
Genová terapie dědičných onemocnění SÍTNICE A ZRAKOVÉHO NERVU: současný stav poznání
Gene Therapy for Inherited RETINAL AND OPTIC NERVE Disorders: Current Knowledge
The aim of this review is to provide a comprehensive summary of current gene therapy clinical trials for monogenic and optic nerve disorders.
The number of genes for which gene-based therapies are being developed is growing. At the time of writing this review gene-based clinical trials have been registered for Leber congenital amaurosis 2 (LCA2), retinitis pigmentosa 38, Usher syndrome 1B, Stargardt disease, choroideremia, achromatopsia, Leber hereditary optic neuropathy (LHON) and X-linked retinoschisis. Apart from RPE65 gene therapy for LCA2 and MT-ND4 for LHON which has reached phase III, all other trials are in investigation phase I and II, i.e. testing the efficacy and safety.
Because of the relatively easy accessibility of the retina and its ease of visualization which allows monitoring of efficacy, gene-based therapies for inherited retinal disorders represent a very promising treatment option. With the development of novel therapeutic approaches, the importance of establishing not only clinical but also molecular genetic diagnosis is obvious.
Key words:
gene therapy, monogenic retinal diseases, optic nerve atrophy, mitochondrial disease
Autoři:
Ľ. Ďuďáková 1*; B. Kousal 1,2*; H. Kolářová 1; L. Hlavatá 1,3; P. Lišková 1,2
Působiště autorů:
Ústav dědičných metabolických poruch, 1. lékařská fakulta, Univerzita Karlova v Praze a Všeobecná fakultní nemocnice, Praha, přednosta prof. MUDr. Viktor Kožich, CSc.
1; Oční klinika, 1. lékařská fakulta, Univerzita Karlova v Praze a Všeobecná fakultní nemocnice, Praha, přednostka doc. MUDr. Jarmila Heissigerová, Ph. D., MBA
2; Oční klinika JL, s. r. o., primář doc. MUDr. Ján Lešták, CSc., FEBO, MBA, LL. A, DBA, FAOG
3
Vyšlo v časopise:
Čes. a slov. Oftal., 72, 2016, No. 4, p. 128-136
Kategorie:
Původní práce
Souhrn
Cíl:
Práce poskytuje souhrn výsledků probíhajících klinických studií, které testují genové terapie monogenně dědičných onemocnění sítnice a zrakového nervu.
Metody:
Literární rešerše zaměřená na principy, doporučené postupy, aplikaci a výsledky genových terapií u primárně sítnicových onemocnění a onemocnění zrakového nervu.
Výsledky:
V současné době probíhají v zahraničních centrech klinické zkoušky testující genové terapie pro Leberovu kongenitální amaurózu typu 2 (LCA2), retinitis pigmentosa typu 38, Usherův syndrom typu 1B, Stargardtovu chorobu, chorioideremii, achromatopsii, Leberovu hereditární neuropatii optiku (LHON) a X-vázanou retinoschízu. Až na terapie vnášející gen RPE65 (LCA2) a MT-ND4 (LHON), které již pokročily do fáze III, jsou všechna klinická hodnocení ve fázích I a II, v nichž se testuje bezpečnost a zjišťuje se účinná dávka léku.
Závěr:
V současné době se vývoj genové terapie zaměřuje na celou řadu nových onemocnění. Sítnice je pro svoji dostupnost z hlediska aplikace a kontroly účinků neobyčejně vhodnou tkání pro tento druh léčby. S rozvojem nových terapeutických postupů stoupá význam znalosti molekulárně genetické příčiny onemocnění, která je i podmínkou zařazení do klinických studií testujících cílené, především genové terapie.
Klíčová slova:
genová terapie, monogenní onemocnění sítnice, atrofie optického nervu, mitochondriální onemocnění
ÚVOD
Dědičná onemocnění sítnice jsou jednou z nejčastějších příčin závažné ztráty zraku v dětském věku a časné dospělosti (49). Terapeutické možnosti jsou v běžné klinické praxi omezeny na léčbu některých doprovodných příznaků, ovšem bez možnosti ovlivnění degenerativních procesů v samotných fotoreceptorech. Genové terapie představují v léčbě této skupiny chorob zcela nový přístup. Jsou zaměřeny na přenos plně funkční kopie genu s cílem nahradit sníženou či nulovou funkci proteinu, který je kódován mutovaným genem, a/nebo na regeneraci či stabilizaci narušené retinální struktury (40). Oko je vzhledem ke snadné dostupnosti, malým rozměrům, imunologickému privilegiu, kompartmentalizaci a možnosti kontralaterální kontroly ideální cílovým orgánem pro genové terapie (37).
CÍL
Cílem naší práce bylo podat souhrnný přehled o probíhajících klinických zkouškách testujících genové terapie hereditárních onemocnění sítnice a zrakového nervu.
METODY
Literární rešerše zaměřená na principy, doporučené postupy, aplikaci a výsledky genových terapií pro monogenní hereditární onemocnění sítnice a zrakového nervu ve stadiu klinických zkoušek. Zdrojem informací o probíhajících klinických studiích byl mezinárodní registr klinických studií „https://clinicaltrials.gov“.
VÝSLEDKY
Základní předpoklady pro aplikaci genové terapie
Genová terapie je léčebný postup, při němž je do genomu buněk vnesen genetický materiál, který nahrazuje nebo ovlivňuje expresi proteinu účastnícího se patogeneze konkrétního onemocnění. Ačkoliv tato léčba byla již v minulosti úspěšně použita např. u závažných imunodeficitů (42), stále se jedná pouze o terapii experimentální, jež s sebou může nést řadu nežádoucích účinků.
K vývoji a aplikaci genové terapie je třeba znát příčinu geneticky podmíněné choroby na molekulární úrovni, tedy určit gen odpovědný za vznik onemocnění. Shromážděné poznatky o genetických příčinách monogenních onemocnění jsou vkládány do databáze OMIM (Online Mendelian Inheritance in Man), která je přístupná na adrese „http://www.omim.org“ a v současnosti obsahuje přes 5 000 záznamů. Dalším krokem je určení konkrétní mutace způsobující patologický proces u daného pacienta. V rámci vývoje se hodnotí, zda po aplikaci genové terapie dojde k vymizení nebo zmírnění projevů onemocnění. Zároveň léčba nesmí mít negativní dopady na životně důležité funkce organismu.
Strategie genové terapie se stanovuje s ohledem na výše uvedené požadavky. Pokud je příčinou onemocnění nedostatek produktu mutovaného genu, stačí začlenit normální (tzv. wild type) sekvenci genu do genomu příslušných buněk (obr. 1A) nebo vnesením terapeutického genu zmírnit projevy onemocnění (obr. 1B). Pokud však patologicky působí pozměněný produkt mutovaného genu charakteru aberantního proteinu, je nutné mutovaný gen buď zablokovat (obr. 1C), nebo ho opravit (obr. 1D) (56).
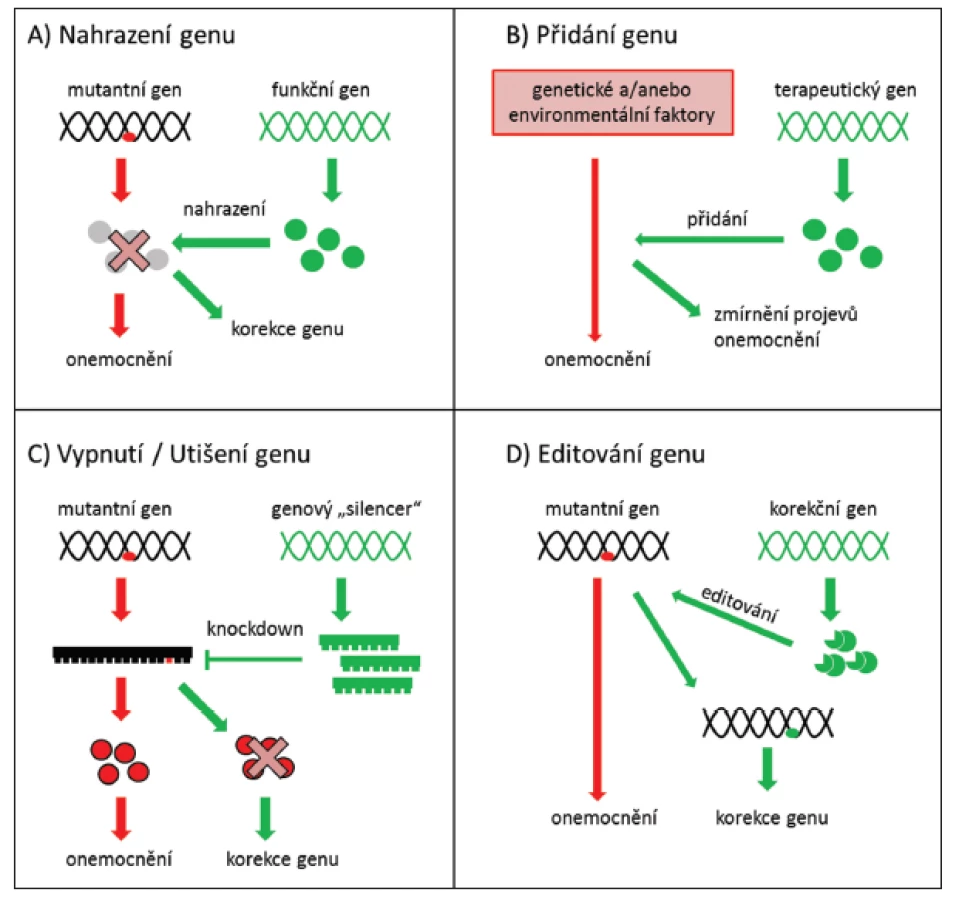
Genovou terapii lze provádět in vivo, kdy jsou cílové buňky po celou dobu léčby součástí organismu, nebo in vitro, kdy jsou cílové buňky z těla organismu odebrány a po provedení léčby vráceny na původní místo (25).
Zařazování pacientů do klinických studií je vždy podmíněno řadou kritérií. Vzhledem k samotné podstatě geneticky podmíněných onemocnění bývá standardním postupem a zároveň i prvním krokem zjištění příčiny onemocnění na molekulárně genetické úrovni. Rozdílné genetické pozadí spolu s možnou přítomností preexistujících protilátek proti virovým vektorům jsou příčinou variability výstupních klinických parametrů po aplikaci genové terapie u jednotlivých pacientů. Vysoká hladina specifických neutralizujících protilátek je jedním z kritérií vylučujících zařazení do studií (30). Mezi současné nevýhody genové terapie sítnicových onemocnění patří vysoká finanční a technologická náročnost, stejně tak i nutnost aplikovat léčbu v tzv. terapeutickém okénku, kdy ještě nedošlo k nevratnému poškození tkáně (5).
Vektory používané pro genovou terapii
Genetická informace se přenáší do cílových buněk pomocí nosičů zvaných vektory. Buňka, do které byl gen vnesen a zároveň v ní probíhá i jeho funkční exprese (vyjádření genetické informace), se označuje jako buňka transdukovaná. Ideální vektor by měl pronikat do velkého počtu cílových buněk a exprese vneseného genu v transdukované buňce by měla probíhat po dostatečně dlouhou dobu, aby bylo dosaženo požadovaného terapeutického účinku. Vektor navíc nesmí být pro cílové buňky toxický či vyvolávat u příjemce nežádoucí účinky, jakými jsou virové infekce či autoimunitní reakce (36). Vektory dělíme na virové a nevirové. Pro řadu nevýhod fyzikálních a chemických vektorů se v současné době v klinických zkouškách, které testují terapie pro onemocnění sítnice u lidí, využívá vektorů virových (tab. 1), zejména adenovirových a retrovirových (36).

Z virových částic jsou odstraněny geny, které souvisí s patogenitou a množením virových částic. Tyto jsou pak nahrazeny expresní kazetou (obr. 2). Připravena je z DNA přepsané do mRNA procesem transkripce, ve které jsou již vystřiženy nekódující sekvence (introny) a ta je zpětně přepsána do cDNA, která obsahuje jen kódující sekvence genu (exony). cDNA je ohraničena na 5´-konci promotorem, který spouští expresi genu, a na 3´-konci terminátorem transkripce a polyadenylačním signálem, které expresi ukončují (26).

Riziko použití virových vektorů spočívá v tom, že genetická informace je do genomu příjemce vkládána víceméně náhodně. Tím může dojít k narušení sekvence jiného genu s funkčními následky, např. změna v protoonkogenu nebo v tumor-supresorovém genu může spustit maligní transformaci buňky. Dalším problémem je nestabilní exprese genu způsobená potenciální imunogenicitou virových vektorů způsobující zánětlivé reakce (40). Další aplikace virového vektoru již nemusí být dostatečně účinná, jelikož proti vektoru jsou vytvořeny protilátky (7). Opakovaná aplikace terapeutika do subretinální oblasti také nese riziko poškození nebo odchlípení sítnice (obr. 3, tab. 2) (8). Problematické je rovněž stanovení terapeutické dávky a množství aplikovaných virových částic. V počátcích genové terapie způsobily vysoké dávky virových vektorů při léčbě deficitu ornitin transkarbamylázy masivní zánětlivou odpověď a úmrtí pacienta (48). Poté došlo ke zpřísnění podmínek pro aplikaci genových terapií a tato závažná nežádoucí událost se již neopakovala.


Způsoby aplikace vektorů
Při léčbě onemocnění sítnice se k aplikaci vektorů nejčastěji využívají intravitreální a subretinální injekce (obr. 3, tab. 2). Aplikace do sklivce je sice pro sítnici méně invazivní, ale transdukce probíhá zejména ve vnitřních vrstvách sítnice, tedy v buňkách Müllerových a gangliových. Bariéru průniku vektorů a léčiv do větší hloubky sítnice tvoří vnitřní limitující membrána a další sítnicové vrstvy. Pro aplikaci virových vektorů do vrstvy fotoreceptorů a vrstvy buněk retinálního pigmentového epitelu (RPE) je vhodnější subretinální aplikace, kdy je vektor injikován do puchýřku mezi uvedenými vrstvami, a je tak s nimi v těsném kontaktu. Během 14 hodin buňky RPE subretinální tekutinu obsahující virový vektor kompletně absorbují. Účinnost je 5-10x vyšší než při podání do sklivce. Nevýhodou je však větší riziko možných komplikací spojených s chirurgickým výkonem (tab. 2) (35).
Genové terapie onemocnění sítnice ve stadiu klinických zkoušek
Klinické testování má několik fází. Ve fázi I se na jedné nerandomizované skupině dobrovolníků stanovuje maximálně tolerovaná dávka a sleduje se bezpečnost. Ve fázi II se již na pacientech zjišťuje účinná dávka léku, opět většinou v rámci nerandomizované studie. Ve fázi III je jedna skupina léčena běžnými postupy, druhé skupině je podávána léčba nová. Porovnává se tedy účinnost nového léku se standardní terapií. Zařazení i hodnocení bývají dvojitě zaslepené. Na základě výsledků fáze III pak může být lék zaregistrován. V poslední fázi IV se sledují nežádoucí účinky léčiva po registraci a při dlouhodobém užívání (https://clinicaltrials.gov/ct2/about-studies/learn) (24).
Leberova kongenitální amauróza 2
Leberova kongenitální amauróza (LCA) je geneticky heterogenní skupina onemocnění podmíněných mutacemi v nejméně 22 genech (23). LCA2 (OMIM #204100) je autozomálně recesivní choroba vznikající na podkladě mutací v genu RPE65 (retinal pigment epithelium-specific 65 kDa protein) (18). Tento gen je téměř výlučně exprimován v RPE, kde se podílí na recyklaci opsinu a rodopsinu tím, že konvertuje all-trans-retinoidy na 11-cis retinal. Nedostatečná funkce nebo absence RPE65 vede k hromadění trans-retinyl esterů s následnou degenerací fotoreceptorů (10).
Přestože výskyt onemocnění je relativně vzácný (méně než 1 postižený na 1 000 000 narozených dětí), byla LCA2 navržena pro genovou terapii, a to hlavně kvůli časné manifestaci onemocnění, relativně dlouho zachované struktuře sítnice a dostupnosti zvířecích modelů (8). První výsledky léčby psího modelu publikované již v roce 2001 ukázaly významné dlouhotrvající zlepšení zrakových funkcí po subretinální aplikaci rekombinantního adeno-asociovaného viru (AAV2) obsahujícího RPE65 cDNA (1). V roce 2007 byly pak registrovány první klinické zkoušky fáze I (NCT00481546, NCT00516477; clinicaltrials.gov) a fáze II (NCT00643747; clinicaltrials.gov), do kterých bylo naplánováno zařadit do května 2015 celkově 39 pacientů s LCA2 (clinicaltrials.gov). V současné době probíhá studie fáze III se subretinálním podáním upraveného AAV2 (NCT00999609; clinicaltrials.gov).
U všech pacientů došlo ke zlepšení zrakové ostrosti léčeného oka, nicméně v dosti rozdílné míře (6, 22, 35). Zlepšila se také orientace léčených při slabém osvětlení. Citlivost sítnice měřená pomocí psychofyziologického testu s modrými a červenými stimuly na bílém a barevném pozadí po adaptaci na tmu (47) se během 1 až 2 měsíců po podání vektoru zvýšila. Další zlepšování pak bylo pozorováno po dobu 6 až 12 měsíců. Následně začala citlivost sítnice opět klesat, přesto však u některých pacientů zůstala po 3 letech vyšší než před podáním genové terapie. Zlepšeny byly i části zorného pole odpovídající umístění subretinální injekce vektoru, přičemž u 40 % pacientů vznikla v těchto oblastech excentrická fixace (8). Podstatnější zlepšení funkce sítnice bylo pozorováno u starších pacientů. Oproti psím modelům však nebyly u lidí detekovatelné změny při elektroretinografickém vyšetření. Poslední publikovaná práce zjistila, že degenerativní procesy fotoreceptorových buněk sítnice se po genové terapii nezastavily a pokračovaly dál. Předpokládá se, že účinek této léčby je vysoce závislý na podaném množství vektoru a že aplikované dávky byly nízké (5).
Retinitis pigmentosa 38
Nesyndromová retinitis pigmentosa (RP) je geneticky heterogenní skupina progresivních onemocnění sítnice podmíněných mutacemi v nejméně 50 genech (https://sph.uth.edu/retnet/). RP38 (OMIM #613862) je velmi vzácná autozomálně recesivní forma RP asociovaná s mutacemi v genu MERTK (Mer proto-oncogene, tyrosine kinase) způsobující 1 % nesyndromových RP (44). MERTK je enzym potřebný k fagocytóze odloučených zevních segmentů fotoreceptorů RPE. Ztráta enzymu podmiňuje poruchu fagocytózy fotoreceptorů s akumulací nedegradovaných metabolitů rhodopsinu mezi nimi, což vede k jejich apoptóze a degeneraci sítnice (12). Charakteristickým znakem časné fáze RP38 jsou tečkovitá autofluorescentní sítnicová depozita. Šeroslepost vzniká již v první dekádě života, následuje postižení funkce čípků, často s atrofií makuly (11).
Již dlouhou dobu jsou studovány krysí modely s mutacemi v genu MERTK. Dochází u nich k velmi rychlé akumulaci metabolitů mezi fotoreceptory, ztrátě fotoreceptorů od 20. dne po narození a jejich úplnému chybění od 60. dne. Proto jsou tyto modely vhodné i pro hodnocení postupů a účinnosti genové terapie u lidí.
V preklinické studii byly do sítnice krysího modelu pomocí lentivirových vektorů vneseny funkční kopie genu MERTK, což vedlo k zachování a u některých zvířat i k částečnému obnovení funkcí sítnice 7 měsíců od aplikace (52). Nedávná studie na krysím modelu ukázala, že aminokyselinová záměna tyrozinu za fenylalanin na pozici 733 v kapsidě AAV vektoru umožnila zachování struktury a funkce sítnice nejméně 1 rok po aplikaci (13).
V klinické studii fáze I registrované v roce 2011 (NCT01482195; clinicaltrials.gov) byl třem pacientům subretinálně aplikován AAV2 vektor obnovující funkci genu MERTK bez závažných nežádoucích účinků (8).
Usherův syndrom 1B
Usherův syndrom (USH) je geneticky heterogenní onemocnění, dosud bylo nalezeno nebo mapováno 11 příčinných genů. Autozomálně recesivní typ 1 je způsoben mutacemi v pěti známých genech (39). Všechny jsou exprimovány ve vláskových buňkách vnitřního ucha a fotoreceptorech sítnice. Forma USH1B (OMIM #276900) je způsobená mutacemi v MYO7A (myosin VIIA), jehož produkt hraje v buňkách fotoreceptorů a RPE důležitou roli v transportu opsinu a je nezbytný pro normální průběh Waldova cyklu (33). Na rozdíl od vláskových buněk ucha, není vývoj fotoreceptorů postižen a k jejich odumírání dochází až v průběhu života v důsledku hromadění metabolitů a následné dysfunkci synapsí (8). USH1B je nejzávažnější formou syndromu s vrozenou ztrátou sluchových funkcí, s postižením zrakových funkcí během první dekády života a s vestibulární areflexií. Jeho prevalence je v evropské populaci odhadována 4,2 případu na 100 000 obyvatel (16).
Z důvodů značné velikosti genu MYO7A byly pro jeho přenos zvoleny vektory lentivirové, které mají větší kapacitu pro přenášený gen, avšak nižší účinnost transdukce ve fotoreceptorech než vektory AAV. Na myším modelu bylo prokázáno obnovení transportu opsinu (21), což vedlo v roce 2012 k zahájení klinické studie fáze I/IIa s lékem UshStat (NCT01505062; clinicaltrials.gov). Předběžné výsledky těchto studií nebyly dosud publikovány. Jelikož u USH1B dochází již od počátku onemocnění k poškození fotoreceptorů, jsou dalším krokem pokusy o sbalení MYO7A do účinnějšího AAV vektoru, který je schopen exprese i v tomto typu buněk (8).
Stargardtova choroba
Stargardtova choroba je geneticky heterogenní onemocnění způsobené mutacemi ve třech genech. Nejčastější autozomálně recesivní typ Stargardtovy choroby (STGD1, OMIM #248200) je podmíněn mutacemi v genu ABCA4 (ATP-binding cassette subfamily A member 4), který kóduje protein podílející se na transportu použitých částí fotoreceptorů do RPE (2). Mutace v této lipáze ovlivňují zpracování vitamínu A, což vede k akumulaci toxického bisretinoidu A2E, tzv. dimeru vitamínu A. Následkem je odumírání buněk RPE a fotoreceptorů (57).
Charakteristickým klinickým nálezem Stargardtovy choroby, kterou je postižen 1 z 10 000 obyvatel, je přítomnost žlutavých skvrn v makule, popř. i difúzně po celém fundu spolu se ztenčováním vrstev sítnice v makule. Postupně vzniká v makule jizva se žlutými skvrnami charakteru lipofuscinových depozit při okrajích nebo i po celém očním pozadí (32, 46).
Protože velikost cDNA genu ABCA4 značně přesahuje kapacitu běžných AAV, byly při vývoji genové terapie použity lentivirové vektory. Na základě pozitivních výsledků na myším modelu (29) byla v roce 2011 registrována klinická studie fáze I/II s lékem StarGen (NCT01367444; clinicaltrials.gov). Její výsledky nebyly doposud publikovány. V další studii z roku 2012 byly pro přenos funkční kopie ABCA4 na myším modelu pro tuto chorobu využity nanočástice. Po aplikaci byla zaznamenána perzistentní exprese ABCA4, zlepšení adaptace na tmu a snížení ukládání lipofuscinu. Tento postup však ještě nebyl registrován pro klinické studie (20).
Chorioideremie
Chorioideremie (OMIM #303100) je onemocnění sítnice s dědičností vázanou na chromozóm X a postihující 1 z 50 000 mužů. Chorobu způsobují mutace v genu CHM (Rab Escort Protein 1), jehož produkt prostřednictvím posttranslační modifikace (prenylace) proteinů reguluje transport vezikul v procesech endocytózy a exocytózy (34). Pacienti s chorioideremií mají snížené nebo nulové množství tohoto enzymu, což vede k poruchám transportu opsinu do zevních segmentů fotoreceptorů, k poruchám migrace melanozomů v buňkách RPE a ke snížené fagocytóze zevních segmentů fotoreceptorů buňkami RPE (3). Následkem je degenerace choriokapilaris, RPE a fotoreceptorů. Není však jasné, která část sítnice je u chorioideremie primárně postižená. Je známo, že nedostatek CHM vede k funkčnímu postižení sítnice ještě před odumíráním RPE, nebylo ale objasněno, zda se jedná o důsledek deficitu nebo nespecifický vliv stresu RPE buněk (14). Vzhledem k tomu, že u chorioideremie dochází k poškození cévnatky i sítnice včetně RPE, je nutné zajistit efektivní transdukci vektorů ve více vrstvách buněk.
Prvním krokem ve vývoji genové terapie byla příprava AAV vektoru, který vpravil celý gen CHM do lymfocytů a fibroblastů získaných od pacientů s chorioideremií (4). V další fázi byl připraven lentivirový vektor, jehož aplikace vedla na myším modelu k částečnému obnovení enzymatické aktivity v buňkách RPE (51). V roce 2011 byla registrována klinická studie (NCT01461213; clinicaltrials.gov), v rámci které byla 6 pacientům aplikována do subretinálního prostoru funkční kopie CHM pomocí AAV vektoru. U všech pacientů bylo za 6 měsíců po léčbě pozorováno zvýšení citlivosti sítnice při mikroperimetrii. Průměrná zraková ostrost se zlepšila o 3,8 písmene na optotypech ETDRS (Early Treatment Diabetic Retinopathy Study). V roce 2015 byly registrovány další 3 genové terapie využívající stejný postup (NCT02341807, NCT02553135, NCT02407678; clinicaltrials.gov).
Leberova hereditární neuropatie optiku
Leberova hereditární neuropatie optiku (LHON; OMIM #535000) je nejčastějším mitochondriálním onemocněním projevujícím se oboustrannou akutní nebo subakutní ztrátou zraku (31, 43). Specifické mutace v mitochondriálních genech způsobují u LHON degeneraci retinálních gangliových buněk a atrofii optického nervu. Ve více než 90 % případů se zjišťuje jedna ze tří mutací v genech MT-ND1 (mitochondrially encoded NADH: ubiquinone oxidoreductase core subunit 1), MT-ND4 a MT-ND6 (43).
Tyto mutace vedou k chybnému uspořádání komplexu I respiračního řetězce v mitochondriích, což ovlivňuje tvorbu ATP a vznik nežádoucího množství reaktivních kyslíkových radikálů. Prozatím není jasné, proč efekt těchto mutací postihuje především zrakový nerv a nikoliv nervovou soustavu jako celek (9).
Transfekování mitochondriální DNA je velice obtížné. Pro vývoj genové terapie bylo potřebné připravit vektor transfekující jadernou DNA se sekvencí, která nasměruje protein vzniklý v cytoplazmě do mitochondrie. Tento postup byl vyzkoušen na myším a krysím modelu (15, 19). Intravitreální aplikace vedla k delšímu zachování retinálních gangliových buněk a ke zlepšení zraku u modelových zvířat.
Průběžné výsledky studie fáze III (NCT01267422; clinicaltrials.gov) s intravitreální aplikací AAV vektoru s genem MT-ND4 u 9 pacientů prokázaly zlepšení zrakové ostrosti a citlivosti zorného pole. Tloušťka vrstvy nervových vláken sítnice se však nezvýšila (55).
Achromatopsie
Achromatopsie (OMIM #262300) je charakterizována poruchou funkce čípků a vyskytuje se s prevalencí 1 na 30 000 obyvatel (50). Z pěti dosud popsaných genů, odpovědných za vznik onemocnění, jsou mutace v genu CNGB3 (cyclic nucleotide gated channel beta 3) příčinou přibližně 50 % případů (27). Narušením tvorby CNG kanálů (cyclic nucleotide-gated ion channel) dochází k influxu kationů do buněk, což vede až k jejich apoptóze. Na rozdíl od čípků se tyto kanály nevyskytují na tyčinkách, skotopické vidění proto zůstává zachováno (38).
Subretinální aplikace AAV vektoru nesoucího gen CNGB3 testovaná na psích modelech prokázala možnost obnovení funkce čípků a fotopického vidění nezávisle na pozici mutace. Terapeutická účinnost a stabilita exprese vneseného genu však byla ovlivněna věkem testovaného zvířete v době podání a typem použitého promotoru (28).
V listopadu 2015 byly zahájeny dvě klinické zkoušky fáze I a II se subretinální aplikací AAV vektorů (NCT02610582, NCT02599922; clinicaltrials.gov).
X-vázaná retinoschíza
X-vázaná retinoschíza (XLRS, OMIM #312700) je monogenně podmíněné oční onemocnění s manifestací v první dekádě života s prevalencí 1 nemocný na 5–25 000 obyvatel (17). Choroba je způsobena mutacemi v genu RS1 (retinoschisin 1), které vedou u pacientů mužského pohlaví k rozštěpu častěji vnitřních vrstev sítnice obvykle v makulární oblasti provázenému poklesem zrakové ostrosti (45). Produkt genu je exprimován ve fotoreceptorech a bipolárních buňkách sítnice. Jeho funkce není úplně objasněna, ale předpokládá se, že se podílí na adhezi uvedených buněk (41).
Na myším modelu byly odzkoušeny různé sérotypy AAV vektorů. Jejich účinnost závisí od použitého promotoru a taky od stáří testovaného zvířete. Injekce AAV vektoru do sítnice myšího modelu ve věku 7 měsíců zlepšila strukturu retiny, ne však elektroretinografické parametry. U mladších zvířat dochází po aplikaci k zachování a částečnému vylepšení struktury a funkce retiny. Tyto studie poukázaly, že před zavedením genové terapie pro XLRS je potřeba pečlivě zvážit způsob doručení, výběr vektoru a typ použitého promotoru (8).
V roce 2015 byly registrovány dvě klinické studie (NCT02317887 a NCT02416622; clinicaltrials.gov) fáze I a II s intravitreální aplikací AAV sérotypu 2 a 8.
Genové terapie v kontextu České republiky
Ani jedna z několika desítek klinických studií testujících genovou terapii pro oční choroby, které v současné době probíhají, není realizována na pracovištích v ČR. Nepříznivá situace má několik důvodů. Zásadní je chybění legislativy stanovující pravidla pro klinické studie aplikující genové terapie, což znemožňuje českým klinickým pracovištím účastnit se mezinárodních multicentrických studií. Svůj podíl má i nedostatek odborníků, kteří mohou nové přípravky kontrolovat a nízká informovanost laické a často i odborné veřejnosti o vzácných chorobách, dědičná onemocnění oka nevyjímaje.
ZÁVĚR
Genové terapie představují nadějný přístup v léčbě onemocnění oka, obzvláště u chorob sítnice, kde zatím nejsou k dispozici žádné fungující léky či terapeutické metody. Potrvá však ještě několik let než budou výsledky v současné době probíhajících klinických studií vyhodnoceny a bude prokázán jejich případný dlouhodobý příznivý účinek.
Práce byla podpořena granty GAUK 4315/2015, SVV UK 260256/2016, UNCE 204011 a AZV 16-32341A.
*Autoři přispěli k práci stejným podílem
Autoři práce prohlašují, že vznik i téma odborného sdělení a jeho zveřejnění není ve střetu zájmu a není podpořeno žádnou farmaceutickou firmou.
Do redakce doručeno dne 5. 2. 2016
Do tisku přijato dne 26. 7. 2016
Doc. MUDr. P. Lišková, M.D., Ph.D.
Ústav dědičných metabolických poruch
a Oční klinika
1. lékařská fakulta, Univerzita Karlova
v Praze a Všeobecná fakultní
nemocnice v Praze
Ke Karlovu 2
128 00 Praha 2
petra.liskova@lf1.cuni.cz
http://ocnigenetika.lf1.cuni.cz
Zdroje
1. Acland, GM., Aguirre, GD., Ray J.,et al.: Gene therapy restores vision in a canine model of childhood blindness. Nat Genet, 28; 2001 : 92–5.
2. Allikmets, R., Shroyer, NF., Singh, N.,et al.: Mutation of the Stargardt disease gene (ABCR) in age-related macular degeneration. Science, 277; 1997 : 1805–7.
3. Alory, C., Balch, WE.: Organization of the Rab-GDI/CHM superfamily: the functional basis for choroideremia disease. Traffic, 2; 2001 : 532–43.
4. Anand, V., Barral, DC., Zeng, Y., et al.: Gene therapy for choroideremia: in vitro rescue mediated by recombinant adenovirus. Vision Res, 43; 2003 : 919–26.
5. Bainbridge, JW., Mehat, MS., Sundaram, V., et al.: Long-term effect of gene therapy on Leber‘s congenital amaurosis. N Engl J Med, 372; 2015 : 1887–97.
6. Bainbridge, JW., Smith, AJ., Barker, SS., et al.: Effect of gene therapy on visual function in Leber‘s congenital amaurosis. N Engl J Med, 358; 2008 : 2231–39.
7. Bessis, N., GarciaCozar, FJ., Boissier MC.: Immune responses to gene therapy vectors: influence on vector function and effector mechanisms. Gene Ther, 11 Suppl 1; 2004: S10–7.
8. Boye, SE., GarciaCozar, FJ., Boissier, MC.: A comprehensive review of retinal gene therapy. Mol Ther, 21; 2013 : 509–19.
9. Carelli, V., La Morgia, C., Valentino, ML., et al.: Retinal ganglion cell neurodegeneration in mitochondrial inherited disorders. Biochim Biophys Acta, 1787; 2009 : 518–28.
10. Cideciyan, AV.: Leber congenital amaurosis due to RPE65 mutations and its treatment with gene therapy. Prog Retin Eye Res, 29; 2010 : 398–427.
11. Conlon, TJ., Deng, WT., Erger, K., et al.: Preclinical potency and safety studies of an AAV2-mediated gene therapy vector for the treatment of MERTK associated retinitis pigmentosa. Hum Gene Ther Clin Dev, 24; 2013 : 23–8.
12. D‘Cruz, PM., Yasumura, D., Weir, J., et al.: Mutation of the receptor tyrosine kinase gene Mertk in the retinal dystrophic RCS rat. Hum Mol Genet, 9; 2000 : 645-51.
13. Deng, WT., Dinculescu, A., Li, Q., et al.: Tyrosine-mutant AAV8 delivery of human MERTK provides long-term retinal preservation in RCS rats. Invest Ophthalmol Vis Sci, 53; 2012 : 1895–1904.
14. Dimopoulos, IS., Chan, S., MacLaren, RE., et al.: Pathogenic mechanisms and the prospect of gene therapy for choroideremia. Expert Opin Orphan Drugs, 3; 2015 : 787–98.
15. Ellouze, S., Augustin, S., Bouaita, A., et al.: Optimized allotopic expression of the human mitochondrial ND4 prevents blindness in a rat model of mitochondrial dysfunction. Am J Hum Genet, 83; 2008 : 373–87.
16. Espinos, C., Millan, JM., Beneyto, M., et al.: Epidemiology of Usher syndrome in Valencia and Spain. Community Genet, 1; 1998 : 223–8.
17. George, ND., Yates, JR., Moore, AT.: X linked retinoschisis. Br J Ophthalmol, 79; 1995 : 697–702.
18. Gu, SM., Thompson, DA., Srikumari, CR., et al.: Mutations in RPE65 cause autosomal recessive childhood-onset severe retinal dystrophy. Nat Genet, 17; 1997 : 194–7.
19. Guy, J., Qi, X., Koilkonda, RD., et al.: Efficiency and safety of AAV-mediated gene delivery of the human ND4 complex I subunit in the mouse visual system. Invest Ophthalmol Vis Sci, 50; 2009 : 4205–14.
20. Han, Z., Conley, SM., Makkia, RS., et al.: DNA nanoparticle-mediated ABCA4 delivery rescues Stargardt dystrophy in mice. J Clin Invest, 122; 2012 : 3221–6.
21. Hashimoto, T., Gibbs, D., Lillo, C., et al.: Lentiviral gene replacement therapy of retinas in a mouse model for Usher syndrome type 1B. Gene Ther, 14; 2007 : 584–94.
22. Hauswirth, WW., Aleman, TS., Kaushal, S., et al.: Treatment of leber congenital amaurosis due to RPE65 mutations by ocular subretinal injection of adeno-associated virus gene vector: short-term results of a phase I trial. Hum Gene Ther, 19; 2008 : 979–90.
23. Chacon-Camacho, OF., Zenteno, JC.: Review and update on the molecular basis of Leber congenital amaurosis. World J Clin Cases, 3; 2015 : 112–24.
24. Kao, LS., Tyson, JE., Blakely, ML., et al.: Clinical research methodology I: introduction to randomized trials. J Am Coll Surg, 206; 2008 : 361–9.
25. Kaufmann, KB., Buning, H., Galy, A., et al.: Gene therapy on the move. EMBO Mol Med, 5; 2013 : 1642–61.
26. Kay, MA., Glorioso, JC., Naldini, L.: Viral vectors for gene therapy: the art of turning infectious agents into vehicles of therapeutics. Nat Med, 7; 2001 : 33–40.
27. Kohl, S., Varsanyi, B., Antunes, GA., et al.: CNGB3 mutations account for 50% of all cases with autosomal recessive achromatopsia. Eur J Hum Genet, 13; 2005 : 302–8.
28. Komaromy, AM., Alexander, JJ., Rowlan, JS., et al.: Gene therapy rescues cone function in congenital achromatopsia. Hum Mol Genet, 19; 2010 : 2581–93.
29. Kong, J., Kim, SR., Binley, K., et al.: Correction of the disease phenotype in the mouse model of Stargardt disease by lentiviral gene therapy. Gene Ther, 15; 2008 : 1311–20.
30. Kotterman, MA., Yin, L., Strazzeri, JM., et al.: Antibody neutralization poses a barrier to intravitreal adeno-associated viral vector gene delivery to non-human primates. Gene Ther, 22; 2015 : 116–26.
31. Leber, T.: Ueber hereditäre und congenital-angelegte Sehnervenleiden [About hereditary and congenital optic nerve disorders]. Albrecht Von Graefes Arch Klin Exp Ophthalmol, 17; 1871 : 249–91.
32. Lois, N., Holder, GE., Bunce, C., et al.: Phenotypic subtypes of Stargardt macular dystrophy-fundus flavimaculatus. Arch Ophthalmol, 119; 2001 : 359–69.
33. Lopes, VS., Gibbs, D., Libby, RT., et al.: The Usher 1B protein, MYO7A, is required for normal localization and function of the visual retinoid cycle enzyme, RPE65. Hum Mol Genet, 20; 2011 : 2560–70.
34. MacDonald IM., Hume S., Chan S., et al.: Choroideremia. GeneReviews® [online]. 2003-2015 [cit. 11. dubna 2016]. Dostupný z WWW: .
35. Maguire, AM., Simonelli, F., Pierce, EA., et al.: Safety and efficacy of gene transfer for Leber‘s congenital amaurosis. N Engl J Med, 358; 2008 : 2240–48.
36. Mali, S.: Delivery systems for gene therapy. Indian J Hum Genet, 19; 2013 : 3-8.
37. Martin, KR., Quigley, HA.: Gene therapy for optic nerve disease. Eye (Lond), 18; 2004 : 1049–55.
38. Meighan, PC., Peng, C., Varnum, MD.: Inherited macular degeneration-associated mutations in CNGB3 increase the ligand sensitivity and spontaneous open probability of cone cyclic nucleotide-gated channels. Front Physiol, 6; 2015 : 177.
39. Millan, JM., Aller, E., Jaijo, T., et al.: An update on the genetics of usher syndrome. J Ophthalmol, 2011; 2011 : 417217.
40. Misra, S.: Human gene therapy: a brief overview of the genetic revolution. J Assoc Physicians India, 61; 2013 : 127–33.
41. Molday, LL., Hicks, D., Sauer, CG., et al.: Expression of X-linked retinoschisis protein RS1 in photoreceptor and bipolar cells. Invest Ophthalmol Vis Sci, 42; 2001 : 816–25.
42. Mukherjee, S., Thrasher, AJ.: Gene therapy for PIDs: progress, pitfalls and prospects. Gene, 525; 2013 : 174–81.
43. Newman, NJ.: Hereditary optic neuropathies: from the mitochondria to the optic nerve. Am J Ophthalmol, 140; 2005 : 517–23.
44. Ostergaard, E., Duno, M., Batbayli, M., et al.: A novel MERTK deletion is a common founder mutation in the Faroe Islands and is responsible for a high proportion of retinitis pigmentosa cases. Mol Vis, 17; 2011 : 1485–92.
45. Prenner, JL., Capone Jr., A., Ciaccia, S., et al.: Congenital X-linked retinoschisis classification system. Retina, 26; 2006: S61–4.
46. Rivera, A., White, K., Stohr, H., et al.: A comprehensive survey of sequence variation in the ABCA4 (ABCR) gene in Stargardt disease and age-related macular degeneration. Am J Hum Genet, 67; 2000 : 800–13.
47. Roman, AJ., Cideciyan, AV., Aleman, TS., et al.: Full-field stimulus testing (FST) to quantify visual perception in severely blind candidates for treatment trials. Physiol Meas, 28; 2007: N51–6.
48. Sibbald, B.: Death but one unintended consequence of gene-therapy trial. CMAJ, 164; 2001 : 1612.
49. Steinkuller, PG., Du, L., Gilbert, C., et al.: Childhood blindness. J AAPOS, 3; 1999 : 26–32.
50. Thiadens, AA., Somervuo, V., van den Born, LI., et al.: Progressive loss of cones in achromatopsia: an imaging study using spectral-domain optical coherence tomography. Invest Ophthalmol Vis Sci, 51; 2010 : 5952–7.
51. Tolmachova, T., Tolmachov, OE., Wavre-Shapton, ST., et al.: CHM/REP1 cDNA delivery by lentiviral vectors provides functional expression of the transgene in the retinal pigment epithelium of choroideremia mice. J Gene Med, 14; 2012 : 158–68.
52. Tschernutter, M., Schlichtenbrede, FC., Howe, S., et al.: Long-term preservation of retinal function in the RCS rat model of retinitis pigmentosa following lentivirus-mediated gene therapy. Gene Ther, 12; 2005 : 694–701.
53. Veleri, S. Lazar, CH., Chang, B., et al.: Biology and therapy of inherited retinal degenerative disease: insights from mouse models. Dis Model Mech, 8; 2015 : 109–29.
54. Verma, IM., Somia N.: Gene therapy - - promises, problems and prospects. Nature, 389; 1997 : 239–42.
55. Wan, X., Fei, H., Zhao, MJ., et. al.: Efficacy and safety of RAAVZ-NDY treatment for leber´s hereditary optic neuropathy. Sci Rep, 19; 2016 : 1–10.
56. Wang, D., Gao, G.: State-of-the-art human gene therapy: part II. Gene therapy strategies and clinical applications. Discov Med, 18; 2014 : 151–61.
57. Weng, J., Mata, NL., Azarian, SM., et al.: Insights into the function of Rim protein in photoreceptors and etiology of Stargardt‘s disease from the phenotype in abcr knockout mice. Cell, 98; 1999 : 13–23.
Štítky
OftalmologieČlánek vyšel v časopise
Česká a slovenská oftalmologie

2016 Číslo 4
- Současná oční operativa – bezpečná investice do výborného zraku
- Selektivní laserová trabekuloplastika nesnižuje nitroční tlak více než argonová laserová trabekuloplastika
- Progresi glaukomu je třeba hodnotit strukturálními i funkčními parametry
- Ztráta centrálního vidění po filtrujících operacích glaukomu
- Od PGF-2 alfa-isopropyl esteru k latanoprostu: přehled vývoje Xalatanu
Nejčtenější v tomto čísle
- Genová terapie dědičných onemocnění SÍTNICE A ZRAKOVÉHO NERVU: současný stav poznání
- Nová diagnostická zobrazovací metoda – shear waves elastografie
- HEMANGIOMY ORBITÁLNÍ KRAJINY U DĚTÍ
- MUTÁCIA BRAF A MOŽNOSTI IDENTIFIKÁCIE PROGNOSTICKÝCH MARKEROV METASTÁZOVANIA UVEÁLNEHO MELANÓMU
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy