
A Drastic Reduction in the Life Span of Cystatin C L68Q Carriers Due to Life-Style Changes during the Last Two Centuries
Hereditary cystatin C amyloid angiopathy (HCCAA) is an autosomal dominant disease with high penetrance, manifest by brain hemorrhages in young normotensive adults. In Iceland, this condition is caused by the L68Q mutation in the cystatin C gene, with contemporary carriers reaching an average age of only 30 years. Here, we report, based both on linkage disequilibrium and genealogical evidence, that all known copies of this mutation derive from a common ancestor born roughly 18 generations ago. Intriguingly, the genealogies reveal that obligate L68Q carriers born 1825 to 1900 experienced a drastic reduction in life span, from 65 years to the present-day average. At the same time, a parent-of-origin effect emerged, whereby maternal inheritance of the mutation was associated with a 9 year reduction in life span relative to paternal inheritance. As these trends can be observed in several different extended families, many generations after the mutational event, it seems likely that some environmental factor is responsible, perhaps linked to radical changes in the life-style of Icelanders during this period. A mutation with such radically different phenotypic effects in reaction to normal variation in human life-style not only opens the possibility of preventive strategies for HCCAA, but it may also provide novel insights into the complex relationship between genotype and environment in human disease.
Published in the journal:
. PLoS Genet 4(6): e32767. doi:10.1371/journal.pgen.1000099
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1000099
Summary
Hereditary cystatin C amyloid angiopathy (HCCAA) is an autosomal dominant disease with high penetrance, manifest by brain hemorrhages in young normotensive adults. In Iceland, this condition is caused by the L68Q mutation in the cystatin C gene, with contemporary carriers reaching an average age of only 30 years. Here, we report, based both on linkage disequilibrium and genealogical evidence, that all known copies of this mutation derive from a common ancestor born roughly 18 generations ago. Intriguingly, the genealogies reveal that obligate L68Q carriers born 1825 to 1900 experienced a drastic reduction in life span, from 65 years to the present-day average. At the same time, a parent-of-origin effect emerged, whereby maternal inheritance of the mutation was associated with a 9 year reduction in life span relative to paternal inheritance. As these trends can be observed in several different extended families, many generations after the mutational event, it seems likely that some environmental factor is responsible, perhaps linked to radical changes in the life-style of Icelanders during this period. A mutation with such radically different phenotypic effects in reaction to normal variation in human life-style not only opens the possibility of preventive strategies for HCCAA, but it may also provide novel insights into the complex relationship between genotype and environment in human disease.
Introduction
Amyloid deposits are found in various diseases, both genetic and sporadic, such as Alzheimer's disease and the prionoses [1]. HCCAA (MIM #105150) has so far only been found in Iceland and it was the first to be described of a group of diseases called hereditary cerebral amyloid angiopathies, characterized by amyloid deposition in brain arteries [2],[3]. The cystatin C gene encodes an extracellular proteinase inhibitor with activity against cysteine proteases of the papain and legumain family [4]. The cystatin C L68Q mutation [5],[6] is highly penetrant and the disease is manifest by an intracerebral hemorrhage (ICH) in young normotensive adults. The ICH is recurrent if the patient survives the first attack and then commonly associated with dementia of variable severity and/or paralysis, leading to death at 30.7 years of age, on average (based on 130 individuals born after 1900 with life spans ranging from 15 to 79 years). Cystatin C is the predominant component [7] of the amyloid which is found in the cerebral arterioles of patients [8]. In addition, wild-type cystatin C is found in amyloid deposits with the Aβ peptide in Alzheimer's patients, modulating cerebral β-amylodosis [9]–[11]. Furthermore, the cystatin C locus (CST3) is one of the candidate susceptibility loci for sporadic Alzheimer's disease [12].
In the present study we collected data on patients diagnosed with HCCAA, traced their family trees and found a progressive reduction in the life span of patients and obigate carriers of the disease mutation in addition to a parent-of-origin effect.
Results
Families
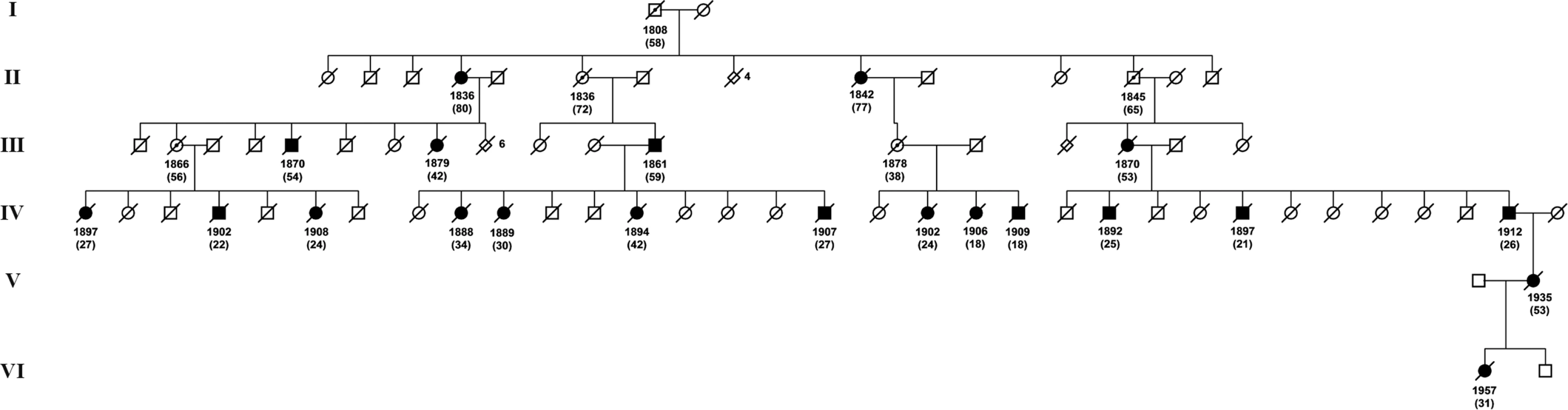
Overall, 15 families were identified with 266 known or inferred carriers of the L68Q mutation. Figure 1 depicts the geographical distribution of all 15 families around the year 1800 and Figure 2 shows an example pedigree for one extended family.

Age of the Mutation
The mean uncorrected single marker estimate was 13.9 generations, which increased to 21.5 after applying the Luria-Delbrück correction for population growth rate (Table S1). Both means are affected by several extreme values obtained for microsatellites where founder alleles are still in very strong LD with the L68Q mutation.
An analysis of all 20 microsatellites simultaneously, using the DMLE+ software, yielded a single age estimate of 17.9 generations (95% C.I. 10.9–28.5). This suggests that the mutation occurred on a chromosome carried by an individual born around the mid 16th century, presumably in Iceland.
Earliest Ancestor
We could not find the common ancestor of all the families using genealogy due to lack of data. However, the earliest known common ancestor was a man, born in 1684 in region B, who moved and founded the two Southern families in region C (see Figure 1).
Studies of Life Span
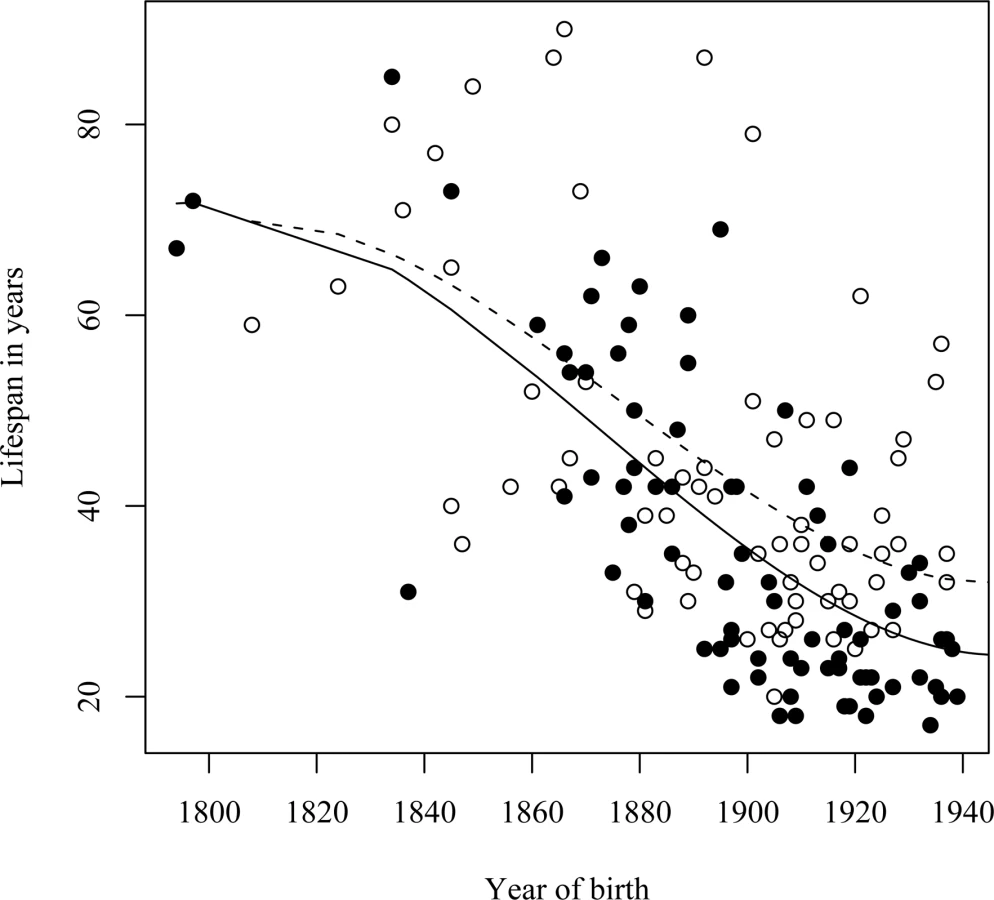
As premature death is such a striking phenotypic feature of the L68Q mutation in confirmed carriers, we next attempted to determine whether their ancestors carrying the mutation also had reduced life span. First, we examined only the 157 confirmed carriers, using their spouses as controls with the rationale that they originated from the same regions [13], show a similar distribution of birth years and led similar lifestyles as the mutation carriers. The dependence of life span on the year of birth was studied by linear regression models including polynomial terms. This analysis revealed that the life span of L68Q carriers, both men and women, underwent a significant reduction (P≤2.2*10−16) during the 19th century (Figure 3A and Table 1). This life span reduction is evident in the pedigrees, an example of which is shown in Figure 2. In comparison, we found no change in the life span of the spouses (n = 84) during this period (b = 0.093, P≤0.1232, Figure 3B). The reduction in life span of L68Q carriers, compared to controls, became evident in individuals born 1825 and after, following a sigmoid curve showing a continuous decrease that leveled out at the present average at the beginning of the 20th century (P≤3.3*10−13, Figure 3C).


A similar reduction in life span was seen in a separate analysis of the assumed carrier group (Figure 4A), whose spouses (n = 31) had a slight but insignificant increase in life span (Figure 4B), thus supporting the notion that those six families were also bona fide HCCAA families. However, due to increasing early death of carriers in the 20th century the L68Q mutation has now become extinct in these families and in five of the nine families where carrier status could be confirmed with genotyping.

Parent-of-Origin Effect
We observed a parent-of-origin effect on the life span of L68Q carriers (Table 1) that is most notable for people born after 1900 (Figure 5). The data indicate that carriers who inherited the L68Q mutation from their mother (n = 53; 29 sons and 24 daughters) lived 27 years on average (standard deviation (S.D.) = 7.78), whereas those who inherited the mutation from their father (n = 51; 23 sons and 28 daughters) lived 36.4 years on average (S.D. = 11.44). This 9.4 year difference in life span is highly significant (t-test, P≤0.001). It should be noted that of the handful of mutation carriers born after 1900 (1–2%) who achieved a “normal” life span [14], all inherited the mutation from their father.
Geographic Difference in Life Span
A significant difference (P≤0.0016) was seen in the timing of life span reduction between carriers from the remote Northwest coastal region A and those from the South and West regions B and C combined (Figures 1 and 6, and Table 1). The life span reduction in region A was delayed by about 20 years compared to the other regions.

Discussion
In this study of HCCAA families with the cystatin C L68Q mutation we show that the life span of L68Q carriers in all the families underwent a reduction in the 19th century reaching the present day average of 30 years around 1900. Concurrently a parent-of-origin effect became evident whereby those who inherited the disease gene from the mother had, on average, a 9.4 years shorter life span compared to those who inherited it from the father.
How can we account for the rather drastic reduction in the life span of L68Q carriers that seems to have occurred three centuries after the mutation arose? It is unlikely that this is due to an additional, but currently undocumented, mutation acting as a modifier of the risk conferred by the L68Q mutation. Indeed, an additional mutation would need to have occurred many times in several different extended families. Alternatively, and even less plausible, a putative modifier mutation would have needed to spread from very low frequency to near fixation in each of the L68Q carrier families in the space of a few generations. Rather, we postulate the involvement of some environmental factor that came into play around 1825 and reached saturation around 1900, possibly a dietary factor such as the increased consumption of carbohydrates (Figure 7) or salt (for food preservation). The notion of an environmental modifier of disease risk conferred by the L68Q mutation is supported by the observation that the decrease in life span was delayed by about 20 years in the remote Northwest coastal region A compared to the West and South regions B and C combined. Thus, the life-style changes associated with economic development in Iceland during the 19th century occurred later in remote regions than in regions closer to the capital Reykjavik.

It is interesting that in the same time period (the 19th century) a parent-of-origin effect of the L68Q mutation is seen in the context of the life span reduction of carriers. This could be explained by a transgenerational epigenetic mechanism [15], such as the one described for the agouti viable yellow mutation in mice [16],[17] where the frequency of disease in offspring can be changed with dietary methyl supplementation of pregnant dams. However, although cystatin C has a large CpG region in the promoter, the gene is not known to be a metastable epiallele or reside in a known imprinting region.
The conclusion that environmental factors alter the phenotypic impact of mutations underlying serious heritable disorders is not novel. A classic example is the dietary treatment of phenylketouria (PKU) patients to prevent cognitive damage [18]. What is unusual about the findings reported here is rather that the detrimental phenotypic impact of the L68Q mutation appears to have emerged in reaction to life-style changes that fall within the normal range of behavior of a single population in the space of a few generations. Even this may not be without parallel, as a reduced life span over time coupled with a maternal effect has also been described in another familial amyloid disease in Sweden, i.e. FAP (familial amyloidotic polyneuropathy) which is caused by a mutation (V30M) in the transthyretin gene [19]. Such diseases challenge simplistic views of Mendelian diseases as solely genetic in nature and insensitive to environmental factors. An understanding of the phenotypic flexibility of the L68Q mutation might provide the possibility for preventive or therapeutic strategies to deal with HCCAA. Such an understanding may also be relevant to other common but pathophysiologically related disorders such as Alzheimer's disease, where cerebral amyloid angiopathy (CAA) is very common. Finally it may also present an opportunity for more general insights into the complex relationship between genotype and environment in human disease.
Materials and Methods
Family Data
A total of 36 individuals from nine families with known histories of ICH were diagnosed as carriers of the L68Q mutation through direct genotyping [6]. The ancestors of these individuals were found by tracing pedigrees, either with the help of Arnason [3], published pedigrees [20], or with the deCODE Genetics genealogy database that contains information about the relationships between 740.000 Icelanders, past and present [13]. Ancestors who were obligate carriers of L68Q were defined as parents or common ancestors of known carriers whose inferred carrier status minimizes the number of transmission events of the mutation in the genealogies. For most individuals, the only available phenotypic information was age at death, as the first signs of HCCAA can be subtle such as personality changes and dementia. Death certificates (first issued in Iceland in 1911) and parish records were also checked and the cause of death was noted when known. The year of birth, and death, of spouses of obligate L68Q carriers were noted when available.
The nine extended families thus defined contain 202 individuals, including 92 ancestors who are obligate carriers. Although the cause of death for these obligate carriers was often consistent with ICH, we also included those who died from other causes such as drowning or infections. In some cases, family members with no offspring or contemporary descendants were defined as obligate carriers, when their death certificate (n = 61) or their parish record (n = 17) indicated brain hemorrhage. Individuals were excluded if no information was available about cause of death. Limiting the analyses to all carriers born before 1940 (to exclude those who have an exceptionally long life span) we hereafter refer to 157 carriers as the confirmed carrier group.
In addition to the aforementioned known and obligate carriers, there are 34 deceased individuals who are strongly suspected of having been carriers of the L68Q mutation, but for whom no DNA samples were available. The inference of carrier status for these individuals is based on death certificates (n = 17) and parish records (n = 17) that indicate brain hemorrhage as the cause of death. Using the same approach as before to trace obligate carrier ancestors, we identified six new families and 30 new obligate carriers. We refer to this set of 64 individuals as the assumed carrier group.
Age of the Mutation
In order to gain insight into the history of the cystatin C L68Q mutation in the Icelandic gene pool, we estimated its age by examining the decay of linkage disequilibrium (LD) between it and 20 surrounding microsatellites in the 36 known carriers and 722 non-carrier controls. The microsatellites spanned the physical map positions 14.7–38.3 Mb (NCBI build 35) on chromosome 20, with the L68Q mutation located at position 23,563,968 (see Table S1).
PCR amplifications were set up, run, and pooled on Gilson Cyberlab robots. The reaction volume was 5 µl, and, for each PCR, 20 ng of genomic DNA was amplified in the presence of 2 pmol of each primer, 0.25 U AmpliTaq Gold, 0.2 mM dNTPs, and 2.5 mM MgCl2 (buffer was supplied by the manufacturer, Applera). Cycling conditions were as follows: 95°C for 10 min, followed by 37 cycles of 94°C for 15 s, annealing for 30 s at 55°C, and 1 min extension at 72°C. One primer of each primer pair was fluorescently labeled. The PCR products were pooled into panels of 8–16 markers, mixed with size standards, and analyzed on ABI 3700 sequencing machines using Genescan (version 3.0) peak-calling software (Applied Biosystems, Foster City, CA). Alleles were automatically called using DAC, an allele-calling program developed at deCODE Genetics [21], and the program DecodeGT was used to fractionate called genotypes, according to quality, and to edit when necessary [22].
First, we established the microsatellite allele states of the chromosome on which the L68Q mutation is most likely to have occurred, referred to hereafter as the founder alleles. This was achieved by picking the microsatellite allele that exhibited the strongest LD (measured by D') with the cystatin C mutation and showed the greatest difference in frequency between the mutation carriers and controls. Haplotype frequencies were estimated using the expectation maximization (EM) algorithm [23]. Age estimates of the mutation were then obtained based on the pattern of LD with each individual microsatellite based on the formula:where t is the age of the mutation in generations, θ is the recombination rate in Morgans between the mutation and the flanking microsatellite, x(t) is the frequency of chromosomes carrying both the cystatin C mutation and the microsatellite ancestral allele and y is the frequency of chromosomes that do not carry the cystatin C mutation but do carry the microsatellite ancestral allele [24]. Age estimates based on individual microsatellites were also calculated using a correction, based on the Luria-Delbrück model, that takes into account the impact of population growth [25]. The corrected age is defined as:using the same notation as before with additional parameters, t, the time estimate based on the previous formula, and r, the rate of past exponential population growth per generation. Based on detailed information about the size of the Icelandic population during the past 250 years [26], we estimated r to have been 0.3. The recombination rates between loci were obtained by interpolating positions from the high-resolution recombination map estimated from the phase II HapMap project data, using physical map locations as the common frame of reference (www.hapmap.org) [27].
As age estimates based on individual loci can be subject to considerable measurement error, and are unreliable between alleles that are still in very strong LD, we also employed the method implemented in the DMLE+ software version 2.3 [28]. Using this approach we were able to use the LD pattern of all 20 microsatellites simultaneously with the L68Q mutation to provide a single age estimate. The DMLE+ software was run using 100,000 burn-in iterations and 100,000 calculation iterations, with r = 0.3.
Web Resources
The URLs for data presented herein are as follows: Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/sites/omim.
Supporting Information
Zdroje
1. BarnhamKJCappaiRBeyreutherKMastersCLHillAF 2006 Delineating common molecular mechanisms in Alzheimer's and prion diseases. Trends Biochem Sci 31 465 472
2. YamadaM 2004 Cerebral amyloid angiopathy and gene polymorphisms. J Neurol Sci 226 41 44
3. ArnasonA 1935 Apoplexie und ihre vererbung. Acta Psychiat Neruol Suppl VII
4. GrubbAO 2000 Cystatin C–properties and use as diagnostic marker. Adv Clin Chem 35 63 99
5. PalsdottirAAbrahamsonMThorsteinssonLArnasonAOlafssonI 1988 Mutation in cystatin C gene causes hereditary brain haemorrhage. Lancet 2 603 604
6. AbrahamsonMJonsdottirSOlafssonIJenssonOGrubbA 1992 Hereditary cystatin C amyloid angiopathy: identification of the disease-causing mutation and specific diagnosis by polymerase chain reaction based analysis. Hum Genet 89 377 380
7. LofbergHGrubbAONilssonEKJenssonOGudmundssonG 1987 Immunohistochemical characterization of the amyloid deposits and quantitation of pertinent cerebrospinal fluid proteins in hereditary cerebral hemorrhage with amyloidosis. Stroke 18 431 440
8. GudmundssonGHallgrimssonJJonassonTABjarnasonO 1972 Hereditary cerebral haemorrhage with amyloidosis. Brain 95 387 404
9. LevyESastreMKumarAGalloGPiccardoP 2001 Codeposition of cystatin C with amyloid-beta protein in the brain of Alzheimer disease patients. J Neuropathol Exp Neurol 60 94 104
10. KaeserSAHerzigMCCoomaraswamyJKilgerESelenicaML 2007 Cystatin C modulates cerebral beta-amyloidosis. Nat Genet 39 1437 1439
11. MiWPawlikMSastreMJungSSRadvinskyDS 2007 Cystatin C inhibits amyloid-beta deposition in Alzheimer's disease mouse models. Nat Genet 39 1440 1442
12. BertramLMcQueenMBMullinKBlackerDTanziRE 2007 Systematic meta-analyses of Alzheimer disease genetic association studies: the AlzGene database. Nat Genet 39 17 23
13. HelgasonAYngvadottirBHrafnkelssonBGulcherJStefanssonK 2005 An Icelandic example of the impact of population structure on association studies. Nat Genet 37 90 95
14. SveinbjornsdottirSBlondalHGudmundssonGKjartanssonOJonsdottirS 1996 Progressive dementia and leucoencephalopathy as the initial presentation of late onset hereditary cystatin-C amyloidosis. Clinicopathological presentation of two cases. J Neurol Sci 140 101 108
15. MorganHDSutherlandHGMartinDIWhitelawE 1999 Epigenetic inheritance at the agouti locus in the mouse. Nat Genet 23 314 318
16. WolffGLKodellRLMooreSRCooneyCA 1998 Maternal epigenetics and methyl supplements affect agouti gene expression in Avy/a mice. Faseb J 12 949 957
17. WaterlandRAJirtleRL 2003 Transposable elements: targets for early nutritional effects on epigenetic gene regulation. Mol Cell Biol 23 5293 5300
18. ScriverCR 2007 The PAH gene, phenylketonuria, and a paradigm shift. Hum Mutat 28 831 845
19. DruggeUAnderssonRChizariFDanielssonMHolmgrenG 1993 Familial amyloidotic polyneuropathy in Sweden: a pedigree analysis. J Med Genet 30 388 392
20. JenssonOGudmundssonGArnasonABlondalHPetursdottirI 1987 Hereditary cystatin C (gamma-trace) amyloid angiopathy of the CNS causing cerebral hemorrhage. Acta Neurol Scand 76 102 114
21. FjalldalJBenediktssonKSigurdssonJEllingsenL 2001 Automated genotyping: combining neural networks and decision trees to perform robust allele calling.
22. PalssonBPalssonFPerlinMGudbjartssonHStefanssonK 1999 Using quality measures to facilitate allele calling in high-throughput genotyping. Genome Res 9 1002 1012
23. ExcoffierLSlatkinM 1995 Maximum-likelihood estimation of molecular haplotype frequencies in a diploid population. Mol Biol Evol 12 921 927
24. SlatkinMRannalaB 2000 Estimating allele age. Annu Rev Genomics Hum Genet 1 225 249
25. LabudaMLabudaDKorab-LaskowskaMColeDEZietkiewiczE 1996 Linkage disequilibrium analysis in young populations: pseudo-vitamin D-deficiency rickets and the founder effect in French Canadians. Am J Hum Genet 59 633 643
26. JonssonGMagnussonM 1997 Hagskinna: Icelandic historical statistics Reykjavik Statistics Iceland
27. 2005 A haplotype map of the human genome. Nature 437 1299 1320
28. ReeveJPRannalaB 2002 DMLE+: Bayesian linkage disequilibrium gene mapping. Bioinformatics 18 894 895
29. JonssonG 1998 Changes in Food Consumption in Iceland 1770–1940. Scandinavian Economic History Review XLVI 24 41
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2008 Číslo 6
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Evaluating Statistical Methods Using Plasmode Data Sets in the Age of Massive Public Databases: An Illustration Using False Discovery Rates
- A Drastic Reduction in the Life Span of Cystatin C L68Q Carriers Due to Life-Style Changes during the Last Two Centuries
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy