
Výstupy multicentrické studie příčin náhlé srdeční smrti (SCD) v České republice a primární prevence srdeční zástavy u příbuzných
Outcomes of a multicenter study of the causes of sudden cardiac death (SCD) in the Czech Republic and primary prevention of cardiac arrest in relatives
Sudden cardiac death (SCD) in individuals younger than 40 years has a heritable cause in a significant part of the cases. Identification of SCD, post mortem genetic analysis along with the cardiological screening examination in first degree represents an important diagnostic tool for the primary prevention of cardiac arrest in victim´s relatives and requires multicentric and multidisciplinary collaboration. Between 2016 and 2021 the complex cardiogenetic analysis was performed in 115 deaths with post mortem diagnosis of cardiomyopathy, acute aortic dissection and cases without morphological finding explaining the cause of death (sudden arrhythmic death or sudden unexplained death). DNA was isolated from post mortem collected tissue samples or relative´s blood and subjected to massively parallel sequencing (Illumina, USA) in extent of 100 to 20 000 genes. Sequencing results were analysed using the SOPHiA GENETICS DDM bioinformatics platform (Switzerland). Genetic counselling and cardiological examinations were carried out in 328 family members. Highly likely or certain molecular aetiology (i.e. based on presence of ACMG.net Class 4 to 5 variants) was disclosed in 19,8 % of analysed cases in RYR2, KCNH2, KCNQ1, SCN5A, FLNC (stop), GLA, TTN, TNNT2, RBM 20, MYBPC3, MYPN, FHL1, TGFBR1, and COL3A1 genes. With cardiogenetic screening we identified 25 % relatives at risk of life threating arrhythmias and offered them an individualised care.
Keywords:
prevention – genetic analysis – guidelines – sudden cardiac death – inherited cardiovascular diseases
Autoři:
Kučerová Pohlová Štěpánka 1; Krebsová Alice 2; Votýpka Pavel 3; Peldová Petra 3; Kulvajtová Markéta 4; Dohnalová Petra 5; Bílek Matěj 5,6; Stufka Veronika 6; Rücklová Kristina 7; Grossová Iva 8; Wünschová Hanka 2; Tavačová Terezia 9; Hašková Jana 2; Segeťová Markéta 2; Gřegořová Andrea 10; Zoubková Veronika 3; Petřková Jana 11,12,13; Dobiáš Martin 14; Makuša Michal 15; Blanková Alžběta 16; Veitr David 16; Řehulka Hynek 17; Šubrt Ivan 18; Pilin Alexander 6; Tomášek Petr 5; Janoušek Jan 9; Kautzner Josef 2; Macek Milan Jr. 3
Působiště autorů:
Ústav soudního lékařství LF UK a FN Hradec Králové
1; Klinika kardiologie, IKEM, Praha
2; Ústav biologie a lékařské genetiky 2. LF UK a FN Motol, Praha
3; Ústav soudního lékařství 3. LF UK a FNKV, Praha.
4; Ústav soudního lékařství 2. LF UK a Fakultní nemocnice Bulovka, Praha
5; Ústav soudního lékařství a toxikologie 1. LF UK a VFN v Praze
6; Klinika dětí a dorostu 3. LF UK a FNKV, Praha
7; Vojenský ústav soudního lékařství, ÚVN, Praha
8; Dětské kardiocentrum 2. LF UK a FN Motol, Praha
9; Ústav klinické a molekulární patologie a lékařské genetiky FN Ostrava
10; I. interní klinika – kardiologická FNOL a LF UP, Olomouc
11; Ústav lékařské genetiky FNOL, Olomouc
12; Ústav patologické fyziologie LF UP, Olomouc
13; Ústav soudního lékařství a medicínského práva FNOL a LF UP, Olomouc
14; Soudnělékařské oddělení Nemocnice České Budějovice
15; Oddělení soudního lékařství a toxikologie Krajské nemocnice Liberec
16; Ústav soudního lékařství LF v Plzni, UK Praha, a FN Plzeň
17; Ústav lékařské genetiky LF v Plzni, UK Praha, a FN Plzeň
18
Vyšlo v časopise:
Soud Lék., 67, 2022, No. 2, p. 10-24
Kategorie:
Původní práce
Souhrn
Náhlá srdeční smrt u jedinců mladších 40 let je ve významném procentu způsobena dědičným kardiovaskulárním onemocněním. Identifikace těchto případů, provedení post mortem genetického vyšetření a kardiologické screeningové vyšetření přímých příbuzných je prvním krokem k primární prevenci srdeční zástavy u pozůstalých. Tato péče vyžaduje multidisciplinární a multicentrickou spolupráci. Jistá až velmi pravděpodobná molekulární příčina onemocnění byla nalezena u 19,8 % analyzovaných případů, u 25 % příbuzných bylo zjištěno riziko významných arytmií.
Klíčová slova:
prevence – genetická analýza – guidelines – doporučený postup – náhlá srdeční smrt – dědičná kardiovaskulární onemocnění
Náhlá smrt je definována jako neočekávané úmrtí z přirozených příčin, které nastává do jedné hodiny od začátku příznaků u doposud zdravého jedince, případně u jedince, jehož onemocnění nebylo natolik závažné, aby se očekávalo jeho úmrtí (1). Vzhledem ke skutečnosti, že tato definice z roku 1982 zcela neodpovídala reálné situaci, např. pokud k úmrtí dojde ve spánku, o samotě, bez svědků, případně se jedná o pitvu, kdy k úmrtí došlo v blíže neurčeném intervalu před nálezem těla (2), byl posléze časový interval, před kterým byl zemřelý naposledy viděn zdráv v definici náhlé smrti rozšířen na 24 hodin (3). Jako náhlá (srdeční) smrt by však měla být klasifikována i úmrtí pacientů resuscitovaných a přežívajících po srdeční zástavě, kteří umírají na nevratné poškození mozku (2).
Existující nejednotnost a variabilita v definici náhlé smrti (sudden death, SD) a náhlé srdeční smrti (sudden cardiac death, SCD) společně s nejednotným systémem autoptické praxe napříč kontinenty i jednotlivými evropskými státy se projevuje v absenci přesných epidemiologických dat, která by reflektovala incidenci obou jednotek (4). S jistotou však incidence náhlé srdeční smrti stoupá s věkem, v korelaci se stoupající incidencí ischemické choroby srdeční. U osob ve věku 60 let se pohybuje kolem 2 : 1000/rok až 200 : 1000/rok. Oproti tomu v nízkých věkových skupinách průměrná incidence náhlé srdeční smrti osciluje kolem 0,01 : 1000/rok (5,6) .
Náhlé úmrtí je ve většině případů způsobeno onemocněním srdce a označováno jako náhlá srdeční smrt (sudden cardiac death – SCD). Na rozdíl od vyšších věkových skupin, kde jako podklad dominují aterosklerotické změny věnčitých tepen, jsou za většinu případů SCD u jedinců mladších 40 let zodpovědná dědičná kardiovaskulární onemocnění typu kardiomyopatií, arytmických syndromů a dědičných aortopatií (2,7,8). S ohledem na skutečnost, že predispozice pro většinu dědičných kardiovaskulárních onemocnění, stejně jako pro familiární hypercholesterolemii, se dědí s pravděpodobností 50 % bez závislosti na pohlaví, existuje významné riziko rozvoje identického onemocnění s rizikem srdeční zástavy u prvostupňových příbuzných (obr. 1).

Morfologický pitevní nález je u většiny případů náhlé srdeční smrti ve vyšším věku na podkladě aterosklerózy věnčitých tepen jednoznačný a nesporný. Úskalím pitevní diagnostiky u diagnostických jednotek, které jsou zodpovědné za většinu případů náhlé srdeční smrti u mladých jedinců, je však mnohdy jejich nepřesvědčivý morfologický obraz (v případě kardiomyopatií), absence jakéhokoliv makroskopicky či mikroskopicky postihnutelného patologického nálezu (v případě arytmických syndromů), případně nejednoznačná příčinná souvislost zjištěných hraničních patologických nálezů s náhlým úmrtím dotyčného jedince (nelimitní ateroskleróza věnčitých tepen, vrozené vývojové vady věnčitých tepen, fokální myokarditida, lipomatóza pravé komory aj.) (2).
S ohledem na složitou diagnostiku uvedených jednotek byly v rámci AECVP (Association for European Cardiovascular Pathology) vytvořeny doporučené postupy, tzv. guidelines, jejichž cílem je harmonizovat pitevní postup a diagnostiku u případů náhlé srdeční smrti a formulovat minimální standard rutinně prováděné pitvy u těchto případů (2,9), vč. spektra doplňujících laboratorních vyšetření.
Dle výsledků pitvy a na podkladě zjištěných makroskopických a mikroskopických nálezů jsou mezinárodně definovány kategorie typů náhlé kardiální smrti ve smyslu kardiomyopatie, náhlá arytmická smrt a náhlá nevyjasněná smrt u jedinců mladších, nebo starších 1 roku. Odděleně je zmiňováno náhlé úmrtí epileptika, kdy epilepsie může být nesprávně stanovenou diagnózou pro bezvědomí provázející setrvalé komorové arytmie, případně některé epilepsie mohou představovat formy mozkových i srdečních kanálopatií (tab. 1).

Doporučené postupy AECVP dále vymezují souhrn suspektních nálezů, u kterých by mělo být provedeno post mortem genetické vyšetření, někdy nazývané jako tzv. molekulární pitva, s cílem přesného určení příčin srdeční smrti a s tím související možnosti primární prevence SCD u prvostupňových příbuzných (2,8) (tab. 2).

Post mortem genetické vyšetření u zemřelého jedince by mělo být následováno, nebo za ideálních podmínek současně doprovázeno klinicko-genetickou konzultací a kardiologickým screeningovým vyšetřením prvostupňových příbuzných (2,11,12).
Zjištění příčin náhlé srdeční smrti tedy představuje multidisciplinární proces, do kterého jsou zapojeni pitvající lékaři, kliničtí genetici, molekulární genetici, kardiologové dětští i dospělí a v neposlední řadě i psycholog, event. neurolog, lipidolog, praktický lékař a další odbornosti dle individuálních kritérií pro jednotlivé případy (8,12) (obr. 2). S ohledem na komplexní problematiku se tento typ diagnostiky soustřeďuje do center terciální péče (tedy především na fakultní a univerzitní pracoviště).

Prvotní snahy o souvislou kardiogenetickou post mortem diagnostiku případů SCD byly zaznamenány již v roce 2009 na Ústavu soudního lékařství FNUSA v Brně ve spolupráci s Interní kardiologickou klinikou FN Brno (13).
Česká společnost soudního lékařství a soudní toxikologie (ČSSLaST) reagovala na mezinárodní odborný vývoj v roce 2018 ustanovením pracovní skupiny Náhlá srdeční smrt a v roce 2020 formulovala a následně schválila vlastní Standardní operační a diagnostický postup na soudnělékařských pracovištích v případech náhlé srdeční smrti u jedinců do 40 let věku.
V předkládaném textu představujeme výsledky multicentrického a multidisciplinárního řešení případů náhlé srdeční smrti v ČR v letech 2016–2021. Jedná se o pilotní projekt finančně zastřešený grantem ministerstva zdravotnictví ČR s reg. č. NV18 - 02-00237. Do projektu se postupně zapojila většina pracovišť soudního lékařství v Česku a na Moravě, klinicko-genetickou konzultaci prováděli genetici v IKEM, FN Motol Praha a FN Olomouc. Kardiologické screeningové vyšetření a dle potřeby individualizovanou péči o příbuzné v riziku provádí pracoviště Kardiologie IKEM, Dětské kardiocentrum FN Motol, Kardiologie FN Olomouc a Pediatrie FN Královské Vinohrady (tab. 3). Molekulárně genetická diagnostika se prováděla centrálně v laboratoři Ústavu biologie a lékařské genetiky FN Motol.

Cílem projektu bylo v rámci nově ustanovené a stále se rozvíjející multicentrické a multidisciplinární spolupráce v ČR identifikovat reprezentativní soubor případů náhlé srdeční smrti. Následně na základě zájmu příbuzných a po získání informovaného souhlasu osob blízkých náhle zemřelého zjistit molekulární příčiny náhlé kardiální smrti a zhodnotit výstupy a dopady tohoto vyšetření na péči o prvostupňové příbuzné s cílem primární prevence život ohrožujících poruch srdečního rytmu u jedinců v riziku.
METODIKA
Pitvající lékař v případě podezření na SCD odebral dle instrukcí tkáň vhodnou pro izolaci DNA (tab. 4). Preferován byl vzorek tkáně bohaté na lymfocyty (slezina, játra, uzliny o velikosti 2 cm3) či vzorek periferní krve odebrané do K3EDTA. Odebraná tkáň byla uložena do hluboko mrazících boxů na -80 °C (v případě, že pracoviště nemělo možnost hlubokého zmražení vzorku, bylo doporučeno zmrazit tkáň v běžném laboratorním mrazáku a zajistit jeho co nejčasnější transport ke genetické analýze). Alternativou bylo skladování vzorku tkáně v roztoku RNA later, a následné uchování až po dobu 4 týdnů v chladícím boxu. V dalším kroku kontaktoval pitvající lékař příbuzné, kterým vysvětlil jeho podezření a předal jim informační dopis, kde je podstata vyšetření zemřelého spolu s nabídkou kardiologického vyšetření příbuzných srozumitelně popsána. V informačním dopise jsou uvedeny důležité telefonické i elektronické kontakty, které mohou pozůstalí využít. Pokud pozůstalí předběžně s vyšetřením zemřelého souhlasili a projevili zájem o vlastní kardiologické vyšetření, zanechali u pitvajícího lékaře podepsaný informovaný souhlas a kontakt na sebe. Současně byli informováni koordinátoři studie, kteří zajistili přepravu materiálu na molekulárně genetické pracoviště a v rozumném intervalu (cca 3 měsíce) po náhlém úmrtí kontaktovali pozůstalé a pozvali je na konzultaci.

Kromě případů náhlé kardiální smrti rozeznaných a odeslaných pitvajícími lékaři, byly v rámci projektu řešeny další případy, na které byli kardiogenetici upozorněni příbuznými, kteří si možnost post mortem vyšetření a jejich vlastní screeningové vyšetření sami vyhledali, nebo jim toto vyšetření bylo doporučeno ošetřujícím lékařem. Některé rodiny s případem náhlého úmrtí kojence byly doporučeny na vyšetření přes specializovanou ambulanci v Thomayerově nemocnici (www.ftn.cz/ambulance-pro-rizikove-deti-a-sids/).
Na podkladě studia pitevních protokolů, soudnělékařských diagnóz a výsledků histologického vyšetření srdce byly případy náhlé srdeční smrti rozděleny do kategorií náhlá arytmická smrt (sudden arrhytmic death syndrome – SADS), náhlá nevysvětlitelná smrt (sudden unexplained death syndrome – SUDS), náhlé úmrtí kojence (sudden infant death syndrome – SIDS), náhlé nevysvětlitelné úmrtí dítěte (sudden unexplained death in infancy – SUDI), aneuryzma/disekce hrudní aorty a kardiomyopatie hypertrofická (HCM), arytmogenní kardiomyopatie (ARVC), dilatovaná (DCM) a nekompaktní (left ventricle non-compact cardiomyopathy – LVNC) (tab. 1, obr. 3).

K izolaci DNA z dodaného biologického vzorku byl použit automatický izolátor MagCore HF16 plus (RBC Bioscience, Taiwan) pracující na principu extrakce na magnetických kuličkách. Kvalita extrahované DNA byla kontrolována na fluorimetru Qubit 2.0 (Invitrogen, USA). Sekvenační knihovna byla připravena s použitím cílených prób pro 100 vybraných genů pro kardiovaskulární onemocnění a prostřednictvím exomových panelů od firmy Sophia Genetics: Sophia Clinical Exome Solution a Sophia Whole Exome Solution, od firmy TwistBioscence panel Twist Comprehensive Exome). Samotná sekvence byla prováděna na NGS (Illumina, USA). Získaná sekvenační data byla hodnocena v programu Sophia DDM (Sophia Genetics, Švýcarsko) s použitím pomocných programů a databází (Alamut, ClinVar, Franklin Genoox aj.). Detekované varianty byly klasifikovány na základě standardů ACMG/ AMP (American College of Medical Genetics and Genomics and the Association for Molecular Pathology) do 5 DNA tříd (class 1–5): benigní = class 1, pravděpodobně benigní = class 2, varianta nejasného významu = class 3, pravděpodobně patogenní = class 4, patogenní = class 5 (14). U variant třídy 3 až 5 byla provedena segregační analýza u příbuzných pomocí sekvenování cíleného úseku Sangerovým sekvenováním.
Pokud se výše uvedenými metodami nepodařilo získat dostatečně kvalitní materiál ze zemřelého, bylo diagnostické genetické vyšetření pomocí NGS zacíleno na jasně postiženého prvostupňového příbuzného, nebo výjimečně na oba žijící rodiče (při předpokladu, že predispozice pro onemocnění musela být od jednoho z nich zděděna). Genetické screeningové vyšetření u zdravých potomků náhle zemřelého při chybění materiálu bylo v iniciální fázi projektu také prováděno, ale později s ohledem na minimální výpovědní hodnotu bylo od tohoto vyšetřování upuštěno.
Příbuzní byli objednáni nejdříve ke komplexní klinicko-genetické konzultaci, kde s nimi byla událost podrobně probrána a zjištěny okolnosti úmrtí. Příbuzným byla vysvětlena povaha pravděpodobné dědičné příčiny, vysvětlen způsob dědičnosti a z toho vyplývající rizika pro další rodinné příslušníky. Během konzultace byl sestaven rodokmen alespoň ve 3 generacích s co nejpodrobnějšími anamnestickými daty. Dále byl získán souhlas s vyžádáním lékařské dokumentace náhle zemřelého i s molekulárně genetickým vyšetřením zemřelého a jich samotných. Toto iniciovalo návazné laboratorní vyšetření. Příbuzní byli následně pozváni na odpovídající kardiologická screeningová vyšetření (8) a konzultaci jejich výsledků spolu s výsledkem molekulárně genetického vyšetření v intervalu cca 4–6 měsíců.
MATERIÁL A PACIENTI
V rámci časového intervalu studie (2016–2021) bylo řešeno celkem 133 případů náhle zemřelých osob (36 žen a 97 mužů, průměrný věk 30,7 let), jejichž úmrtí po provedení zdravotní pitvy odpovídalo náhlé srdeční smrti s indikací genetického vyšetření. Tyto případy pocházely z celkem 128 rodin, v 5 rodinách se jednalo o vícenásobné úmrtí (obr. 3). V 75/133 případů (56,4 %) se jednalo o přímou indikaci pitvajících lékařů. U zbylých 58 případů byli řešitelé přímo kontaktováni pozůstalými na základě jejich osobní iniciativy nebo doporučení ošetřujícího lékaře.
S genetickým vyšetřením zemřelého a kardiologickým screeningovým vyšetřením nevyslovili příbuzní či blízcí zemřelého primárně souhlas ve 4/133 (3,0 %) případů. Svůj prvotní souhlas s vyšetřením později odvolalo či nepotvrdilo dalších 9/133 příbuzných (6,8 %). V 5/133 případech (3,8 %) po podrobné analýze pitevního nálezu a okolností úmrtí jsme nakonec vyloučili nebo neprokázali jistou dědičnou kardiální příčinu a molekulárně genetické vyšetření nebylo indikováno (obr. 3). Celkem se tedy naše klinická a anamnestická analýza náhlých srdečních úmrtí s indikací genetického vyšetření týkala 115 případů (34 žen a 81 mužů).
Ze skupiny rodin, které si možnost vyšetření vyžádaly samostatně, se v 7/58 případů (12,1 %) nepodařilo získat materiál ze zemřelého ani od jeho obou rodičů, nejčastěji z důvodu již příliš dlouhého intervalu od události. V 5/7 případů jsme provedli molekulárně genetické vyšetření u nepostižených prvostupňových příbuzných. Později jsme od tohoto postupu upustili a příbuzným dle platných standard doporučili kardiologickou dispenzarizaci s kardiologickým screeningovým vyšetřením v pravidelných intervalech do 50. – 60. roku života (8,15,16). Z této kohorty jsme vyšetřovali dva případy fibromuskulární dysplasie koronárních cév, tedy onemocnění zatím s neznámou genetickou příčinou (17). S rodinami jsme se proto v obou případech, kdy nebyla provedena molekulární diagnostika, dohodli na jen kardiologickém screeningovém vyšetření. Z výše uvedeného vyplývá, že molekulárně genetické vyšetření bylo provedeno celkem ve 106 případech (obr. 3, tab. 5).

Kardiologické screeningové vyšetření proběhlo u 328 přímých příbuzných formou klidového EKG a transthorakální echokardiografie v případě vysloveného podezření na dědičnou kardiomyopatii u zemřelého vyšetřovaného blízkého příbuzného. Tato vyšetření byla doplněna Holter EKG a námahovým EKG, pokud bylo u zemřelého vysloveno podezření na dědičný arytmický syndrom jako příčinu úmrtí (zejména případy SADS/SUDS). V případě post mortem podezření na ARVC jsme dále doplnili vyšetření pozdních potenciálů. Příbuzní náhle zemřelých s akutní disekcí byli vyšetřeni echokardiograficky a aorta znázorněna v celém jejím průběhu pomocí CT/MR. Pokud u vyšetřených byly nalezeny známky některého z dědičných kardiovaskulárních onemocnění, byla doplněna další vyšetření dle individuální diagnózy k lepšímu odhadu rizika život ohrožujících arytmií (LTVA) formou magnetické rezonance srdce (MRI), programované stimulace komor, nebo farmakologických zátěžových testů. Indikace výše uvedených vyšetření odpovídají doporučeným postupům (8).
VÝSLEDKY
Po iniciální stratifikaci jsme komplexní kardiogenetické vyšetření případů náhlé srdeční smrti provedli u 115 osob (34 žen, 81 mužů). Z důvodů chybění materiálu nebo smysluplnosti molekulárně genetického vyšetření byla laboratorní molekulárně genetická analýza provedena ve 106 případech. Průměrný věk zemřelých byl 31,8 let, při vyjmutí kategorie SIDS a SUDI dosáhl průměrný věk zemřelých 34,2 let.
Vyhodnocení anamnestických dat (tab. 5).
Rodinná anamnéza
V rodinách náhle zemřelých jsme zaznamenali náhlé úmrtí u prvostupňového nebo druhostupňového příbuzného ve 22/115 (19,1 %) případů. Ve vyšetřovaných rodinách byl ještě před zahájením kardiologického kaskádového screeningu jeden nebo více příbuzných v odborné péči pro onemocnění srdečního svalu ve 35/115 případů (30,4 %).
Osobní anamnéza
Z hlediska anamnestických údajů byl údaj o dlouhodobých potížích (např. kolaps či synkopa v anamnéze, epilepsie v anamnéze, dlouhodobá nepřirozená únava, dušnost, palpitace apod.) dohledán v 57/115 případů (49,5 %). V 51/115 případů (42,6 %) náhle zemřelý jedinec dlouhodobými potížemi netrpěl, ale náhlému úmrtí předcházely obdobné obtíže v intervalu kratším než 24 hodin, v 7 případech nebyly tyto údaje dostupné. Horečnatý infekt předcházel úmrtí v 19/115 případů (16,5 %).
Denní doba
Z hlediska úmrtí v určité denní době se jednalo u 59/115 případů (51,0 %) o úmrtí ve dne, u 35/115 případů (30,4 %) o úmrtí v noci a u 21/115 případů (18,2 %) nebyla denní doba známa či zjistitelná. Ve všech kategoriích pitevních diagnóz kromě SIDS a disekce převyšoval počet úmrtí ve dne počet úmrtí v noci.
Místo úmrtí
Nejčastějším místem úmrtí byl domov, kde došlo k úmrtí celkem v 74/115 případů (64,3 %). Následovalo veřejné místo, kde došlo k úmrtí ve 14/115 případů (12,2 %). V lůžkovém zdravotnickém zařízení, na jiném neurčeném místě a kategorii „místo úmrtí neznámo“ naplnilo shodně vždy 6/115 případů (7,0 %). Na jiném blíže určeném místě, např. ve volné přírodě došlo k úmrtí v 7 případech (6,1 %), v práci v 6 případech (5,2 %).
Denní činnost a fyzická aktivita
Z hlediska činnosti a tělesné aktivity docházelo nejčastěji k úmrtí ve spánku, a to ve 41/115 případů (35,7 %). Ve 20/115 případů (17,4 %) došlo ke kolapsu či úmrtí při běžné denní činnosti, v 18 případech (15,7 %) při bdělém odpočinku, v 9 případech (8,7 %) při lehké fyzické aktivitě, v 6 případech (6,1 %) při těžké fyzické aktivitě (dlouhodobý běh, těžká fyzická práce), ve 4 případech (2,6 %) při středně těžké fyzické aktivitě a v 15 případech nebyly údaje o činnosti či aktivitě, během které došlo ke kolapsu, známy.
Molekulárně genetické vyšetření
Molekulárně genetické vyšetření bylo provedeno přímo z izolované DNA náhlé zemřelého v 75/106 případů (70,8 %). Pro nekvalitní DNA bylo u dalších 21/106 případů (19,8 %) analýza NGS provedena u jasně postiženého prvostupňového příbuzného a v 10/106 případů (9,4 %) u obou rodičů, kdy v případě nálezu suspektní varianty byla tato cíleně potvrzena u zemřelého. Vysoce pravděpodobná nebo jistá molekulární etiologie (DNA class 4 nebo 5) byla ze 106 molekulárně geneticky vyšetřených případů SCD zjištěna ve 21/106 případů
Kategorie úmrtí dle výsledků pitvy
Dle pitevních diagnóz jsme řešili celkem 22 případů SUDS/ SUDI (7 žen a 15 mužů), 20 případů SADS (8 žen a 12 mužů), 7 případů SIDS (2x ženské pohlaví a 5x mužské pohlaví), 12 případů HCM, 13 případů DCM, 22 případů ARVC, 1 případ LVNC a celkem 11 případů náhlého úmrtí na disekci aorty nebo koronárních cév (obr. 2, tab. 5 a 6).
SADS
Do kategorie SADS bylo zařazeno 20 případů, 8 žen a 12 mužů, jejichž průměrný věk byl 31 let (4–55 let). 15 případů bylo dodáno z pracovišť soudního lékařství, v jednom případě bylo vyšetření doporučeno ošetřujícím kardiologem a 4 případy byly zařazeny na základě zájmu rodiny po úmrtí v rodině. Ve 2/20 případů (10,0 %) byla identifikována varianta DNA class 4 nebo 5 v genu KCNH2 a genu RYR2. Genetická stratifikace tedy stanovila diagnózu dědičného arytmického syndromu typu dlouhého QTc intervalu typu 2 (LQT2) v 1/20 případů (5,0 %) a katecholaminergní polymorfní komorovou tachykardii (CPVT) v 1/20 případů (5,0 %). Vyšetřeno bylo celkem 62 příbuzných, přičemž u 13 z nich (20,6 %) byl zjištěn rizikový genotyp nebo fenotyp dědičného onemocnění a nabídnuta kardiologická dispenzarizace s individualizovanou péčí (tab. 6 a 7).

× 2 příbuzní byli vyšetřeni již jen klinicky, molekulárně genetické vyšetření “naslepo” nebylo doporučeno
# prvostupňoví příbuzní zemřeli na SUDS (2 případy), SADS (3 případy), HCM (1 případ), akutní disekce aorty (1 případ)

SUDS + SUDI
Do této kategorie bylo zařazeno 22 případů, 7 žen a 15 mužů, jejichž průměrný věk byl 30,4 let (2 měsíce – 54 let). 18 případů bylo dodáno z pracovišť soudního lékařství, ve 4 případech bylo vyšetření doporučeno ošetřujícím kardiologem. 4/22 (18 %) případů spadalo na základě genetické analýzy do DNA class 4 nebo 5 (geny TTN, FLCN, RYR2 a TNNT2). Genetická stratifikace stanovila diagnózu dilatační/arytmogenní kardiomyopatie ve 3/22 případů (13,6 %), diagnózu katecholaminergní polymorfní komorové tachykardie (CPVT) v 1/22 případů (4,5 %). Vyšetřeno bylo celkem 67 příbuzných, přičemž u 20 z nich (33,3 %) byl zjištěn rizikový genotyp nebo fenotyp dědičného onemocnění a nabídnuta kardiologická dispenzarizace s individualizovanou péčí (tab. 6 a 7).
HCM
Do kategorie HCM bylo zařazeno 12 případů. Celkem se jednalo o 1 ženu a 11 mužů, jejichž průměrný věk byl 37,8 let (14–59 let). 7 případů bylo dodáno z pracovišť soudního lékařství, ve 3 případech doporučil vyšetření spádový kardiolog příbuzného a 2 případy byly zařazeny na základě zájmu rodiny po úmrtí v rodině. 4/12 (33 %) případy spadaly na základě genetické analýzy do DNA class 4 nebo 5 (geny MYBPC3, FHL1, MYPN, GLA – KCNQ1). U náhlého úmrtí ženy ve věku 59 let bylo dle genetické stratifikace zjištěno nerozeznané střádavé onemocnění typu sfingolipidózy ve smyslu Fabryho choroby (GLA gen). Současně měla i dědičný arytmický syndrom typu dlouhého QTc intervalu typu 1 (LQT1 – gen KCNQ1). Tato rodina se přihlásila sama, neboť se v ní udála dvě další náhlá úmrtí a sestra zemřelé měla dlouhodobé srdeční obtíže bez dosud přesně stanovené diagnózy. Post mortem vyšetření její sestry vedlo ke stanovení diagnózy u žijící příbuzné. Toto umožnilo nabídnout jí a dalším dvěma genotyp pozitivním rodinným příslušníkům péči ve specializovaném centru, včetně enzymové substituční terapie tohoto onemocnění, jejíž efektivita je prokázaná ve vícero studiích (18,19). Vyšetřeno bylo celkem 44 příbuzných, přičemž 16 z nich (37,2 %) má fenotyp dědičného kardiovaskulárního onemocnění a/nebo nese rizikový genotyp a byla jim nabídnuta kardiologická dispenzarizace s individualizovanou péčí (tab. 6 a 7).
DCM + 1 případ LVNC
Do této kategorie bylo zařazeno 13 případů s pitevním obrazem DCM a 1 případ s morfologickým nálezem LVNC. Celkem se jednalo o 3 ženy a 11 (1 LVNC) mužů, jejichž průměrný věk byl 33,4 let (8–48 let). 5 případů bylo dodáno z pracovišť soudního lékařství, 5 případů bylo vyšetřeno na základě doporučení ošetřujícího kardiologa jednoho z žijících příbuzných a 4 případy byly zařazeny na základě zájmu rodiny po úmrtí v rodině. 5/14 (35,7 %) případů spadalo na základě genetické analýzy do DNA class 4 nebo 5 (geny TTN (3x), FLNC stop, RBM20). Všechny geny jsou rozeznanými molekulárními příčinami dilatační kardiomyopatie s rizikem významných poruch srdečního rytmu. Vyšetřeno bylo celkem 41 příbuzných, přičemž u 11 z nich (29,7 %) byl zjištěn rizikový genotyp a/nebo fenotyp kardiomyopatie a byla jim nabídnuta kardiologická dispenzarizace s individualizovanou péčí (tab. 6 a 7).
ARVC
K diagnóze ARVC bylo přiřazeno 22 případů, 10 žen a 12 mužů, jejichž průměrný věk byl 36,0 let (17–56 let). 14 případů bylo dodáno z pracovišť soudního lékařství, v jednom případě doporučil k vyšetření ošetřující kardiolog nemocného příbuzného a 8 případů bylo zařazeno na základě zájmu rodiny po úmrtí v rodině. 2 případy spadaly na základě genetické analýzy do DNA class 4 nebo 5 (pozitivita SCN5A, FLNC stop). Oba geny patří mezi rozeznanou příčinou tohoto typu kardiomyopatie s rizikem život ohrožujících arytmií. Vyšetřeno bylo celkem 73 příbuzných, z nichž u 11 (22,5 %) byl zjištěn rizikový genotyp a/nebo fenotyp dané kardiomyopatie a byla jim nabídnuta kardiologická dispenzarizace s individualizovanou péčí (tab. 6 a 7).
Akutní disekce
Mezi tuto kategorii bylo zařazeno celkem 11 případů, přičemž ve 2 případech se jednalo o fibromuskulární dysplazii koronárních tepen (oba případy muži), v jednom případě o spontánní disekci koronární tepny (žena) a zbylých 8 případů spadalo mezi akutní disekce aorty. Celkem se jednalo o 1 ženu a 10 mužů, jejichž průměrný věk byl 33,3 let (16–49 let). 5 případů bylo dodáno z pracovišť soudního lékařství, ve 3 případech doporučil vyšetření ošetřující kardiolog jednoho z příbuzných a 3 případy byly zařazeny na základě zájmu rodiny po úmrtí v rodině. 3 případy spadaly na základě genetické analýzy do DNA class 4 nebo 5 (geny COL3A1 2x, TGFBR1). Genetická stratifikace určila diagnózu vaskulárního Ehrles-Danlosova syndromu (EDS) u dvou vyšetřených náhle zemřelých mladých mužů, u třetího z nich diagnózu Loyes-Dietzova syndromu typu 1 (LDS1). Vyšetřeno bylo celkem 31 příbuzných, přičemž 9 z nich (29,0 %) je v riziku akutní disekce nebo již s dilatací aorty a byla jim nabídnuta kardiologická dispenzarizace s individualizovanou péčí (tab. 6 a 7).
SIDS
Do kategorie SIDS bylo zařazeno 7 případů. Celkem se jednalo o 2 dívky a 5 chlapců, jejichž průměrný věk byl 0,3 let (3 týdny – 5 měsíců). 4 případy byly dodány z pracovišť soudního lékařství, zbylé případy byly zařazeny na základě zájmu rodiny po úmrtí v rodině a doporučení specializované ambulance. Jistá příčinná varianta nebyla v žádném z případů nalezena. V jedné rodině byla nalezena varianta nejasného významu, která souvisí s fenotypem a v rodině u 5 klinicky postižených, prvo - i druhostupňových příbuzných segreguje. Tato varianta je popsána v kapitole VUS níže v textu (tab. 6 a 8).
Vyšetření zdravých potomků náhle zemřelých
Molekulárně genetické vyšetření s cílem vyloučení známé formy dědičného onemocnění u prvostupňových příbuzných – zdravých potomků, kteří nevykazovali žádné jisté známky dědičného kardiovaskulárního onemocnění, nepřineslo nález žádné jisté příčinné varianty DNA (tab. 6). V těchto případech se jednalo o pitevní diagnózu hypertrofické kardiomyopatie nebo akutní disekce aorty ve vždy jednom případě, v dalších 2 případech dle pitevní zprávy se jednalo o SUDS a ve 3 případech o SADS prvostupňového příbuzného.
LQT5 lite
Ve skupinách SUDS, SADS, ARVC a DCM jsme vždy u jednoho zemřelého identifikovali známou rizikovou variantu DNA v genu pro draslíkový kanál: NM_000219.5(KCNE1):c.253G>A p. (Asp85Asn). Přítomnost této varianty definuje diagnózu syndromu dlouhého QTc intervalu typu 5 s lehkým průběhem a neúplnou penetrancí (20,21).
DNA varianty neznámého významu (VUS, class 3)
V rámci molekulárně genetického vyšetření jsme nalezli 29/106 (27,3 %) variant nejasného významu. Jednalo se o DNA varianty v genech, jejichž změny jsou rozeznanou příčinou jednotlivých kardiomyopatií či dědičných arytmických syndromů. Nicméně i přesto, že se jednalo dle ACMG kritérií o varianty velmi vzácné (kritérium PM2) a dle funkčních programů významně měnící funkci genového produktu (kritérium PP3), jedná se dle uznaných diagnostických kritérií o varianty nejasného významu (VUS) (tab. 8). Tuto klasifikaci nepozměnil ani fakt, že některé z nich segregovaly s onemocněním u prvo - i druhostupňových příbuzných (kritérium PP1) dle ACMG: Genetic Variant Interpretation Tool University of Maryland School of Medicine (umaryland.edu) (14). V případě náhlého úmrtí novorozence jsme nalezli variantu neznámého významu v genu KCNT1, která ale v rozsáhlé rodině segregovala s fenotypem a tento souvisí s popisovanými projevy onemocnění v literatuře (22) (tab. 8). Protože se jedná o onemocnění, které je dle literatury možné specificky léčit, tuto variantu jsme rodině reportovali a odkázali je na specializované pracoviště.
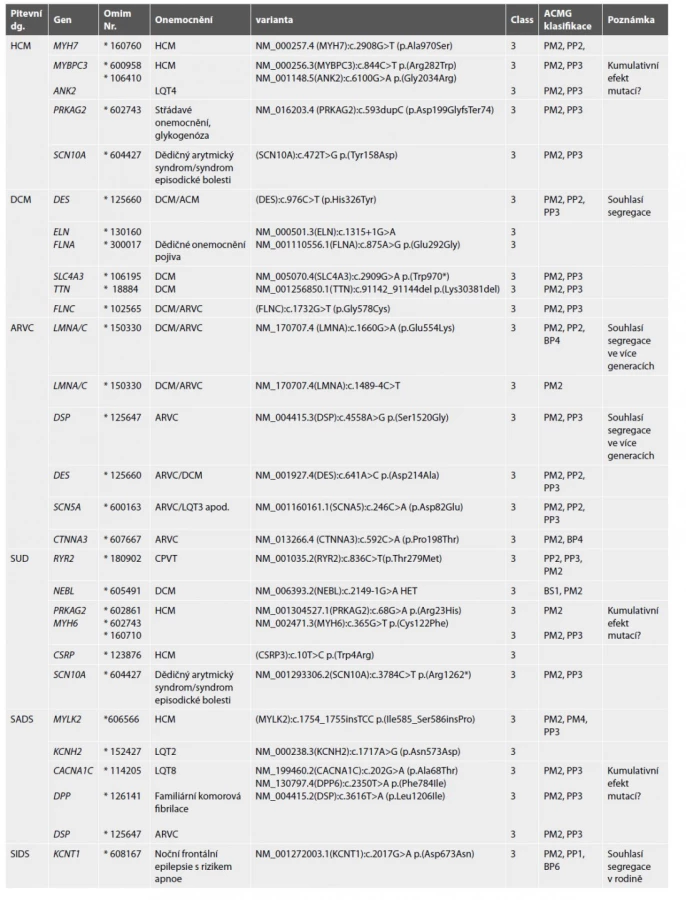
Kardiogenetické vyšetření příbuzných
Z celkem 328 vyšetřených příbuzných mělo pozitivní genotyp, nebo fenotyp dědičného kardiovaskulárního onemocnění 83 vyšetřených příbuzných (25,3 %). Tento počet zahrnuje i příbuzné, kteří nesou variantu VUS a současně mají známky dědičného onemocnění. V jednom případě byl na základě kardiologické rizikové stratifikace implantován defibrilátor. V případě LDS1 syndromu podstoupila v rámci primární prevence akutní disekce sestra zemřelého kardiochirurgickou náhradu aorty ascendens. V ostatních případech obdrželi příbuzní se známkami dědičného onemocnění s klinickými příznaky nebo jasným rizikovým genotypem odpovídající doporučení pro úpravu životního stylu a/ nebo terapii jako primární prevenci náhlého úmrtí. Dále jsme příbuzným nabídli dlouhodobou kardiologickou dispenzarizaci. Pravidelná kardiologická vyšetření jsme doporučili všem prvostupňovým rodinným příslušníkům, kde aktuálně nebyl nalezen jasný fenotyp ani genotyp dle současných doporučení (8,14).
DISKUZE
Prezentujeme doposud nejrozsáhlejší studii molekulárních příčin náhlého úmrtí v České republice. Tato pilotní studie byla zpočátku omezena na fakultní pracoviště v Praze a Olomouci. Soustavnou přednáškovou činností a komunikací se podařilo postupně významně rozšířit povědomí o této problematice nejen v kardiologické, ale i soudnělékařské společnosti v ČR (tab. 3). Tím se velmi navýšil počet spolupracujících odborníků a následně i počet vyšetřených případů oprávněných k tomuto typu diagnostiky.
Studie ověřila, že naprostá většina rodin (120/128, 93,7 %) má zájem o vyšetření příčiny úmrtí u jejich příbuzného i o kardiologické screeningové vyšetření sebe a nezletilých potomků. Ze získaných anamnestických dat vyplývá, že až 1/3 případů náhlé smrti se udála v rodinách s předchozími případy náhlého úmrtí nebo s příbuznými, kteří již byli ve specializované kardiologické péči. Tento fakt podporuje indikaci kaskádového rodinného screeningu u pacientů s jinak nevysvětlitelným srdečním selháním s redukovanou i zachovanou systolickou funkcí či závažnými poruchami srdečního rytmu nebo rozšířením aorty bez anamnézy hyperlipoproteinemie nebo dlouhodobě neléčené arteriální hypertenze.
Naprostá většina náhlých úmrtí se udála ve dne a v domácím prostředí při běžné činnosti nebo ve spánku, což odpovídá mezinárodním studiím (23). Pouze 6,1 % zemřelých zemřelo při intenzivní sportovní nebo fyzické aktivitě. Do studie nebyl zařazen ani jeden případ náhlého úmrtí vrcholového sportovce.
Velkou výhodu vidíme v centralizaci molekulárně genetického vyšetření, čímž jsme zajistili jednotné metodické postupy a v neposlední řadě i posuzování identifikovaných DNA variant se standardizovaným přidělením odpovídajících diagnostických kritérií dle ACMG, která se jinak mohou v molekulárně genetické praxi lišit mezi jednotlivými laboratořemi.
Celkové výsledky molekulárně genetického vyšetření u zemřelých, včetně nálezů v jednotlivých podskupinách dle pitevní diagnózy v naší studii odpovídají publikovaným mezinárodním studiím obdobného rozsahu (7,24–26). Na základě našich zkušeností a v souladu s mezinárodními studiemi lze tedy očekávat, že genetická stratifikace vede k odhalení jednoznačně dědičného onemocnění u zhruba 20–25 %, tedy asi v 1/5–1/4 případů.
Tento poznatek je klíčový pro komunikaci s pozůstalými, aby jejich očekávání od tohoto typu vyšetření byla reálná. Zároveň je důležité zdůraznit, že post mortem diagnostika je potřebná především pro pozůstalé, pro jejich primární prevenci život ohrožujících arytmií. Bez jejich souhlasu a zájmu nemá smysl takto profesionálně, finančně i časově náročné vyšetření zahajovat. V tomto smyslu je klíčová role pitvajícího lékaře, který informuje pozůstalé o závěrech pitvy, i konzultujícího klinického genetika, který musí s pozůstalými detailně probrat všechny možné výstupy komplexního vyšetření.
V rámci molekulárně genetické analýzy jsme potvrdili výrazné proarytmické působení trunkujících variant v genu pro filamin C (FLNC) zejména u mužského pohlaví (27). V naší studii jsme ji identifikovali jako jasnou molekulární příčinu u 3 jedinců ze skupiny SUDS, DCM a ARVC. Různorodé pitevní nálezy korelují s popsanými klinickými projevy FLNC genu a ukazují také na známý fakt, že arytmické komplikace mohou předcházet rozvoji jednoznačných strukturálních změn srdečního svalu (28,29).
V rámci naší studie jsme vyšetřovali významně více postižených mužů (81 versus 34 žen). Rozdíly v závažnosti průběhu kardiomyopatií i počtu náhlých úmrtí mužů byly opakovaně popisovány (30,31) a v našem souboru vícekrát pozorovány, zejména pro příčinné varianty v genu titin (TTN) a filamin C (FLNC). Jako názorný příklad uvádíme případ náhlé zemřelého 13letého chlapce, který zemřel během distanční výuky doma. Před smrtí si opakovaně stěžoval na rychlé bušení srdce, nikdy ale neupadl do bezvědomí. Protože i starší sestra zemřelého měla palpitace, v minulosti diagnostikované jako život neohrožující supraventrikulární arytmie a podstoupila úspěšnou radiofrekvenční ablaci, nepřikládali rodiče obtížím takovou závažnost a chlapce nenechali časně vyšetřit. Genetické vyšetření nakonec prokázalo příčinnou trunkující variantu v genu pro FLNC, kterou nesla sestra, matka a její matka. Podrobné kardiologické screeningové vyšetření u žijících příbuzných žen neprokázalo žádnou jistou formu strukturálního onemocnění, opakovaně nebyly v dlouhodobé monitoraci a při zátěži prokázány významné poruchy srdečního rytmu. Sestra i matka dostaly preventivě betablokátory, byla jim nabídnuta dlouhodobá kardiologická dispenzarizace a prozatím jim nebyl implantován defibrilátor (ICD) pro převyšující rizika implantačního výkonu a jeho přítomnosti nad rizikem arytmií (10) (obr. 4).

Na druhou stranu se kardiomyopatie u genotyp pozitivních žen mohou projevit až ve věku nad 50 let (32), a proto je vhodné zvažovat kaskádový rodinných screening nebo post mortem genetické vyšetření i v těchto případech. V rámci studie jsme tuto zkušenost ověřili v případě náhlé zemřelé 59leté ženy s hypertrofickou kardiomyopatií a geneticky určenou Fabryho chorobou, nebo v případě 56leté ženy s post mortem diagnózou fibrózy srdce a geneticky určenou familiární dilatační kardiomyopatií s defektem v genu pro titin (TTN). V obou případech k post mortem vyšetření vedl zájem příbuzných pro výskyt náhlých úmrtí a onemocnění srdce u více rodinných příslušníků.
Naopak riziko pro LTVA je v případě syndromu dlouhého QTc intervalu identické pro obě pohlaví, ale liší se výskytem v určitých věkových obdobích (33).
Ve srovnání s výsledky molekulárně genetického vyšetření u žijících jedinců s identickou diagnózou dědičného kardiovaskulárního onemocnění byl v případech HCM, DCM a akutní disekce téměř identický záchyt příčinných variant (32,34–36). Post mortem případy SADS a SUDS by odpovídaly souboru pacientů po přežité srdeční zástavě, kteří byli úspěšně resuscitováni a nebyla u nich diagnostikována jednoznačná příčina, tedy stanovena diagnóza tzv. idiopatické fibrilace komor (iVF). Záchyt příčinných variant je téměř identický v souboru zemřelých (SADS a SUDS) a přeživších (36).
Naopak v souboru post mortem diagnózy ARVC jsou míra detekce jistých příčinných variant i spektrum genů zásadně odlišné – výrazně nižší, než u žijících jedinců s touto diagnózou (37). Není vyloučeno, že skutečné příčiny náhlého úmrtí jsou jiné, než příčiny ARVC u žijících/přeživších pacientů. Pravděpodobnějším vysvětlením je však spíše nespolehlivost post mortem diagnózy, která se zakládá na detekci tukové degenerace a fibrózy myokardu pravé komory. Tyto nálezy mohou být zkresleny díky obecné tendenci k nadváze u současné populace. Pro spolehlivé zhodnocení by bylo nutné analyzovat tělesnou hmotnost a výšku u této kohorty náhle zemřelých, avšak tyto údaje nebyly k dispozici zdaleka u všech případů pro nejednotné postupy na různých pracovištích soudního lékařství.
Nález časté varianty v genu pro draslíkový kanál KCNE1 definující dědičný arytmický syndrom LQT5 lite je obtížně interpretovatelný u zemřelých i žijících. Na základě dostupné literatury jsme jej neoznačili jako jednoznačnou molekulární příčinu náhlého úmrtí (21,38). Rodinám jsme ale nález sdělili, detekovaným nosičům varianty doporučili betablokátory v maximálně tolerované dávce a upozornili na další úpravy životního stylu, které jsou důležité pro prevenci významných poruch srdečního rytmu (např. vyhnout se výkyvům minerálů, vyhnout se lékům uvedeným v seznamu na www.crediblemeds.org) (10).
Limitace molekulárně genetických metod lze spatřovat v průkazu DNA variant neznámého významu (DNA class 3, tzv. VUS) u významné skupiny SCD, v našem souboru u 30/106 případů (28 %). Vzhledem k současně nejednoznačné pitevní morfologii se ani kombinace obou metod nezdá být dostatečná ke stanovení jednoznačné příčiny smrti. V několika případech jsme u zemřelých nalezli i více variant neznámého významu a není vyloučen jejich kumulativní efekt. Vliv je ale nejasný a za současných podmínek neprokazatelný. Tento fenomén byl popsán i jinými autory (39). Významným interpretačním problémem je vztah tohoto molekulárně genetického nálezu k možnostem prevence u přímých příbuzných. Východiskem je dlouhodobá kardiologická dispenzarizace rodinných příslušníků a re-evaluace nalezených variant v pravidelných časových intervalech (40,41).
Někteří autoři diskutují výskyt větších genomových insercí a delecí u genů pro kardiomyopatie nebo dědičné arytmické syndromy a jejich limitní detekční možnosti (42). Nicméně námi zvolená molekulárně genetická metoda sekvenování nové generace (NGS) od firmy Sophia Genetics zaručuje spolehlivou detekci právě těchto změn, které raritně nacházíme (vlastní nepublikovaná data).
V rámci studie jsme nabídli molekulárně genetické vyšetření 5 prvostupňovým příbuzným bez jistých známek dědičného onemocnění, kde tkáň z náhle zemřelého nebo jeho obou rodičů nebyla dostupná. Tato diagnostika nepřinesla žádný výsledek, který ale v principu přítomnost genetického onemocnění u vyšetřených nevylučuje. Dle našich zkušeností je pravděpodobně racionální těmto rodinným příslušníkům nabídnout kardiologickou dispenzarizaci do 50. – 60. roku života (15). Molekulárně genetické vyšetření je oprávněné provést až v případě přítomnosti jasných známek určitého dědičného onemocnění (jasného fenotypu).
Při screeningovém vyšetření jsme prokázali, že až 1/3 příbuzných má známky dědičného kardiovaskulárního onemocnění a je tedy v principu v riziku náhlého úmrtí, bereme-li v úvahu různou penetranci a expresivitu jednotlivých genových variant. U některých z nich přispěla genetická stratifikace ke stanovení přesné diagnózy, při již existujícím onemocnění s hraničním fenotypem. Individualizovaná kontinuální péče v těchto případech přispěje k primární prevenci závažných poruch srdečního rytmu u mladých ekonomicky aktivních individuí. V některých, obzvláště závažných případech s vysokou penetrancí v rodině, se postižené rodiny rozhodují o primární prevenci onemocnění u potomků formou asistované reprodukce a preimplantační diagnostiky. Tím mohou zamezit dalšímu šíření tohoto konkrétního onemocnění do budoucích generací.
Zavedení molekulárně genetických metod přineslo řadu nových úkolů a výzev do jinak striktně morfologického oboru, jakým je soudní lékařství. Primárním úkolem soudních lékařů je zajistit záchyt suspektních případů SCD. Mezi kategorie nálezů, u kterých je doporučeno indikovat post mortem genetické vyšetření patří SUDS, SADS, kardiomyopatie, předčasná ateroskleróza věnčitých tepen, disekce aorty, náhlé úmrtí epileptiků (SUDEP) a v případě náhlého úmrtí v dětském věku také SIDS a SUDI (2) (tab. 2). Při záchytu případu spadajícího do jedné z těchto kategorií je nezbytné zajistit materiál z pitvy a uchovat ho po nezbytně dlouhou dobu (tab. 4).
Zásadním krokem v celém procesu diagnostiky je potom navázání kontaktu s pozůstalými a předání informací o pitevním nálezu, o možnosti post mortem kardiogenetické analýzy u jejich zemřelého příbuzného, vč. nutnosti podepsání informovaného souhlasu s touto analýzou a o možnosti kardiologického, ev. kardiogenetického vyšetření přímých příbuzných. Pokud je možné pozůstalé dohledat, popřípadě sami kontaktují soudnělékařské pracoviště a s pokračováním diagnostiky cestou kardiogenetického vyšetřování souhlasí, nastává prostor pro zahájení analýz. V té souvislosti je třeba upozornit na skutečnost, že pokud se nepodaří pozůstalé dohledat, případně rodina další vyšetřování (kardiogenetické i kardiologické) odmítá, nemá ve většině případů, s ohledem na nemožnost další prevence, smysl provádět post mortem genetické vyšetření u zemřelého jedince. Konečná diagnóza nadto vychází z komplexního posouzení nálezů soudnělékařských, molekulárně genetických a klinických, a především pak z posouzení jejich vzájemného kontextu.
Na základě mezinárodních doporučení (2) by měl být uvedený diagnostický postup aplikován i u případů náhlé srdeční zástavy s úspěšnou resuscitací a oddáleným úmrtím pacienta během hospitalizace. Interval mezi zástavou a smrtí se může pohybovat od několika hodin až po několik dnů až týdnů. S ohledem na indikace pitev dle aktuálně platného Zákona o zdravotních službách se mohou někteří takto postižení pacienti dostat na patologicko-anatomickou pitvu na ústavy a oddělení patologie. V té souvislosti je třeba vyslovit apel na provedení pitvy i u těchto zemřelých, ať už v režimu patologicko-anatomické pitvy či po dohodě se soudnělékařským pracovištěm v režimu zdravotní pitvy (dle zvyklostí zdravotnického zařízení) a zajištění materiálů k post mortem diagnostice i u těchto případů.
Výstupem provedené pitvy by měl být vždy pitevní protokol a soudnělékařská diagnóza. Složitost diagnostiky u případů SCD předpokládá nezbytnou komunikaci mezi všemi zúčastněnými diagnostickými pracovišti, a proto by výstupy měly být k dispozici též vyšetřujícímu kardiologovi a kardiogenetikovi, společně s popisem histologického nálezu na srdci (ev. dalších orgánech) (12). Esenciální je v takovém případě vyjádření pitvajícího lékaře o morfologickém nálezu na myokardu, který by měl být pro další kardiogenetickou analýzu jednoznačně zařazen do jedné z následujících kategorií: SADS – sudden arrhytmic death syndrome (zcela negativní pitevní nález), SUDS – sudden unexplained death syndrome (nespecifické nálezy nesplňující kritéria pro diagnózu kardiomyopatie), kardiomyopatie (HCM, ACM, DCM vč. suspektních nálezů), předčasná ateroskleróza věnčitých tepen, disekce aorty, náhlé úmrtí epileptika (SUDEP), SIDS – sudden infant death syndrome, SUDI – sudden unexplained death in infancy (tab. 1 a 2). Možnost efektivní prevence u rodinných příslušníků se dále nabízí u případů SCD z důvodu disekce aorty, a to formou indikace kardiochirurgické intervence u obdobného stupně postižení aorty jaký byl zjištěn při pitvě u jejich přímého příbuzného. Pitevní diagnostika umožňuje relativně přesné informace o stupni postižení (dilatace, výduť) aorty na základě prostého měření obvodu aorty. Problémem se však jeví nepřesnosti v pitevních protokolech vyplývající z neustálené lokalizace místa měření, nedůslednosti v přesnosti měření (do jisté míry vyplývající např. z konkavity, resp. konvexity rozstřižené aorty) a vlivu posmrtných změn na dilataci stěny aorty.
I přes individuální a lokální zvyklosti ve formulování soudnělékařských diagnóz zdůrazňujeme nutnost komunikace mezi pitvajícím lékařem a vyšetřujícím kardiogenetikem s cíleným zařazením odesílaného případu do jedné z výše uvedených kategorií.
Pro zlepšení komunikace mezi jednotlivými profesemi navrhujeme dle obecných doporučení evropských kardiologických, patologických a genetických společností (2,8,12) sestavení lokálních multidisciplinárních týmů, které se pravidelně schází (obr. 5). Tyto týmy se potom mohou na národní úrovni setkávat, vyměňovat si zkušenosti a sdílet data v rámci pravidelných národních kongresů jednotlivých odborných společností. Molekulárně genetická analýza by měla být nicméně v budoucnu sjednocena do jednoho, maximálně 2 specializovaných center (obr. 5)

ZÁVĚR
Post mortem genetická analýza při náhlé srdeční smrti (SCD) představuje důležitý diagnostický nástroj pro primární prevenci srdeční zástavy u příbuzných obětí a vyžaduje multicentrickou a multidisciplinární spolupráci. Grantový projekt NV18-02-00237 podpořený Ministerstvem zdravotnictví ČR umožnil dosud bezprecedentní navázání spolupráce mezi klinickými kardiogenetickými a kardiologickými pracovišti s pracovišti soudního lékařství v Čechách a na Moravě.
Obor soudní lékařství (případně patologie) sehrává nezastupitelnou roli v celém procesu rozpoznávání potenciálně dědičného kardiovaskulárního onemocnění u zemřelého pacienta, v zajištění materiálu z pitvy ke genetické analýze a v informování a nasměrování rodiny k možnosti preventivního kardiologického vyšetření.
Pozůstalí jsou dle dosavadních zkušeností převážně ochotni podstoupit genetickou a kardiologickou péči, zatímco centralizovaná molekulárně genetická analýza v ČR umožňuje získat spolehlivé výsledky, které jsou v souladu s jinými světovými multicentrickými studiemi zhruba u 20 % všech případů. Tento postup vede k primární prevenci náhlého úmrtí až u 25 % příbuzných, v naprosté většině v produktivním věku.
Výsledky grantového projektu objektivně významně pomáhají zlepšit primární prevenci náhlého úmrtí u mladých jedinců a jsou důležitým podkladem pro možnost právního i finančního ukotvení post mortem diagnostiky v ČR.
Po ukončení grantového projektu pokračuje analýza případů náhlé srdeční smrti v rámci zavedené multioborové spolupráce i nadále. Finančně náročné post mortem genetické vyšetření lze dle aktuálních zkušeností hradit až šest měsíců po smrti, případně lze náklady hradit ze zdravotního pojištění rizikového a náhlou srdeční smrtí potencionálně ohroženého přímého příbuzného.
Seznam použitých zkratek
SCD – sudden cardiac death – náhlá srdeční smrt
SUD(S) – sudden unexplained death (syndrome) – náhlá nevysvětlená smrt u jedinců starších 1 rok
SUDI – sudden unexplained death in infancy – náhlá nevysvětlená smrt u jedinců mladších 1 roku
SADS – sudden arrhythmic death syndrome – náhlá arytmická smrt bez strukturálních změn a toxikologického nálezu u jedinců starších 1 rok
SIDS – sudden infant death syndrome – syndrom náhlého úmrtí kojence
SUDEP – sudden unexplained death in epilepsy – náhlá nevysvětlitelná smrt u jedince s preexistující epilepsií
HCM – hypertrophic cardiomyopathy – hypertrofická kardiomyopatie
ARVC – arrythmogenic right ventricular cardiomyopathy – arytmogenní kardiomyopatie pravé komory
LVNC – non kompaktní kardiomyopatie
DCM – dilated cardiomyopathy – dilatovaná kardiomyopatie
NGS – next generation sequencing – sekvenování nové generace
DNA – deoxyribonukleová kyselina
LTVA – life threattining ventricular arrhytmia – život ohrožující komorová arytmie
EDS – Ehlersův-Danlosův syndrom
LDS – Loyesův-Dietzův syndrom
LQT – syndrom dlouhého QTc intervalu (dle genetiky typu 1–17)
CPVT – catecholaminerg polymorphic ventricular tachycardia – katecholaminergní polymorfní komorová tachykardie
QTc – korigovaný QT interval v EKG
ACMG – American College of Medical Genetics
iVF – idiopathic ventricular fibrillation – idiopatická fibrilace komor
AECVP – Association of European Cardiovascular Pathology
DEDIKACE
Podpořeno grantovým projektem Ministerstva zdravotnictví ČR NV18-02-00237 Diagnostika příčin náhlé srdeční smrti u lidí ve věku 0-35 let pomocí molekulárně genetických metod – pilotní studie.
PROHLÁŠENÍ
Autor práce prohlašuje, že v souvislosti s tématem, vznikem a publikací tohoto článku není ve střetu zájmů a vznik ani publikace článku nebyly podpořeny žádnou farmaceutickou firmou. Toto prohlášení se týká i všech spoluautorů.
Received: 03. 02. 2022
Accepted: 27. 02. 2022
Adresa pro korespondenci:
MUDr. Bc. Štěpánka Pohlová Kučerová, Ph.D.
Ústav soudního lékařství LF UK a FN Hradec Králové
Sokolská 581, 500 05, Hradec Králové
Tel.: +420495836832, Fax: +420495836833
e-mail: kucerovas@lfhk.cuni.cz
Zdroje
1. Goldstein S. The necessity of a uniform definition of sudden coronary death: witnessed death within 1 hour of the onset of acute symptoms. Am Heart J 1982; 103(1): 156-159.
2. Basso C, Aguilera B, Banner J, et al. Guidelines for autopsy investigation of sudden cardiac death: 2017 update from the Association for European Cardiovascular Pathology. Virchows Arch 2017; 471(6): 691-705.
3. Virmani R, Burke AP, Farb A. Sudden cardiac death. Cardiovasc Pathol 2001; 10(6): 275-282.
4. Kong MH, Fonarow GC, Peterson ED, et al. Systematic review of the incidence of sudden cardiac death in the United States. J Am Coll Cardiol 2011; 57(7): 794-801.
5. Myerburg RJ, Junttila MJ. Sudden cardiac death caused by coronary heart disease. Circulation 2012; 125(8): 1043-1052.
6. Myerburg RJ, Kessler KM, Castellanos A. Sudden cardiac death: Structure, function, and time-dependence of risk. Circulation 1992; 85(1 Suppl): I2-10.
7. Raju H, Parsons S, Thompson TN, et al. Insights into sudden cardiac death: exploring the potential relevance of non-diagnostic autopsy findings. Eur Heart J 2019; 40(10): 831 - 838.
8. Stiles MK, Wilde AAM, Abrams DJ, et al. 2020 APHRS/HRS expert consensus statement on the investigation of decedents with sudden unexplained death and patients with sudden cardiac arrest, and of their families. Heart Rhythm 2021; 18(1): e1-e50.
9. Brinkmann B. Harmonisation of medico-legal autopsy rules. Int J Legal Med 1999; 113(1): 1-14.
10. Priori SG, Blomström-Lundqvist C, Mazzanti A, et al. 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: The Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC)Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Europace 2015; 17(11): 1601-1687
11. Stiles MK, Wilde AAM, Abrams DJ, et al. 2020 APHRS/HRS expert consensus statement on the investigation of decedents with sudden unexplained death and patients with sudden cardiac arrest, and of their families. J Arrhythm 2021; 37(3): 481-534.
12. Fellmann F, van El CG, Charron P, et al. European recommendations integrating genetic testing into multidisciplinary management of sudden cardiac death. Eur J Hum Genet 2019; 27(12): 1763-1773.
13. Zeman M, Sepši M, Vojtíšek T, Šindler M. Suddenly deceased young individuals autopsied at the Department of forensic medicine, Brno – analysis. Soud Lek 2012; 57(3): 44-47.
14. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015; 17(5): 405-424.
15. Charron P, Arad M, Arbustini E, et al. Genetic counselling and testing in cardiomyopathies: a position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J 2010; 31(22): 2715-2728.
16. Verhagen JMA, Kempers M, Cozijnsen L, et al. Expert consensus recommendations on the cardiogenetic care for patients with thoracic aortic disease and their first-degree relatives. Int J Cardiol 2018; 258 : 243-248.
17. Khoury MH, Gornik HL. Fibromuscular dysplasia (FMD). Vasc Med 2017; 22(3): 248-252.
18. Wanner C, Arad M, Baron R, et al. European expert consensus statement on therapeutic goals in Fabry disease. Mol Genet Metab 2018; 124(3): 189-203.
19. Hughes DA, Nicholls K, Shankar SP, et al. Oral pharmacological chaperone migalastat compared with enzyme replacement therapy in Fabry disease: 18-month results from the randomised phase III ATTRACT study. J Med Genet 2017; 54(4): 288-296.
20. Lane CM, Giudicessi JR, Ye D, et al. Long QT syndrome type 5-Lite: Defining the clinical phenotype associated with the potentially proarrhythmic p.Asp85Asn-KCNE1 common genetic variant. Heart Rhythm 2018; 15(8): 1223-1230.
21. Garmany R, Giudicessi JR, Ye D, Zhou W, Tester DJ, Ackerman MJ. Clinical and functional reappraisal of alleged type 5 long QT syndrome: Causative genetic variants in the KCNE1 - encoded minK β-subunit. Heart Rhythm 2020; 17(6): 937-944.
22. Kuchenbuch M, Barcia G, Chemaly N et al., KCNT1 epilepsy with migrating focal seizures shows a temporal sequence with poor outcome, high mortality and SUDEP. Brain 2019; 42(10): 2996-3008.
23. Thakur RK, Hoffmann RG, Olson DW, et al. Circadian variation in sudden cardiac death: Effects of age, sex, and initial cardiac rhythm. Ann Emerg Med 1996; 27(1): 29-34.
24. Larsen MK, Christiansen SL, Hertz CL, et al. Targeted molecular genetic testing in young sudden cardiac death victims from Western Denmark. Int J Legal Medicine 2020; 134(1): 111-121.
25. Lahrouchi N, Raju H, Lodder EM, et al. Utility of Post-Mortem Genetic Testing in Cases of Sudden Arrhythmic Death Syndrome. J Am Coll Cardiol 2017; 69(17): 2134-2145.
26. Lahrouchi N, Raju H, Lodder EM, et al. The yield of postmortem genetic testing in sudden death cases with structural findings at autopsy. Eur J Hum Genet 2020; 28(1): 17-22.
27. Ortiz-Genga MF, Cuenca S, Dal Ferro M, et al. Truncating FLNC Mutations Are Associated With High-Risk Dilated and Arrhythmogenic Cardiomyopathies. J Am Coll Cardiol 2016; 68(22): 2440-2451.
28. Isbister JC, Nowak N, Butters A, et al. “Concealed cardiomyopathy” as a cause of previously unexplained sudden cardiac arrest. Int J Cardiol 2021; 324 : 96-101.
29. Verdonschot JAJ, Vanhoutte EK, Claes GRF, et al. A mutation update for the FLNC gene in myopathies and cardiomyopathies. Hum Mutat 2020; 41(6): 1091-1111.
30. Butters A, Arnott C, Sweeting J, Winkel BG, Semsarian C, Ingles J. Sex Disparities in Sudden Cardiac Death. Circ Arrhythm Electrophysiol 2021; 14(8).
31. Kim SK, Bennett R, Ingles J, Kumar S, Zaman S. Arrhythmia in Cardiomyopathy: Sex and Gender Differences. Curr Heart Fail Rep 2021; 18(5): 274-283.
32. Mazzarotto F, Tayal U, Buchan RJ, et al. Reevaluating the Genetic Contribution of Monogenic Dilated Cardiomyopathy. Circulation 2020; 141(5): 387-398.
33. Zareba W, Moss AJ, Locati EH, et al. Modulating effects of age and gender on the clinical course of long QT syndrome by genotype. J Am Coll Cardiol 2003; 42(1): 103-109.
34. Bonaventura J, Veselka J. Genetic testing in patients with hypertrophic cardiomyopathy. Vnitr Lek 2019; 65(10): 652-658.
35. Bonaventura J, Norambuena P, Tomašov P, et al. The utility of the Mayo Score for predicting the yield of genetic testing in patients with hypertrophic cardiomyopathy. Arch Med Sci 2019; 15(3): 641-649.
36. Marschall C, Moscu-Gregor A, Klein HG. Variant panorama in 1,385 index patients and sensitivity of expanded next-generation sequencing panels in arrhythmogenic disorders. Cardiovasc Diagn Ther 2019; 9(Suppl 2): S292-S298.
37. Gandjbakhch E, Redheuil A, Pousset F, Charron P, Frank R. Clinical Diagnosis, Imaging, and Genetics of Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia: JACC State-of-the - Art Review. J Am Coll Cardiol 2018; 72(7): 784-804.
38. Roberts JD, Asaki SY, Mazzanti A, et al. An International Multicenter Evaluation of Type 5 Long QT Syndrome: A Low Penetrant Primary Arrhythmic Condition. Circulation 2020; 141(6): 429-439.
39. Grassi S, Campuzano O, Coll M, et al. Genetic variants of uncertain significance: How to match scientific rigour and standard of proof in sudden cardiac death? Legal Med (Tokyo) 2020; 45 : 101712.
40. Campuzano O, Sarquella-Brugada G, Fernandez - Falgueras A, et al. Reanalysis and reclassification of rare genetic variants associated with inherited arrhythmogenic syndromes. EBioMedicine 2020; 54 : 102732.
41. Sarquella-Brugada G, Fernandez-Falgueras A, Cesar S, et al. Clinical impact of rare variants associated with inherited channelopathies: a 5-year update. Human Genet 2021; 21: Online ahead of print.
42. Mates J, Mademont-Soler I, Fernandez-Falgueras A, et al. Sudden Cardiac Death and Copy Number Variants: What Do We Know after 10 Years of Genetic Analysis? Forensic Sci Int Genet 2020; 47 : 102281.
Štítky
Patologie Soudní lékařství ToxikologieČlánek vyšel v časopise
Soudní lékařství

2022 Číslo 2
Nejčtenější v tomto čísle
- Výstupy multicentrické studie příčin náhlé srdeční smrti (SCD) v České republice a primární prevence srdeční zástavy u příbuzných
- Kritické zhodnocení rekodifikace znaleckého práva v České republice po prvním roce účinnosti
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy