakiK
SonodaE
YamashitaYM
HaradaK
2005 Dual roles for DNA polymerase eta in homologous DNA recombination and translesion DNA synthesis. Mol Cell 20 793 799
82. CannistraroVJ
TaylorJS
2007 Ability of polymerase eta and T7 DNA polymerase to bypass bulge structures. J Biol Chem 282 11188 11196
83. RothDB
ChangXB
WilsonJH
1989 Comparison of filler DNA at immune, nonimmune, and oncogenic rearrangements suggests multiple mechanisms of formation. Mol Cell Biol 9 3049 3057
84. YoungSD
MarshallRS
HillRP
1988 Hypoxia induces DNA overreplication and enhances metastatic potential of murine tumor cells. Proc Natl Acad Sci U S A 85 9533 9537
85. CoquelleA
ToledoF
SternS
BiethA
DebatisseM
1998 A new role for hypoxia in tumor progression: induction of fragile site triggering genomic rearrangements and formation of complex DMs and HSRs. Mol Cell 2 259 265
86. SubarskyP
HillRP
2003 The hypoxic tumour microenvironment and metastatic progression. Clin Exp Metastasis 20 237 250
87. BindraRSS
ChafferPJ
MengA
WooJ
MåseideK
2004 Down-regulation of Rad51 and decreased homologous recombination in hypoxic cancer cells. Mol Cell Biol 24 8504 8518
88. BindraRS
GlazerPM
2007 Repression of RAD51 gene expression by E2F4/p130 complexes in hypoxia. Oncogene 26 2048 2057
89. HuangLE
BindraRS
GlazerPM
HarrisAL
2007 Hypoxia-induced genetic instability–a calculated mechanism underlying tumor progression. J Mol Med 85 139 148
90. BindraRS
CrosbyME
GlazerPM
2007 Regulation of DNA repair in hypoxic cancer cells. Cancer Metastasis Rev 26 249 260
91. McVeyM
AdamsM
Staeva-VieiraE
SekelskyJJ
2004 Evidence for multiple cycles of strand invasion during repair of double-strand gaps in Drosophila. Genetics 167 699 705
92. BindraRS
GlazerPM
2007 Co-repression of mismatch repair gene expression by hypoxia in cancer cells: role of the Myc/Max network. Cancer Lett 252 93 103
93. MihaylovaVT
BindraRS
YuanJ
CampisiD
NarayananL
2003 Decreased expression of the DNA mismatch repair gene Mlh1 under hypoxic stress in mammalian cells. Mol Cell Biol 23 3265 3273
94. MyungK
ChenC
KolodnerRD
2001 Multiple pathways cooperate in the suppression of genome instability in Saccharomyces cerevisiae. Nature 411 1073 1076
95. LombardoM-J
AponyiI
RosenbergSM
2004 General stress response regulator RpoS in adaptive mutation and amplification in Escherichia coli. Genetics 166 669 680
96. Fishman-LobellJ
HaberJE
1992 Removal of nonhomologous DNA ends in double-strand break recombination: the role of the yeast ultraviolet repair gene RAD1. Science 258 480 484
97. MortensenUH
BendixenHC
SunjevaricI
RothsteinR
1996 DNA strand annealing is promoted by yeast Rad52 protein. Proc Natl Acad Sci U S A 93 10729 10734
98. TsukamotoY
KatoJ
IkedaH
1996 Effects of mutations of RAD50, RAD51, RAD52, and related genes on illegitimate recombination in Saccharomyces cerevisiae. Genetics 142 383 391
99. WuY
KantakeN
SugiyamaT
KowalczykowskiSC
2008 Rad51 protein controls Rad52-mediated DNA annealing. J Biol Chem 283 14883 14892
100. LeeK
LeeSE
2007 Saccharomyces cerevisiae Sae2 - and Tel1-dependent single-strand DNA formation at DNA break promotes microhomology-mediated end joining. Genetics 176 2003 2014
101. LupskiJR
2007 An evolution revolution provides further revelation. Bioessays 29 1182 1184
102. OhnoS
1970 Evolution by gene duplication Berlin, New York Springer-Verlag 160
103. HurlesM
2004 Gene duplication: the genomic trade in spare parts. PLoS Biol 2 e206 doi:10.1371/journal.pbio.0020206
104. HittingerCT
CarrollSB
2007 Gene duplication and the adaptive evolution of a classic genetic switch. Nature 449 677 681
105. SpenceJE
PerciaccanteRG
GreigGM
WillardHF
LedbetterDH
1988 Uniparental disomy as a mechanism for human genetic disease. Am J Hum Genet 42 217 226
106. LeeJA
LupskiJR
2006 Genomic rearrangements and gene copy-number alterations as a cause of nervous system disorders. Neuron 52 103 121
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2009 Číslo 1
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- A -Acting Diversification Activator Both Necessary and Sufficient for AID-Mediated Hypermutation
- Order and Disorder during Divergence
- Mouse Genome-Wide Association Mapping Needs Linkage Analysis to Avoid False-Positive Loci
- Why Is the Correlation between Gene Importance and Gene Evolutionary Rate So Weak?
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy