
Statiny a jejich specifika
Datum publikace: 11. 9. 2017
Úprava životosprávy v prevenci kardiovaskulárních onemocnění
Navzdory běžně rozšířenému názoru o zásadním významu diety a úpravy životního stylu v prevenci kardiovaskulárních onemocnění dokládají strohá fakta velmi omezený význam dietních opatření a zvýšení pohybové aktivity. Zcela recentní závěry US Preventive Services Task Force Recommendation Statement (JAMA. 2017; 318 [2]: 167–195) konstatují „velmi málo dokladů o dlouhodobém pozitivním či negativním účinku dietních opatření a zvýšení pohybové aktivity“ v rámci primární prevence. Metaanalýza 88 validních kontrolovaných studií (> 120 tis. probandů sledovaných 3 až 15 let) sice dokládá statisticky významný pokles cholesterolu v nízkodenzitních lipoproteinech (LDL-C), ale v absolutním procentu nízký (pouze 0,07 mmol/l); stejný byl i pokles celkového cholesterolu (TC) – o 0,07 mmol/l. Obdobně došlo ke statisticky významnému, ale minimálnímu poklesu systolického TK i diastolického TK – o 1,26 mm Hg, resp. o 0,49 mm Hg. Bohužel do analýzy nebyla zahrnuta poslední studie PREDIMED se středozemní dietou saturovanou olivovým olejem/ořechy, kde výskyt velkých vaskulárních příhod poklesl v primární prevenci o téměř 30 %. Problémem je, že účinné intervence dosahujeme při „západním životním stylu“ velmi obtížně. Není tak pochyb, že dominantní postavení v prevenci kardiovaskulárních onemocnění, zejména v prevenci sekundární, má farmakoterapie. Nicméně úprava životosprávy významně efekt podporuje.
V profylaxi aterotrombotických příhod a ve snížení mortality má farmakologická léčba dyslipidemie zásadní význam. Např. v primárně preventivní studii HOPE 3 u osob s mírným rizikem měla léčba malou dávkou statinu (10 mg rosuvastatinu) při fyziologickém či jen mírně nad 3 mmol/l LDL-C podstatně větší prognostický dopad než léčba hypertenze s vyšším, normálním či mírně zvýšeným TK. Tyto suboptimální hodnoty LDL-C či krevního tlaku patří přitom v populaci mezi nejčastější. I v rámci preventivních zásahů má tedy farmakologická léčba dyslipidemie výsostné postavení.
Možnosti farmakologické léčby dyslipidemie
1) Co je cílem intervence?
Cílem intervence není primárně úprava dyslipidemie, cílem je zlepšení prognózy a kvality života nemocného. Neléčíme „lipidogram“, ale intervenujeme komplexní rizikový profil, tj. vedle dyslipidemie léčíme též hypertenzi, kuřácký návyk, diabetes či obezitu. Na každou strategii tak musíme pohlížet z tohoto prizmatu.
V rámci jednotlivých lipidů i lipoproteinů jsou z pohledu aterogenicity i z pohledu strategie intervence významné rozdíly. Lipidy (triglyceridy, fosfolipidy, cholesterol aj.) jsou v plazmě přítomny ve formě lipoproteinů a chylomiker. Ty jsou tvořeny zpravidla povrchovou hydrofilní vrstvou (zejm. fosfolipidy), lipofilním jádrem (zejm. triglyceridy, cholesterol a jeho estery) a jedním či více biologicky aktivními proteiny (apolipoproteiny), které fungují jako ligandy a zajišťují vazbu lipoproteinu na membránové receptory cílových tkání (s možností internalizace lipoproteinu a předání obsahu do cílové buňky). V aterogenním nízkodenzitním lipoproteinu (LDL), který zajišťuje transport cholesterolu do cílových tkání, je apolipoprotein B100 (apoB100), naopak v antiaterogenním vysokodenzitním lipoproteinu HDL, který přijímá nadbytečný cholesterol a zprostředkuje jeho reverzní transport, je apolipoprotein A1 (apoA1). Další funkcí apolipoproteinů, působících jako kofaktory, je aktivace řady enzymů a transkripčních faktorů. Tímto způsobem lipoproteiny ovlivňují např. reparační procesy, napětí cévní stěny či hemostázu.
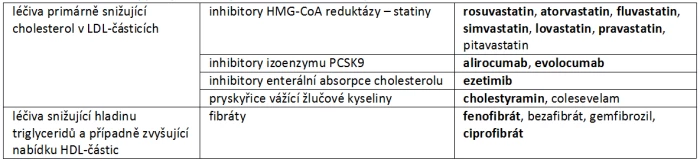
Léčba dyslipidemie tak neovlivňuje pouze vlastní aterogenezi, ale má větší přesah, např. na úrovni ovlivnění cévní perfuze či aktivace trombotických pochodů. Léčebné postupy jsou zaměřeny jak na snížení koncentrace LDL-částic (resp. snížení cholesterolu v LDL), na snížení koncentrace triglyceridů (obsažených v řadě lipoproteinů), tak na zvýšení koncentrace lipoproteinu typu HDL (nikoli však na zvýšení obsahu cholesterolu v těchto částicích, tj. HDL-C). V ČR dostupná základní léčiva (ne vždy však hrazená) uvedená v tabulce 1 jsou vyznačena tučně.
Tab. 1: Rozdělení hypolipidemik podle mechanizmu působení.
2) Strategie cílená na snížení nízkodenzního lipoproteinu (LDL)
Lipoproteiny typu LDL obsahují velké množství cholesterolu a jeho esterů. Jejich úlohou je transport cholesterolu do všech tkání, zejména tkání s jeho vyšší spotřebou – kůry nadledvin či pohlavních orgánů nebo do jater. Tento cholesterol, předaný z LDL přímo intracelulárně (cestou LDL-receptorů, LDL-R), není podkladem aterogeneze a je potřebný k zajištění funkce organizmu. Jak bude uvedeno dále, pouze aterogenní lipidy (zejm. cholesterol) uložené extracelulárně v subendoteliálním prostoru mohou iniciovat a progredovat aterogenezi.
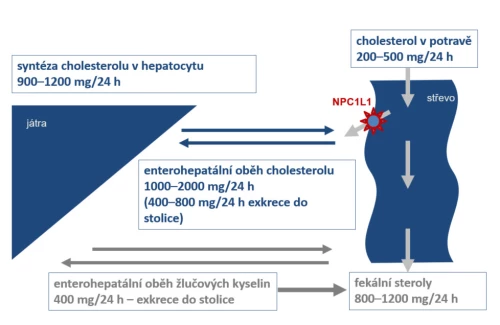
Syntéza aterogenních lipidů je mimo jiné závislá na nabídce cholesterolu v hepatocytu. Cyklus příjmu, syntézy a eliminace cholesterolu je znázorněn na obr. 1. Množství cholesterolu přijatého z potravy je relativně menší v porovnání s cholesterolem syntetizovaným v játrech či cirkulujícím v enterohepatálním cyklu. Na úrovni enterocytu je absorpce cholesterolu facilitována transportérem – proteinem NPC1L1 (Niemann–Pick C1 like 1). Jeho blokáda sníží exogenní příjem cholesterolu i reabsorpci cholesterolu v enterohepatálním cyklu. Syntéza v hepatocytu je regulována zejména nabídkou cholesterolu v buňce. Vyšší konverze cholesterolu na žlučové kyseliny (např. při snížení jejich reabsorpce z enterohepatálního cyklu) sníží jeho koncentraci v hepatocytu.
Obr. 1: Vztah mezi příjmem cholesterolu, jeho syntézou a enterohepatálním oběhem. Významnější část cholesterolu je syntetizována v játrech, menší část je přijímána z potravy. Velké množství cholesterolu je uvolněno do žluče a obíhá v enterohepatálním cyklu. Reabsorpce cholesterolu z potravy i ze žluče je facilitována transportérem NPC1L1 (Niemann–Pick C1 like 1). Paralelně cirkulují žlučové kyseliny. Snížení jejich reabsorpce a jejich deplece vede k resyntéze a ke snížení zásob cholesterolu.
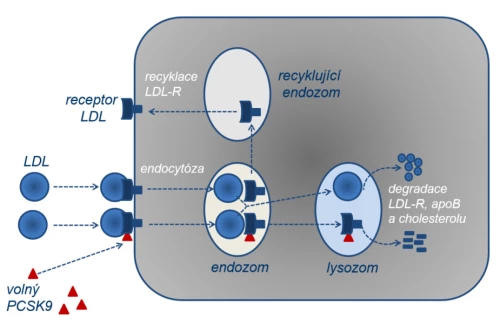
V koloběhu LDL mají důležitou funkci cílové receptory vázající jednotlivé apolipoproteiny a zprostředkující jejich biologické funkce, konkrétně LDL-receptor, který umožňuje vazbu LDL na buňky cílových tkání a internalizaci lipoproteinu do buňky cílového orgánu (obr. 2).
Intracelulárně absorbovaný LDL podléhá lysozomální degradaci a předává svůj obsah do cílové buňky (hepatocytu, steroidogenní buňky aj.). Tak je umožněna utilizace cholesterolu v tkáni. Výsledkem eliminace LDL z plazmy do tkání je pokles plazmatické koncentrace LDL-cholesterolu (LDL-C). Rychlost hepatální syntézy velmi nízkodenzitního lipoproteinu (VLDL), prekurzoru LDL a rychlost eliminace LDL vazbou na LDL-R rozhoduje o koncentraci cholesterolu v nízkodenzitním lipoproteinu, tedy LDL-C. Laboratorně stanovujeme frakci cholesterolu v LDL, tedy LDL-cholesterol, tato hodnota nás dobře informuje o aterogenním riziku nemocného a její snížení spolehlivě odráží pokles rizika aterotrombotických komplikací. Farmakologicky účinně ovlivňujeme jak absorpci cholesterolu ze střeva (z potravy či z enterohepatálního cyklu), syntézu aterogenních lipoproteinů v hepatocytu, tak jejich eliminaci z plazmy.
Obr. 2: Schéma přesunu cholesterolu do buněk tkání utilizujících cholesterol (fyziologický pochod) a průniku LDL do subendoteliálních prostor (patologický proces – aterogeneze).
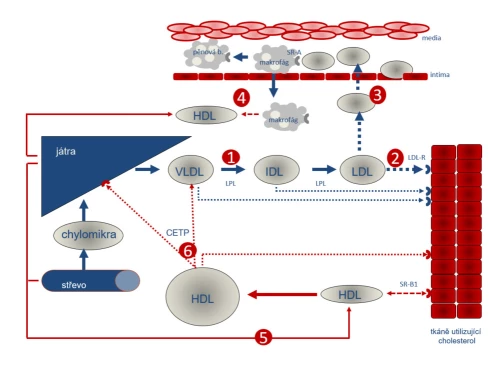
Játra produkují lipoproteiny typu VLDL (obsah převážně triglyceridů), působením lipoproteinové lipázy (LPL) jsou triglyceridy štěpeny a mastné kyseliny utilizovány. Výsledkem je snížení poměru triglyceridů k cholesterolu, lipoproteiny přecházejí do fáze intermediálních částic (IDL), resp. výsledně vznikají na cholesterol bohaté lipoproteiny typu LDL (1). Většina LDL je internalizována pomocí LDL-receptoru (LDL-R) do buněk spotřebovávajících cholesterol (zejm. kůra nadledvinek, pohlavní žlázy, ale v menší míře prakticky všechny tkáně). Nespotřebovaný cholesterol je zpětně vychytáván v játrech (2). Malá část – zejména malých částic LDL – proniká do subendoteliálních prostor cévní stěny, kde jsou pomocí scavengerového receptoru A (SR-A) fagocytovány makrofágy. Nadměrná fagocytóza modifikovaného cholesterolu, zejména cholesterolu oxidovaného, brání zpětnému vycestování makrofágů do cirkulace. Ty pak zůstávají v subendoteliálním prostoru jako pěnové buňky a jsou podkladem vývoje aterosklerotického plátu. Za fyziologických podmínek předává makrofág svůj obsah, zejména cholesterol, do lipoproteinu typu HDL (4). Tento pochod lze označit jako antiaterogenní působení HDL. Játra vytvářejí HDL s nízkým obsahem cholesterolu. Tato diskoidní forma je plně schopná cholesterol z makrofágů převzít a dále transportovat (5). Právě do tohoto typu HDL-částic je přebytek cholesterolu předáván z tkání pomocí scavengerového receptoru B (SR-B1). Po naplnění se z diskoidních partikulí stávají částice sférické, již cholesterolem naplněné. Z nich je cholesterol předáván pomocí proteinu CETP (cholesterol-ester transport protein) zpět do jater či do aterogenních lipidů, zejména VLDL (6).
Syntéza LDL, resp. jeho prekurzoru VLDL, v játrech je závislá na nabídce lipidů (cholesterolu, triglyceridů aj.) a apolipoproteinů, zejm. apoB100. Vlastní VLDL obsahuje velké množství triglyceridů, tyto jsou lipoproteinovou lipázou degradovány, mastné kyseliny metabolizovány a zůstává cholesterol a jeho estery. Výsledná částice LDL má jednu molekulu apolipoproteinu B100, málo triglyceridů a hodně cholesterolu.
Naprostá většina LDL je utilizována v tkáních zpracovávajících cholesterol. Jen nepatrná část, zejména částice malé velikosti, se dostává do subendoteliálního prostoru. Extracelulárně uložené lipidy jsou za pomoci scavengerového receptoru typu A fagocytovány makrofágy. Ty za fyziologických podmínek vycestují do cirkulace, kde jsou retikuloendoteliálním systémem eliminovány. Malé partikule LDL jsou aterogenní; nejenže lépe pronikají do cévní stěny, ale i jejich obsah snáze podléhá oxidaci či glykosylaci. Oxidované a jinak modifikované lipidy omezují mobilitu makrofágů a ty mohou zůstat v subendoteliálním prostoru jako tzv. pěnové buňky. Akumulace pěnových buněk pak je podkladem aterosklerotické léze.
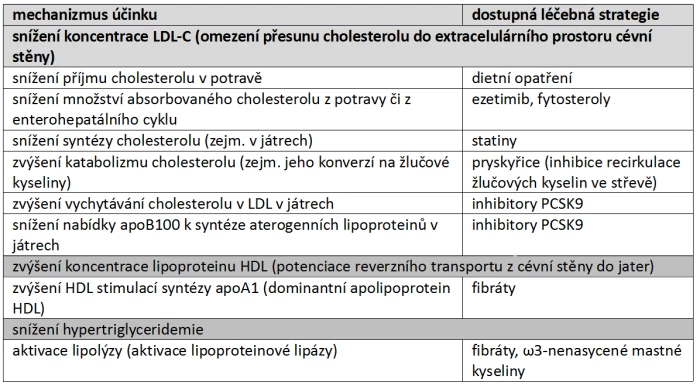
Tab. 2: Možnosti snížení obsahu cholesterolu v LDL, zvýšení koncentrace HDL či snížení koncentrace triglyceridů. Vysvětlivky: apoB100 – apolipoprotein B 100; LDL – low density lipoprotein, lipoproteiny o nízké hustotě; HDL – high density lipoprotein, lipoproteiny o vysoké hustotě; PCSK9 – proproteinová konvertáza subtilisin/kexinového typu 9.
Osvědčeným terapeutickým postupem je snížení množství cirkulujících LDL-částic, eliminovat preferenčně malé denzní molekuly LDL neumíme. Strategie je v podstatě dvojí – snížení syntézy LDL a zvýšení eliminace LDL.
Dominantní cestou je snížení nabídky cholesterolu k syntéze aterogenních lipoproteinů. Můžeme omezit exogenní příjem, potlačit syntézu, zvýšit odpad do žluče či zvýšit přeměnu cholesterolu na jiné látky (tab. 2). Snížení exogenního příjmu cholesterolu dosáhneme buď dietním opatřením, či redukcí absorpce cholesterolu ve střevě blokádou specifického transportního systému. Efekt snížení příjmu je omezen, organizmus deficit hradí zvýšenou syntézou cholesterolu v játrech. Další možností je snížení syntézy cholesterolu v hepatocytu. Tato cesta je účinná zejména v kombinaci s dietou. Slabým místem však může být pokles zásobování tkání cholesterolem s vývojem nežádoucích účinků. Třetí možností je zvýšení přeměny cholesterolu na žlučové kyseliny a tím dosažení jeho relativní deplece v játrech.
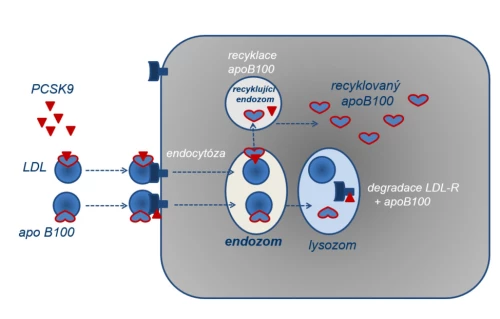
Druhá strategie snížení LDL spočívá ve stimulaci vychytávání LDL v cílových tkáních (v játrech a steroidogenních orgánech). Jak bylo řečeno, internalizace LDL je zajištěna vazbou apoB100 na LDL-R, kde exprese receptoru reguluje rychlost clearance lipoproteinu. Po internalizaci LDL dojde v endozomu k odloučení vlastní lipidové části od LDL-R a od apoB100. Cholesterol a ostatní lipidy jsou zpracovány v buňce, naopak obě proteinové části jsou opětovně využity, LDL-R je např. recyklován 100 - až 200krát (obr. 3). Vlastní recyklace je pod kontrolou specifického proteinu – PCSK9 (proproteinové konvertázy subtilisin/kexinového typu). Volný izoenzym PCSK9 se váže na LDL-R a zabrání jeho recyklaci. Snížená nabídka LDL-R na buněčných membránách tak zpomalí vychytávání lipoproteinu LDL v tkáních, a koncentrace tohoto aterogenního lipoproteinu tak roste. Vzhledem k tomu, že v plazmě je izoenzym PCSK9 vázán na cirkulující LDL, konkrétně na apoB100, a tak je inaktivní, vzestup koncentrace LDL sníží nabídku volného PCSK9 a degradace LDL-R se zpětnovazebně utlumí. Změna poměru volné a vázané konvertázy PCSK9 jemně reguluje recyklaci LDL-R a tím i plazmatickou koncentraci lipoproteinů typu LDL.
Paralelně s kontrolou nabídky LDL-R ovlivňuje tento regulátor PCSK9 i nabídku apoB100. Zde však komplex apoB100/PCSK9 není signálem pro degradaci, ale naopak pro další recyklaci. Na úrovni LDL-R tak konvertáza PCSK9 působí jako „polibek smrti“, na úrovni apoB100 jako „polibek života“. Inhibice PCSK9 specifickou monoklonální protilátkou je využívána v léčbě těžkých forem hypercholesterolemie. Tento léčebný postup patří co do poklesu LDL k nejúčinnějším. Výhodou je, že nedochází ke snížení saturace tkání cholesterolem. Nevýhodou je pak nutnost parenterální aplikace a zcela zásadním způsobem je užití omezeno velmi vysokými náklady na léčbu.
Obr. 3: Regulační funkce izoenzymu PCSK9 – kontrola recyklace LDL-R a apoB100.
a) Receptor LDL umožní vazbu apoB100 a internalizaci lipoproteinu LDL. Bez přítomnosti volného izoenzymu PCSK9 opakovaně LDL-R recykluje. Dostatečná nabídka LDL-R na povrchu buňky utilizující cholesterol snižuje plazmatickou koncentraci lipoproteinu LDL (a LDL-C) při zachování dostatečného přísunu cholesterolu do tkání.

b) Po vazbě LDL-R s izoenzymem PCSK9 nedojde k regeneraci receptoru, ale k jeho lysozomální degradaci. Nižší nabídka LDL-R na membráně sníží vychytávání LDL, a koncentrace LDL (i LDL-C) tak stoupá.
c) Volný izoenzym PCSK9 se váže též na apoB100. Tak je na jedné straně snížena jeho nabídka – vyšší koncentrace LDL sníží podíl volného PCSK9, což umožní recirkulaci LDL-R, a koncentrace LDL se tak snižuje. Funkčně se jedná o autoregulaci koncentrace LDL. Na straně druhé vazba apoB100 s PCSK9 zabrání lysozomální degradaci apolipoproteinu a ten recirkuluje a je v játrech k dispozici pro syntézu dalších lipoproteinů typu VLDL.
3) Strategie cílená na zvýšení vysokodenzního lipoproteinu (HDL)
Lipoprotein HDL má řadu funkcí, v metabolizmu lipidů působí jako transportér přenášející nadbytečný cholesterol a jiné tuky z tkání zpět do jater či do ostatních tkání utilizujících cholesterol. Z hlediska aterogeneze má tento lipoprotein důležitou úlohu, neboť v subendoteliálním prostoru přebírá cholesterol z makrofágů a tím působí antiaterogenně (obr. 2).
Vedle vysoké hladiny lipoproteinu LDL je dalším typem aterogenní dyslipidemie snížení plazmatické koncentrace lipoproteinů s vysokou hustotou (HDL), resp. lépe řečeno snížení kapacity těchto lipoproteinů pro reverzní transport cholesterolu (zejm. z makrofágů) do jater. Zde je nutno zdůraznit, že běžně přijatá praxe, tj. stanovení cholesterolu obsaženého v HDL-částicích, tedy HDL-cholesterolu (HDL-C), není optimální hodnotou, která by nás spolehlivě informovala o kardiovaskulárním riziku. Ani léčebné snahy o zvýšení koncentrace HDL-C nevedou k poklesu rizika aterotrombotických příhod.
Možností zvýšení vysokodenzitního lipoproteinu HDL je méně. Farmakologické postupy, které zvyšovaly koncentraci HDL-C snížením přesunu cholesterolu do aterogenních lipoproteinů či inhibicí vychytávání HDL-částic v játrech, nebyly účinné. Efekt inhibitorů transportního proteinu esteru cholesterolu – CETP či niacinu – se nedostavil, výskyt aterotrombotických příhod naopak vzrostl. V současné době je dostupná pouze léčba deriváty kyseliny fibrové – fibráty, které mají komplexní účinek. Mírný vzestup HDL-C je dán zvýšením syntézy apoA1, dominantního apolipoproteinu HDL i zpomalením přesunu cholesterolu do jater a aterogenních lipidů (inhibicí CEPT). Efekt jednotlivých představitelů skupiny fibrátů na HDL je variabilní, v ČR užívaný fenofibrát zvyšuje koncentraci lipoproteinu HDL minimálně, asi o 2 %. Naopak dominuje efekt na pokles hypertriglyceridemie. Aplikace analog apolipoproteinu A1 získaných rekombinantní cestou, jako je apoA1 Milano, je stále ve vývoji.
4) Strategie cílená na snížení hypertriglyceridemie
Výrazně menší aterogenní potenciál mají triglyceridy. Jejich vyšší hodnota odráží celkovou metabolickou poruchu – zpravidla v rámci metabolického syndromu či diabetu. Izolovaná intervence hypertriglyceridemie má, v porovnání s intervencí vysoké hladiny LDL-C, menší význam a farmakologické postupy v této oblasti mají velmi omezené možnosti. Léčebně využíváme zejména aktivaci lipoproteinové lipázy (LPL), enzymu štěpícího triglyceridy a umožňujícího utilizaci mastných kyselin (obr. 2). Obsah triglyceridů v chylomikrech a ve VLDL tak významně klesá. Aktivace LPL je jedním z mnoha účinků fibrátů. Dopad snížení hypertriglyceridemie na pokles aterotrombotických příhod je podstatně menší než u strategií směřovaných na pokles LDL.
Řada nemocných nemá izolovanou poruchu lipidového metabolizmu, ale objevují se u nich kombinace jednotlivých odchylek. Typickými příklady jsou metabolický syndrom s hypertriglyceridemií a s nízkou plazmatickou koncentrací HDL či smíšená hyperlipidemie s vysokými plazmatickými koncentracemi LDL i triglyceridů. U těchto nemocných, stejně jako při nedostatečném efektu jedné strategie, můžeme hypolipidemika kombinovat.
Statiny (inhibitory HMG-CoA reduktázy) a jejich postavení v rámci hypolipidemické léčby
Statiny jsou v současné době nejvýznamnějšími hypolipidemiky. Snižují zejména koncentraci LDL-cholesterolu, efekt na vzestup HDL-C či pokles triglyceridemie je menší a kolísá v závislosti na užitém statinu a na konkrétním typu dyslipidemie. Statiny mají jednoznačně doložený nejen efekt na úpravu lipidogramu, ale i na zlepšení prognózy (snížení výskytu aterotrombotických příhod i pokles kardiovaskulární i celkové mortality). Snížení celkové mortality mají v současné době doložené jako jediná skupina hypolipidemik. Platí to u nemocných léčených v rámci primární i sekundární prevence. Efekt je přítomen napříč hodnotami LDL-C, tj. nejen u nemocných s hypercholesterolemií, ale i s hodnotami LDL-C v pásmu „fyziologických“ hodnot. Poměr mezi účinností a bezpečností je velmi příznivý. Výhodou je i to, že se ekonomicky jedná o málo nákladnou léčbu. Z těchto vlastností vyplývá i rozšířenost léčby, v ekonomicky vyspělých zemích (včetně ČR) je statiny dlouhodobě léčeno více než 10 % populace.
Mechanizmus působení a farmakodynamický účinek statinů
Statiny inhibují syntézu cholesterolového jádra kompetitivní blokádou hydroxymetylglutaryl koenzym A reduktázy (HMG-CoA reduktázy), enzymu, který je integrální součástí membrány endoplazmatického retikula (ER) hepatocytu. Tento „rate-limiting“ enzym se účastní steroidogeneze již v prvém kroku, kdy postupně kondenzují dvojuhlíkaté produkty β-oxidace mastných kyselin ve formě acetyl-CoA na hydroxymetylglutaryl koenzym A (HMG-CoA). Redukce hydroxymetylglutarylového zbytku na mevalonát je statiny inhibována (obr. 4). Významná je skutečnost, že HMG-CoA se nehromadí, je konvertován zpět na acetyl-CoA. Díky tomu je blokáda redukce HMG-CoA, na rozdíl od zásahu na vyšších etážích, tolerována dobře. Výsledkem inhibice syntézy cholesterolu je jeho deplece v hepatocytu. Následkem toho dochází k expresi LDL-R a zvýšení hepatální clearance lipoproteinu LDL. Plazmatická koncentrace LDL a LDL-C klesá. Snížení nabídky cholesterolu vede též k poklesu syntézy apoB100 a tím i VLDL – lipoproteinu bohatého na triglyceridy. Tímto mechanizmem dochází též k poklesu koncentrace triglyceridů.
Vlastní pokles lipoproteinu typu LDL je komplexní proces. Statiny indukovaná deplece cholesterolu v entoplazmatickém retikulu hepatocytu aktivuje regulační proteiny SREBP-1 a SREBP-2 (sterol response element binding protein 1 a 2), které iniciují transkripci genu kódujícího LDL-receptor. Zvýšení koncentrace tohoto receptoru na membráně hepatocytu umožní vstup LDL-částic do buňky a sníží plazmatickou koncentraci tohoto aterogenního lipoproteinu.
Obr. 4: Mechanizmus účinku statinů. Inhibice HMG-CoA reduktázy je výhodná, neboť nevzniká žádný toxický meziprodukt. Nevýhodou je snížení syntézy farnesyl pyrofosfátů, prekurzorů dalších biologicky aktivních lipidů. Prověřuje se efekt statinů s inhibitory squalen-syntázy, které by zvýšily nabídku farnesylu a mohly by snížit výskyt nežádoucích účinků. Vedle inhibice HMG-CoA reduktázy je ve fázi klinického hodnocení prověřován účinek inhibice acyl-CoA syntázy.
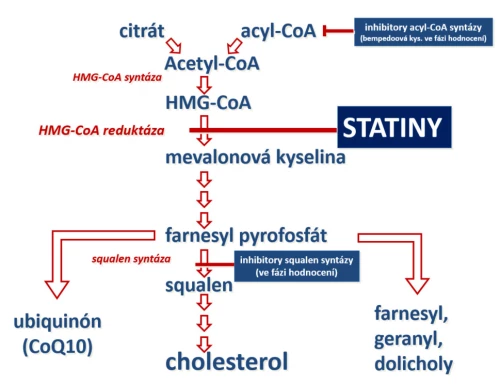
Inhibice HMG-CoA reduktázy netlumí pouze syntézu cholesterolu. Mevalonát je prekurzorem farnesyl pyrofosfátu, ze kterého je syntetizován jak cholesterol, tak řada dalších lipidů – geranyl, farnesyl či ubiquinon. Geranyl a farnesyl jsou výchozími látkami k tzv. prenylaci proteinů (zejm. regulačních proteinů a enzymů), která umožní jejich uchycení na buněčnou membránu přispívající k jejich optimální funkci. Inhibice syntézy farnesyl pyrofosfátu může vést k depleci geranylu a farnesylu. Fyziologický význam inhibice prenylace proteinů není zatím jasný, předpokládá se úloha v rámci extralipidového účinku statinů či v rámci nežádoucích účinků. Ubiquinon (koenzym Q10) je zapojen do energetického metabolizmu, tj. oxidativní fosforylace na membráně mitochondrie. Není pravděpodobné, že by deplece ubiquinonu byla příčinou myalgií a myopatií spojených s léčbou statiny. Substituce koenzymu Q10 v rámci klinických studií nevedla ke snížení výskytu nežádoucích účinků tohoto typu.
Farmakodynamickým efektem statinů je v prvé řadě snížení plazmatické koncentrace LDL-C (o 20–55 %), mírný vzestup HDL-C (o 2–10 %) a individuálně variabilní pokles triglyceridemie (o 5–25 %). Doloženy jsou i tzv. „extralipidové“ účinky – např. zlepšení funkce endotelu s potenciací vazodilatace, snížení trombogenní pohotovosti, regulace imunitní odpovědi, ovlivnění kostního metabolizmu aj. Tyto „funkční“ změny jsou klinicky významné, pokles aterotrombotických příhod totiž výrazně předchází úpravě změn „morfologických“, tj. regresi plátu. Do jaké míry se jedná o efekt zásahu do extralipidového metabolizmu a jakou úlohu hraje změna nabídky multifunkčních apolipoproteinů (zejména apoB100), není zřejmé.
1) Rozdíly mezi statiny v efektu na redukci LDL-cholesterolu
V efektu na pokles LDL-cholesterolu jsou jednotlivé statiny různě potentní. Důvodem je variabilní koncentrace v čase v místě působení, tj. na membránách endoplazmatického retikula hepatocytu. Nedostatečná nabídka statinu na konci dávkového intervalu vede k obnovení syntézy cholesterolu a k poklesu hypolipidemického efektu. Nikoliv náhodou patří mezi nejpotentnější statiny ty s dlouhým poločasem plazmatické eliminace (rosuvastatin, atorvastatin a pitavastatin). U fluvastatinu vedlo užití retardované lékové formy k výraznějšímu účinku. Že by podávání krátkodobě působícího statinu (simvastatinu, lovastatinu a pravastatinu) ve dvou denních dávkách zlepšilo hypolipidemický účinek, není známo.
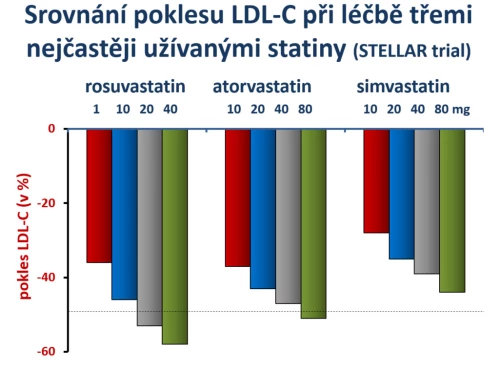
Vlastní srovnání účinku různých dávek rosuvastatinu, atorvastatinu, simvastatinu a pravastatinu na pokles LDL-cholesterolu bylo formou dvojitě zaslepené studie sledováno ve studii STELLAR (Jones P. H., Am J Cardiol 2003; 92 : 152–160). Výsledky srovnání tří v ČR užívaných statinů jsou prezentovány na obr. 5.
Obr. 5: Srovnání poklesu LDL-cholesterolu při léčbě třemi nejčastěji užívanými statiny ve studii STELLAR. Je patrné, že poklesu o více než 50 % bylo dosaženo již střední dávkou rosuvastatinu či maximální dávkou atorvastatinu. Naopak simvastatin byl významně méně účinný. Dále je patrné, že minimální dávka 1 mg rosuvastatinu denně měla významný efekt na pokles LDL-cholesterolu, srovnatelný s nižší dávkou atorvastatinu či střední dávkou simvastatinu.
2) Rozdíly mezi statiny v efektu na vzestup HDL-cholesterolu
Na význam cholesterolu obsaženého ve vysokodenzitních lipoproteinech (HDL-cholesterolu) se v posledních letech vyvíjí názor. Jeho význam je menší než stanovení volné transportní kapacity HDL pro cholesterol. Nicméně též koncentrace HDL-cholesterolu má prognostický význam.
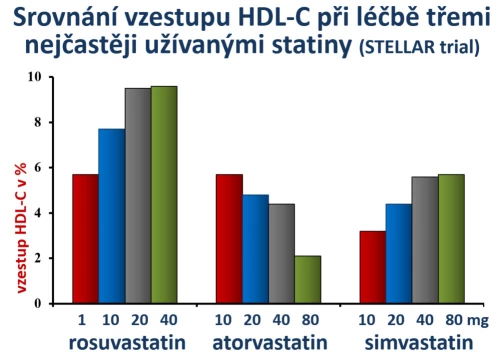
Statiny, stejně jako fibráty, hladinu HDL-cholesterolu zvyšují. I zde však z nejasného důvodu existují též rozdíly mezi jednotlivými molekulami. Nutno však zdůraznit, že nemáme žádný doklad o tom, že by tyto rozdíly měly klinický význam. Podobně jako tomu bylo v případě LDL-cholesterolu, sledovala studie STELLAR efekt různých statinů v různých dávkách na vzestup plazmatické koncentrace HDL-cholesterolu (obr. 6). Z pohledu hodnocení laboratorních hodnot byl nejpotentnější rosuvastatin, vyšší dávky hladinu HDL-cholesterolu zvyšovaly. Paradoxně u atorvastatinu byl pozorován opačný vztah, vyšší dávky měly na vzestup HDL-cholesterolu menší dopad.
Obr. 6: Srovnání vzestupu HDL-cholesterolu při léčbě třemi nejčastěji užívanými statiny ve studii STELLAR.
3) Rozdíly mezi statiny při redukci hypertriglyceridemie
Statiny nesnižují pouze koncentraci cholesterolu v aterogenních lipoproteinech, ale snižují též hladinu triglyceridů (triacylglycerolů). Zde je však efekt závislý méně na konkrétním statinu, ale více na typu dyslipidemie. Největší je účinek u nemocných s hypertriglyceridemií. Například léčba hypertriglyceridemie rosuvastatinem u nemocných s dyslipidemií IV. a V. typu snížila koncentraci proti placebu o 21 až 46 % (Hunninghake D. B., Coronary Artery Disease: 2004, Volume 15, Issue 2, s. 115–123).
Farmakokinetické vlastnosti a rozdíly mezi statiny
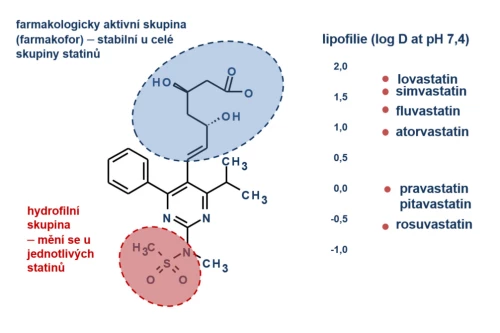
Farmakokinetické vlastnosti statinů se mění v závislosti na jejich lipofilii a afinitě k transportním a metabolickým systémům. Srovnání vlastností základních statinů je uvedeno v tabulce (tab. 3). Absorpce (a biologická dostupnost), koncentrace v místě působení (v hepatocytu), biotransformace a bioeliminace statinů jsou dány jejich fyzikálně-chemickými vlastnostmi, mezi nimiž je významným faktorem rozpustnost v tucích či vodě. Přítomnost hydrofilní skupiny u rosuvastatinu, pravastatinu či pitavastatinu zcela mění vlastnosti molekuly. Naopak farmakologicky aktivní skupina (farmakofor) se u jednotlivých statinů prakticky nemění (obr. 7).
Tab. 3: Srovnání farmakokinetických a farmakodynamických vlastností statinů.
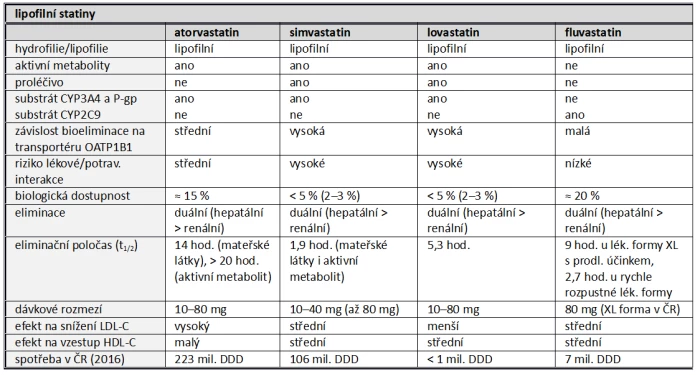
a) Lipofilní statiny
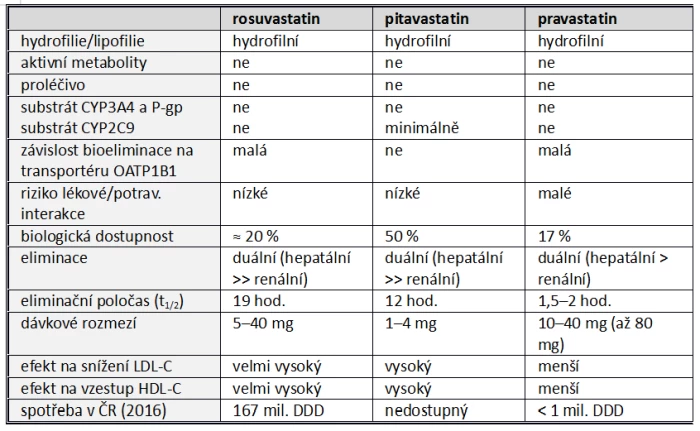
b) Hydrofilní statiny
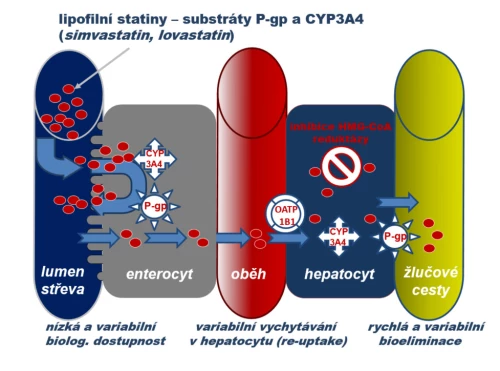
Hlavní rozdělení na statiny lipofilní a hydrofilní má své opodstatnění. Lipofilní statiny, zejména simvastatin a lovastatin, podléhají během absorpce v enterocytu a při průchodu játry transformaci izoenzymy CYP a některé jsou z enterocytu eliminovány transportérem P-glykoproteinem (P-gp). Jejich dostupnost se tak v různé míře snižuje, například u simvastatinu na 2–3 %. Lékovými interakcemi na této úrovni se zvyšuje riziko nežádoucích účinků.
Obr. 7: Vlastnosti statinů se mění v závislosti na lipofilii/hydrofilii. Farmakologicky účinná skupina (farmakofor) se nemění, významné rozdíly však jsou v řetězci, který rozhoduje o vlastnostech molekuly (podle: McTaggart F., et al. Am J Cardiol. 2001; 87 : 28B–32B).
Rovněž před eliminací musí být těmito oxidázami transformovány na hydrofilní metabolity. Též na této úrovni dochází k lékovým interakcím. Při seřazení podle lipofilie jsou na vrcholu simvastatin a lovastatin. Méně lipofilní jsou fluvastatin a atorvastatin.
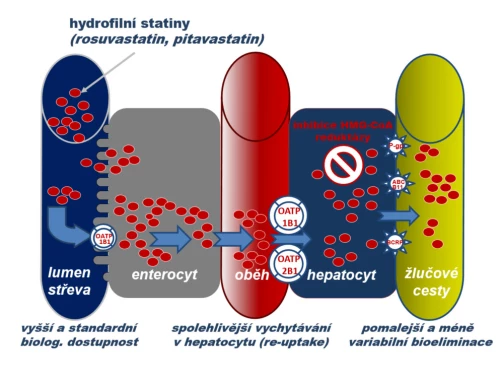
Hydrofilní statiny představují rosuvastatin a pitavastatin, které nejsou významnými substráty metabolických systémů a více než 95 % je eliminováno jako mateřská látka.
Způsob absorpce, transformace a eliminace má zásadní význam pro riziko lékových interakcí. To je u této lékové skupiny důležité, neboť zvýšení expozice statinu významně snižuje jeho toleranci a nezřídka vede k ukončení léčby. Kardiovaskulární riziko po vysazení statinu stoupá na hodnoty před léčbou a prognóza pacienta se zhoršuje. Zajištění optimální tolerance je tak zásadně důležité.
Jak v absorpci (na úrovni enterocytu), tak v eliminaci (na úrovni biliárního pólu hepatocytu) je přesun intracelulárně kontrolován eliminačními pumpami (zejm. glykoproteinem P, P-gp) i influxními pumpami (zejm. rodiny OATP [organic aniont transporting polypeptid]). Absorpce simvastatinu a lovastatinu, které jsou substráty P-gp, je nízká (< 5 %), glykoprotein je eliminuje zpět do lumen. Snížení aktivity P-gp (např. při lékových interakcích) pak významně zvýší dostupnost statinu. Naopak rosuvastatin či pitavastatin jsou substráty řady influxních transportérů, a jejich vstřebávání je tak významně vyšší (až k 50 %).
Pro farmakodynamický účinek statinu není tak podstatná jeho koncentrace v plazmě, ale koncentrace v místě účinku, tj. na membráně endoplazmatického retikula hepatocytu. Důvodem je skutečnost, že enzym HMG-CoA reduktáza je na membránu endoplazmatického retikula vázán. Právě zde, na membránách endoplazmatického retikula hepatocytu, syntéza cholesterolu probíhá. Transmembranózní přenos jednotlivých statinů do hepatocytu (hepatální uptake) je v různé míře facilitován transportní pumpou OATP1B1 (organic aniont transporting polypeptid 1B1) či dalšími influxními transportéry ze stejné rodiny. Tato pumpa je polymorfní, podle funkčnosti obou alel rozeznáváme špatné přenašeče (obě afunkční alely), střední a dobré přenašeče (s oběma funkčními alelami). Při dobré funkci transportéru je zajištěna dostatečná koncentrace statinu v místě efektu i spolehlivá eliminace do žluče (obr. 8a, 8b a 8c). Naopak při afunkční pumpě (přítomna asi u 15 % populace) se setkáváme s nízkým hypolipidemickým účinkem a s mnohonásobně vyšším výskytem nežádoucích účinků. Závislost vstupu jednotlivých statinů do hepatocytu na aktivitě OATP1B1 je různá, nejvíce je stimulován vstup simvastatinu a lovastatinu, nejméně fluvastatinu a rosuvastatinu. Fluvastatin či rosuvastatin jsou substráty též dalších izotransportérů. Výpadek jedné pumpy tak má klinicky málo významný dopad, a tolerance léku je proto významně lepší.
Obr. 8: Absorpce a eliminace jednotlivých typů statinů:
a) Substrátů oxidázy CYP3A4 a transportéru glykoproteinu P (simvastatin, lovastatin a částečně i atorvastatin). Po perorálním podání tohoto typu statinů je větší část eliminována zpět do lumen střeva na úrovni enterocytu izoenzymem CYP3A4 a transportérem P-gp. U simvastatinu či lovastatinu, které mají vysokou afinitu k tandemu CYP3A4/P-gp, je absorbováno jen méně než 5 % léčiva, u atorvastatinu s nižší afinitou k tomuto systému kolem 15 %. Z cirkulace je transmembranózní přestup do hepatocytu (hepatální uptake) facilitován influxní pumpou OATP1B1 (organic aniont transporting polypeptid 1B1). Tak je zajištěna dostatečná koncentrace statinu v hepatocytu i jeho eliminace do žluče pomocí efluxních transportérů, zejména P-gp. Při nízké aktivitě OATP1B1 eliminace statinu vázne, účinek klesá a naopak se zhoršuje tolerance.
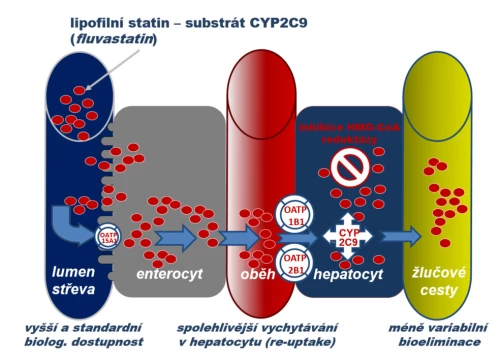
b) Substrátu oxidázy CYP2C9 a řady influxních transportérů rodiny OATP (fluvastatin). Po perorálním podání fluvastatinu je na úrovni enterocytu absorpce zvýšena aktivitou influxní pumpy typu OATP, stejně tak více těchto transportérů zvyšuje hepatální uptake. Jak absorpce, tak eliminace jsou tak daleko méně ovlivněny lékovými interakcemi či farmakogenetickou výbavou. Tolerance fluvastatinu je proto ve skupině statinů nejlepší.
c) Hydrofilních statinů typu rosuvastatinu a pitavastatinu. Tento typ není substrátem izoenzymů CYP, absorpce hydrofilních statinů i hepatální uptake jsou facilitovány řadou influxních transportérů rodiny OATP. Jejich biologická dostupnost je tak vysoká, stejně jako je vysoká koncentrace v hepatocytu. Díky řadě transportérů účastnících se na eliminaci je vylučování stabilní a riziko lékových interakcí a farmakogenetických vlivů je malé. Vysoká koncentrace na membránách endoplazmatického retikula zajišťuje vyšší efekt.
1) Simvastatin a lovastatin
Vysoce lipofilní statiny simvastatin a lovastatin jsou substráty (s vysokou afinitou) eliminační pumpy P-gp a oxidázy CYP3A4. Tato dvojice (P-gp/CYP3A4) tvoří funkční celek. U lipofilních molekul izoenzym CYP3A4 katalyzuje oxidaci, tím zvyšuje afinitu k P-gp a umožňuje eliminaci z buňky: na úrovni enterocytu eliminaci do střeva, na úrovni hepatocytu eliminaci do biliárního systému či na úrovni tubulární buňky nefronu eliminaci do moče. Vysoká afinita simvastatinu k transportnímu a metabolickému systému snižuje biologickou dostupnost dané molekuly na 2–5 % a závislost na „metabolicko-eliminačnímu tandemu“ současně zvyšuje riziko lékových a potravinových interakcí. Poločas plazmatické eliminace je krátký, kolem 2 hodin, při standardní aplikaci v jedné denní dávce tak rychle klesá účinnost a na konci dávkového intervalu je inhibice nedostatečná a cholesterol je částečně resyntetizován. Hypolipidemický účinek je tak nižší. Interindividuální variabilita expozice simvastatinu i jeho účinek jsou výrazně závislé na aktivitě transportéru OATP1B1. Tato skutečnost je příčinou nejvyššího výskytu nežádoucích účinků u simvastatinu z celé skupiny statinů.
2) Atorvastatin
U atorvastatinu, který je rovněž substrátem P-gp a CYP3A4, je díky nízké afinitě k tandemu jeho dostupnost středně vysoká, pohybuje se kolem 12–15 %, a interakční potenciál je tak relativně nízký. Je rozdíl, zvýšíme-li dostupnost z 2–5 % na 80 % (u simvastatinu) či z 15 % na 80 % (u atorvastatinu). Eliminační poločas atorvastatinu je dlouhý – 14 hod., čímž je zajištěna účinnost po dobu 24 hod., resp. při vynechání dávky i na 48 hod. Atorvastatin tak patří mezi nejpotentnější statiny. Závislost účinku a eliminace na influxní pumpě OATP1B1 je menší, interindividuální variabilita expozice i riziko nežádoucích jsou významně menší než u simvastatinu či lovastatinu.
3) Fluvastatin
Fluvastatin je velmi slabým substrátem P-gp a středně silným substrátem izoenzymu CYP2C9. Jeho absorpce na úrovni enterocytu je facilitována influxními transportéry OATP – biologická dostupnost je tak vyšší, asi 20 %. Hepatální uptake je zvýšen hned několika pumpami typu OATP, a závislost transmembranózního přesunu na aktivitě OATP1B1 je tak malá. Riziko lékových interakcí a farmakogenetických vlivů není významné. Též interakční potenciál izoenzymu CYP2C9 (který v hepatocytu degraduje molekulu fluvastatinu) je nízký. Izoenzym CYP2C9 je polymorfní, rozlišujeme metabolizátory rychlé (s expozicí nižší), střední a pomalé (s expozicí vyšší). Tyto rozdíly mají malý klinický význam z pohledu rizika lékových interakcí, ovlivňují však účinnost. Rozdíly v efektu fluvastatinu byly sledovány v české populaci – u rychlých metabolizátorů byla hladina LDL-cholesterolu snížena o 22 %, u středních o 25 % a u pomalých o 40 % (Buzková H., Med Sci Monit. 2012; 18 [8]: CR512–517).
Tolerance fluvastatinu je pak ve skupině statinů nejlepší. Pro krátký eliminační poločas (kolem 3 hod.) je podáván ve formě s prodlouženým účinkem, která tento handicap snižuje. Fluvastatin tak patří mezi středně účinné statiny.
4) Rosuvastatin a pitavastatin
Rosuvastatin a pitavastatin jsou statiny hydrofilní. Nejsou významnými substráty P-gp.
Rosuvastatin je CYP2C9 částečně transformován na metabolit s menší biologickou aktivitou, blokáda izoenzymu CYP2C9 zvyšuje expozici rosuvastatinu asi na dvojnásobek, tj. z klinického pohledu málo významně. Jak enterální absorpce, tak hepatální uptake jsou facilitovány více transportéry typu OATP. Závislost na polymorfním transportéru OATP1B1 je díky tomu malá, a výskyt nežádoucích účinků tak není ovlivněn polymorfizmem OATP1B1. Hepatální influx je facilitován zejména obdobnou pumpou OATP2B1, její vyšší aktivita (na základě polymorfizmu) je spojena s větším hypolipidemickým účinkem. Biologická dostupnost rosuvastatinu je asi 20 %. Eliminační poločas je dlouhý, kolem 20 hodin. Hypolipidemický efekt díky tomu přetrvává několik dnů. Významný efekt na pokles LDL-C (asi o třetinu) je doložen již při dávce 1 mg rosuvastatinu denně. Stejného efektu dosáhneme 10 mg atorvastatinu či 30 až 40 mg simvastatinu. Z tohoto pohledu je racionální podávání rosuvastatinu i v dávce 5 mg obden (v případě snížené tolerance). Rosuvastatin je nejúčinnějším a po fluvastatinu nejlépe tolerovaným statinem. Riziko lékových interakcí je velmi nízké. Ve farmakoekvivalentních dávkách je významně lépe tolerován než simvastatin, lovastatin a atorvastatin.
Pitavastatin je rovněž statinem hydrofilním. Není transformován izoenzymy CYP, resp. je nevýznamně oxidován CYP2C9. Hepatální uptake je výrazně facilitován, stejně jako ostatní statiny, influxní pumpou OATP jen v případě pitavastatinu OATP2A1 a OATP2B1. Díky tomu nejsou eliminace a výskyt nežádoucích účinků ovlivněny polymorfizmem pumpy OATP1B1. Eliminován je v nezměněné podobě žlučí, výrazná je enterohepatální recirkulace. Díky ní je dosaženo vysoké biologické dosažitelnosti – 50 %. Poločas biologické eliminace je dlouhý, kolem 12 hodin. Pitavastatin patří k potentnějším statinům. Riziko lékových interakcí je minimální. Ač je v EU registrován, není běžně dostupný.
Posledním hydrofilním statinem je pravastatin. Pro malou účinnost není v ČR prakticky užíván.
5) Klinický význam farmakokinetických rozdílů mezi statiny
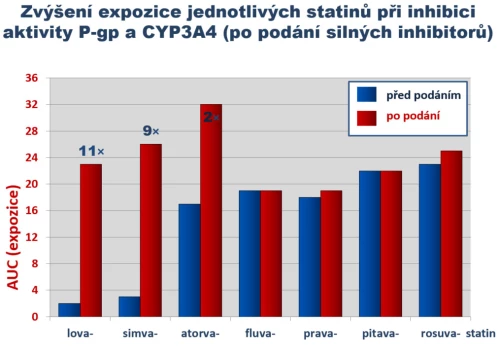
Rozdíly ve farmakokinetice mají praktický dopad, asi nejvýznamnější jsou rozdíly v riziku lékových či potravinových interakcí. Nejčastější jsou interakce na úrovni systému P-gp/CYP3A4. Tento eliminační systém kontroluje absorpci, transformaci a eliminaci simvastatinu, lovastatinu a atorvastatinu. U simvastatinu a lovastatinu, které mají velmi nízkou biologickou dostupnost, hraje inhibice systému velkou roli. Zvýší-li se dostupnost z 3 až 5 % na 30 až 50 % (což jsou reálná data) a současně se zpomalí eliminace, pak expozice vroste o řád. U atorvastatinu s biologickou dostupností kolem 15 % se při zvýšení dostupnosti na 30 až 50 % expozice zvýší maximálně trojnásobně. U ostatních statinů ke zvýšení expozice nedochází (obr. 9).
Obr. 9: Zvýšení expozice statinů po inhibici eliminačního systému P-gp/CYP3A4. Podání silných či středně silných inhibitorů (verapamil, diltiazem, amiodaron, některá makrolidová antibiotika, některá azolová antimykotika, řada psychofarmak či flavonoidy citrusových plodů) zvyšují expozici simvastatinu či lovastatinu troj - až desetinásobně. Podle Vaquero M. P. et al., Nutr Hosp 2010, 25 (2) 193.
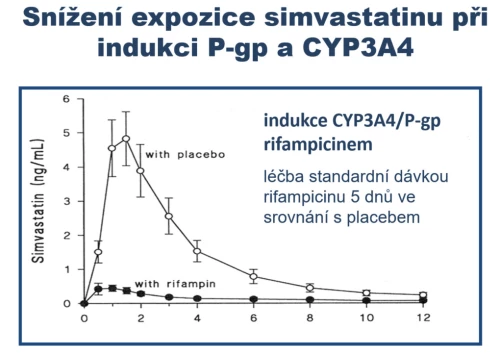
U simvastatinu či lovastatinu může nastat i opačná situace – indukce CYP3A4/P-gp naopak expozici léčiva významně sníží. Příkladem je indukce jak oxidázy CYP3A4, tak transportéru P-gp antibiotikem rifampicinem (obr. 10). Zvýšenou aktivitou eliminačního systému klesá absorpce léčiva. Expozice simvastatinu již po několika dnech souběžného podávání klesá výrazně pod terapeutickou hladinu. Obdobný efekt má např. též fenobarbital, dexametazon či extrakt z třezalky.
Obr. 10: Snížení expozice (součinu koncentrace a času) simvastatinu při indukci CYP3A4 a P-gp rifampicinem. Podle Kyrklund et al., Clin. Pharmacol. Ther. 68 : 592–597, 2000.
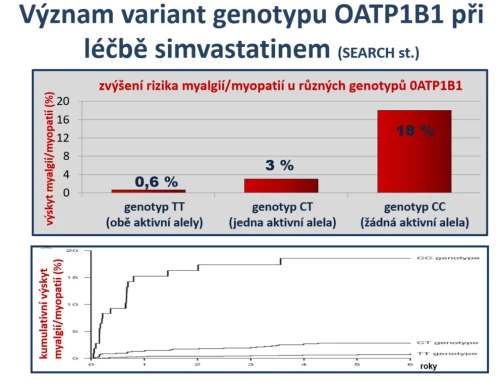
Podobně je riziko nežádoucích účinků ovlivněno rozdíly v eliminaci statinů. Přestup do hepatocytu transportérem OATP1B1 je významný zejména u simvastatinu, kde se na přesunu do jaterní buňky (hepatálním uptake) významně nepodílí jiné transportéry a spontánní přestup je nízký. Influxní pumpa OATP1B1 je polymorfní – asi u 15 % indoevropské populace není transportér aktivní. U jedinců s oběma defektními alelami regulačního genu je uptake nízký, koncentrace v místě působení (na membráně endoplazmatického retikula, kde je inhibována aktivita HMG-CoA reduktázy) je nedostatečná, hypolipidemický efekt klesá a stejně tak klesá eliminace simvastatinu do žluče a následně se zvyšuje výskyt nežádoucích účinků typu myalgie a myopatie. Též u nosičů jedné defektní alely je hepatální uptake snížen, zvýšení výskytu nežádoucích účinků však je, v porovnání s nosiči obou funkčních alel, výrazně menší (obr. 11). V afinitě k transportéru OATP1B1 jsou mezi statiny výrazné rozdíly, největší byla doložena u simvastatinu, nejmenší u fluvastatinu a rosuvastatinu (simvastatin > atorvastatin > pravastatin > rosuvastatin > fluvastatin). Proto je závislost mezi aktivitou transportéru a výskytem nežádoucích účinků největší u simvastatinu a nejmenší u fluvastatinu (Voora D., J Am Coll Cardiol 2009; 54 : 1609–1616).
Obr. 11: Výskyt myalgie a myopatie při léčbě simvastatinem v závislosti na aktivitě influxního transportéru OATP1B1. Hepatální uptake statinů je facilitován transportéry typu OATP, v případě simvastatinu a lovastatinu pouze jediného OATP1B1. Při polymorfizmu s velmi nízkou aktivitou transportéru (genotyp CC s absencí funkční alely) se simvastatin nedostane do hepatocytu a jeho biliární eliminace je nízká. Vzestup koncentrace simvastatinu stoupá a paralelně se zvyšuje výskyt myalgie a myopatie. V porovnání s genotypem TT s oběma funkčními alelami je výskyt tohoto typu nežádoucích účinků více než třicetinásobný: 0,6 % vs. 18 % (SEARCH collaborative group, N Engl J Med 2008; 359 : 789–799).
Obdobně může být aktivita OATP1B1 inhibována některými léčivy (cyklosporinem, některými makrolidovými antibiotiky aj.). Vedle přímé blokády transportéru je klinicky významná též kompetice o přenos. Praktický dopad má současné podávání fibrátů (zejména gemfibrozilu), kde je soutěžení o přenos největší. Užívání cerivastatinu, u něhož byl výskyt nežádoucích účinků (zejména rhabdomyolýzy) při současné léčbě fibráty největší, bylo před lety ukončeno.
U ostatních statinů má tato pumpa (OATP1B1) menší význam, facilitovaný vstup do hepatocytu je zprostředkován více typy transportérů a vyšší riziko nežádoucích účinků na podkladě inaktivity OATP1B1 nebylo doloženo.
Nežádoucí účinky statinů, rozdíly mezi jednotlivými statiny
1) Přehled klinicky významných nežádoucích účinků statinů
Statiny jsou, při respektování lékových a potravinových interakcí, velmi dobře tolerovanou skupinou léků. Nejčastějším nežádoucím účinkem jsou myalgie, tj. bolesti svalové skupiny (nejčastěji stehenní), které nejsou provázeny poškozením, jež by vedlo k uvolnění nitrosvalových enzymů – konkrétně kreatinkinázy (CK). Ekvivalentem myalgií mohou být svalové křeče. Při aplikaci nižších a středních dávek dobře tolerovaných statinů (fluvastatinu či rosuvastatinu) se myalgie objevují u 1–3 % léčených (po korekci na efekt placeba). Podávání vyšších dávek méně tolerovaných statinů, užití nevhodných lékových kombinací či vysoká fyzická aktivita výskyt myalgií zvyšují. Po převedení na jiný statin, po redukci dávky či vynechání léků s rizikem interakce se tolerance zpravidla zlepší. Po vysazení statinu myalgie do několika málo dnů až týdnů odeznívají. Platí, že zdaleka ne všechny myalgie jsou na vrub statinů, jejich výskyt je běžný i při podávání placeba, velké studie dokládají výskyt myalgií při léčbě placebem kolem 5 %, při léčbě statinem kolem 7 %. Významnějším postižením svalstva je myopatie definovaná přítomností svalových bolestí spolu se zvýšením CK. Její incidence je asi 1/1 000 až 1/10 000 léčených standardními dávkami statinu. I tato forma je po vysazení léčby či redukci dávky plně reverzibilní. Mírné zvýšení CK (do desetinásobku horní hranice) není nutně důvodem vysazení léku, zkontrolujeme případné lékové či potravinové interakce, redukujeme dávku či změníme statin. Samotné zvýšení CK při léčbě statiny, které není provázeno myalgiemi, není považováno za projev nežádoucích účinků, a sledování CK je proto indikováno jen při klinických svalových projevech. Nejzávažnějším projevem postižení svalstva při léčbě statiny je rhabdomyolýza, stav kdy dochází k poškození buněčných membrán a úniku myoglobinu do cirkulace. Masivní myoglobinurie může vést až k renálnímu selhání a k úmrtí. Myolýza je naštěstí vzácná, její výskyt se pohybuje mezi 1 a 10 případy na milion léčených. Klinický význam myalgií/myopatií je zejména v tom, že často vedou k vysazení léčby statinem. Nedostatečné léčení významného rizikového faktoru po aterotrombotické příhodě pak zvýší riziko recidivy příhody a mortalitu asi o čtvrtinu.
Relativně často (asi u 0,5 % léčených) se setkáváme s asymptomatickým vzestupem jaterních enzymů (zejména transamináz). Samotné zvýšení nemá velký klinický význam a při léčbě statiny není ani doporučeno sledování jaterních testů.
Často diskutovaným nežádoucím účinkem (s incidencí kolem 1 na 1 000 léčených ročně) je zvýšení incidence nově zachyceného diabetu. Tato skutečnost není z klinického pohledu tak významná, uvědomíme-li si, že v sekundární prevenci se předejde 20–80 aterotrombotickým příhodám ročně na tisíc léčených, resp. 10–20 příhodám v primární prevenci, pak nový záchyt diabetu se vyskytne 1–2× na tisíc léčených, tedy o 1 až 2 řády méně často. Není jasné, zda aterogenní potenciál tohoto typu „statiny manifestovaného“ diabetu je stejný jako diabetu vzniklého na podkladě inzulinorezistence (tedy DM 2. typu). Riziko diabetu při léčbě statiny kolísá, při léčbě málo či středně potentními statiny (simvastatin, lovastatin, fluvastatin či pravastatin) se zvyšuje méně než při léčbě vysoce potentními statiny (rosuvastatin, atorvastatin či pitavastatin). Podobně vyšší dávky statinu riziko rovněž zvyšují. Avšak po přepočtení rizika manifestace diabetu na pokles LDL-C jsou rozdíly mezi statiny i rozdíly závislé na dávce statinu smazány. Je pravděpodobné, že příčinou tohoto fenoménu je zásah do lipidového metabolizmu, obdobný vzestup výskytu diabetu byl pozorován i při léčbě fibráty či niacinem. Při dodržování nízkocholesterolové diety či při inhibici absorpce cholesterolu ezetimibem nebylo pozorováno zvýšení rizika diabetu.
2) Rozdíly mezi statiny ve výskytu nežádoucích účinků
Výskyt nežádoucích účinků je variabilní statin od statinu, největší výskyt je doložen pro simvastatin. Zde je důvodem pravděpodobně vysoký potenciál pro lékové interakce na podkladě zvýšení dostupnosti při inhibici CYP3A4 a P-gp i závislost eliminace na polymorfním transportéru OATP1B1. Naopak nejlépe tolerovaným statinem je fluvastatin, nepatří však mezi statiny nejúčinnější.
Jsou tyto rozdíly v toleranci klinicky významné? Odrazily se poznatky ze sledování farmakokinetiky a farmakogenetických studií na toleranci? Překvapivě málo studií je zaměřeno na rozdíly ve výskytu nejčastějšího nežádoucího účinku při léčbě statiny – myalgií a myopatií.
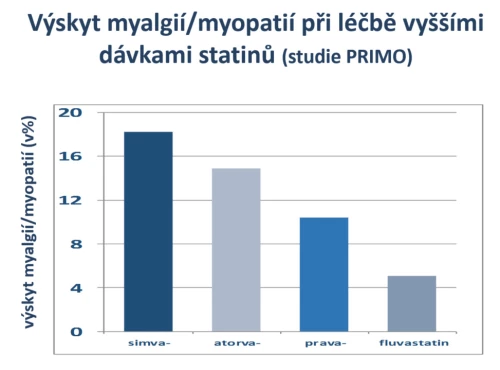
Z metodického hlediska je nejvhodnějším porovnáním studie PRIMO, neboť monitorovala výskyt myalgií/myopatií v reálné praxi (Bruckert E., Cardiovasc Drugs Ther 2005; 19 : 403–14). Z 8 tisíc probandů, léčených tehdy dostupnými statiny (simvastatinem, atorvastatinem, pravastatinem a fluvastatinem), se myalgie/myopatie objevily u 10 %. Při léčbě simvastatinem (v dávce 40–80 mg denně) se objevují v 18,2 %, v porovnání s fluvastatinem (v dávce 80 mg denně) byly téměř čtyřikrát častější (obr. 12). Rozdíly ve výskytu svalových potíží byly při léčbě simvastatinem i atorvastatinem (40 až 80 mg denně) významné také v porovnání s pravastatinem (40 mg denně) či fluvastatinem. Dávky užité při léčbě simvastatinem a fluvastatinem přitom byly – stran poklesu LDL-cholesterolu – přibližně farmakoekvivalentní, naopak obě dávky atorvastatinu byly významně účinnější.
Obr 12: Výskyt nejčastějšího nežádoucího účinku při léčbě statiny – myalgií a myopatií. Je patrné, že při farmakoekvivalentních dávkách je léčba fluvastatinem daleko lépe tolerována než léčba simvastatinem. Srovnání fluvastatinu s atorvastatinem je obtížné, dávky nebyly stejně účinné.
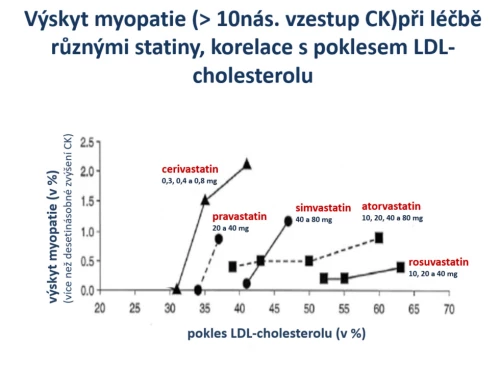
Jiná analýza se zabývala výskytem dobře objektivně doložitelným nežádoucím účinkem – myopatií. Ta byla definována více než desetinásobným zvýšením kreaktinkinázy (CK). Porovnávány byly jak různé statiny (cerivastatin, pravastatin, simvastatin, atorvastatin a rosuvastatin) v různých dávkách, tak korelace s poklesem LDL-cholesterolu (Brewer H. B., Am J Cardiol 2003; 92 [supl.]: 23K–29K). Tedy korelována účinnost a bezpečnost u jednotlivých statinů (obr. 13). Je patrné, že nejméně účinné a přitom nejhůře tolerované a nejméně bezpečné byly cerivastatin, pravastatin a vyšší dávky simvastatinu. Naopak nejúčinnější a přitom i nejbezpečnější byly rosuvastatin a vyšší dávky atorvastatinu. Tato studie doložila největší účinnost a současně největší bezpečnost rosuvastatinu (v malých i vysokých dávkách) a potvrdila větší účinnost i bezpečnost atorvastatinu (ve středních a vyšších dávkách) než ekvivalentních dávek simvastatinu.
Obr. 13: Korelace účinnosti (pokles LDL-C) a bezpečnosti (výskyt myopatie definované vzestupem CK). Cerivastatin, pravastatin a simvastatin byly relativně méně účinné a přitom byla léčba zatížena vyšší incidencí myopatie (vzestup CK nad desetinásobek).
Při sledování ostatních nežádoucích účinků, např. vzestupu jaterních testů či mírného zvýšení rizika manifestace diabetu, se statiny mezi sebou výrazně neliší. Například riziko vzniku diabetu se zvyšovalo při léčbě všemi statiny, konkrétně se nárůst incidence pohyboval okolo jednoho případu nově zjištěné významné hyperglykemie na tisíc léčených po dobu jednoho roku. Riziko je tedy významně nižší, než s jakým se setkáváme např. při léčbě diuretiky. Rovněž riziko zvýšení jaterních testů nemá velký význam, např. FDA inovovala svůj názor a upustila od doporučení pravidelných laboratorních kontrol jaterních testů v průběhu léčby statiny.
Kontraindikací podávání statinů je alergie (bývá pouze individuální na jeden konkrétní statin), gravidita a laktace. Preexistující myopatie či významné jaterní poškození je kontraindikací pro nasazení statinu.
3) Jak postupovat při výskytu nejčastějšího nežádoucího účinku statinů – myalgií/myopatií?
Výskyt nežádoucích účinků typu myalgie není důvodem k přerušení léčby, ale k její úpravě a odhalení případných negativních vlivů, které k myalgiím přispěly. U většiny nemocných lze po úpravě léčebné strategie v léčbě pokračovat. Po vyloučení lékových či potravinových interakcí zvyšujících koncentraci statinu, stavů vedoucích k myalgiím (nadměrná fyzická zátěž, iontová nerovnováha apod.) či nesprávného užívání léku zvážíme buď změnu statinu (simvastatin či atorvastain za fluvastatin či rosuvastatin), či redukci dávky. Význam má pokračovat v léčbě statinem i v dávce minimální – dávka 5 mg rosuvastatinu obden má stále účinek srovnatelný s 20–40 mg simvastatinu! Efekt na pokles LDL-cholesterolu potencujeme inhibitorem absorpce cholesterolu – ezetimibem. Z vlastní zkušenosti vím, že dobrá motivace nemocného umožní u naprosté většiny léčených pokračovat v terapii statinem. Při redukci dávky je možno podávat dlouhodobě účinné statiny, jakými jsou rosuvastatin či atorvastatin i obden, krátkodobě působící simvastatin, lovastatin či fluvastatin (i ve formě XL) je nutno aplikovat výhradně denně.
Důležitá je i otázka, zda jsou statiny skutečně příčinou všech myalgií/myopatií, které se v průběhu hypolipidemické léčby objeví. Není část problému jen strategií PR agentur? Občas se setkáváme s tím, že se před zavedením nové skupiny léčiv nenápadně objevuje kampaň proti léku již zavedenému. Vzpomeňme jen problém „aspirinové rezistence“ při příchodu klopidogrelu. Nyní přichází skupina inhibitorů PCSK9. Myalgie a myopatie jistě jsou komplikací léčby statiny, jsou však skutečně tak zásadním problémem?
Z tohoto pohledu jsou zajímavá data ze studie GAUSS-3, analyzující stav u nemocných s „intolerancí“ statinů, u nichž došlo k vysazení léčby. Opětovné podání 20 mg atorvastatinu vedlo k výskytu myalgií u méně než poloviny (43 %) léčených, podání placeba bylo spojeno naopak s výskytem myalgií u 27 % nemocných. Ukazuje se tak, že opětovné nasazení statinu je schůdné, že velká část myalgií není důsledkem léčby statiny. V porovnání s efektem placeba se u nemocných s předchozí intolerancí vedoucí k vysazení hypolipidemické léčby myalgie objevily po opětovném nasazení statinu jen o 16 % častěji než po podání placeba. Jinak řečeno, u většiny nemocných léčených statiny se jednalo o tzv. „nocebový“ efekt, kdy pacienti pouze předpokládali, že užívají statin, a anticipovali myalgie.
Uvědomíme-li si, že vysazení statinové léčby je spojeno se vzestupem KV mortality a morbidity, měli bychom se pokusit i tzv. „intolerantním“ nemocným znovu léčbu nasadit. Zkusit jiný statin, jinou dávku, podívat se na možné lékové interakce či hledat jinou příčinu myalgií.
Klinický význam statinů
Statiny jsou lékem prvé volby v léčbě nemocných se zvýšenou plazmatickou koncentrací LDL-cholesterolu. Vzhledem k tomu, že hranice „zvýšení“ je relativní a závisí na riziku aterotrombotické příhody, je možno říci, že statiny jsou indikovány ke snížení kardiovaskulárního rizika. Jejich indikací jsou zejména nemocní léčení v rámci sekundární prevence, tj. po proběhlé aterotrombotické příhodě či s přítomností aterosklerotického postižení (po infarktu myokardu, po cévní mozkové příhodě, při ischemické chorobě dolních končetin či např. s přítomností plátu v tepenném řečišti) či s diabetem mellitem. V primární prevenci jsou indikovány statiny u nemocných se zvýšenou koncentrací LDL-cholesterolu, rozhodující pro podávání není jen výše LDL-cholesterolu, ale zejména celkové kardiovaskulární riziko.
Dávka statinu se odvíjí od efektu, přihlížíme k cílové hodnotě LDL-cholesterolu pro absolutní riziko příhody: u nejrizikovějších nemocných je cílem snížení LDL-cholesterolu pod 1,8 mmol/l, u všech ostatních nemocných v sekundární prevenci pod 2,6 mmol/l a v primární prevenci pod 3,0 mmol/l. Léčbu zahajujeme dávkou spíše nižší a posléze podle efektu event. zvyšujeme. Pouze u nemocných bezprostředně po akutní koronární příhodě volíme již zpočátku dávku vysokou, předpokládáme totiž uplatnění extralipidového efektu.
Nedosáhneme-li cílové hodnoty, dávku zvyšujeme, volíme účinnější statin či kombinujeme s další léčebnou strategií (nejčastěji s blokádou absorpce cholesterolu). Pro zvýšení dávky zhruba platí, že každé její zdvojnásobení (stejně jako redukce na polovinu) sníží (zvýší) hladinu LDL-cholesterolu asi o 6 %. Platí také, že zdvojnásobení dávky významně sníží toleranci statinu. Proto je v řadě případů vhodnější kombinace např. s blokátorem absorpce cholesterolu ezetimibem nebo u nemocných s nejvyšším rizikem s inhibitory PCSK9, stimulujícími vstup LDL do buněk. Při souběhu zvýšené hladiny LDL-cholesterolu s nízkou hladinou HDL-cholesterolu a hypertriglyceridemií je možno zvážit kombinaci statinu s fibrátem, pozitivní účinek na snížení velkých kardiovaskulárních příhod však není jednoznačně doložen. V odůvodněných případech je možno užít statiny i u dětí.
Jsou rozdíly mezi statiny v ovlivnění prognózy?
V efektu na zlepšení prognózy nemocných jsou mezi statiny nepochybně rozdíly, nicméně přímé srovnání v rámci skupiny s více statiny nemáme. Přijmeme-li skutečnost, že riziko aterotrombotických příhod je v přímé úměře ke koncentraci LDL-cholesterolu a snížení této koncentrace prognózu zlepšuje, pak per analogiam při rozdílné potenci na snížení LDL-cholesterolu bude přítomen i rozdíl v klinickém efektu mezi statiny. Doklad o rozdílném účinku na základě kontrolovaných studií však chybí.
V současné době je k dispozici sedm statinů, podle klesající účinnosti je lze seřadit následovně: rosuvastatin, atorvastatin, pitavastatin, simvastatin, fluvastatin, lovastatin a pravastatin. Jejich farmakoekvivalentní dávky (měřeno srovnatelným poklesem LDL-C) jsou uvedeny v tab. 4. V ČR se užívají prakticky pouze čtyři statiny – rosuvastatin, atorvastatin, simvastatin a fluvastatin. Porovnáme-li poměr jejich účinnosti a bezpečnosti, favoritem je rosuvastatin, jen o něco méně účinný je atorvastatin. Nejbezpečnější a nejlépe tolerovaný je fluvastatin. Nejméně výhodným – pro horší toleranci, menší účinnost a vysoké riziko lékových interakcí – je simvastatin. Ostatní statiny nejsou u nás pro nízkou účinnost či nedostupnost užívány.
Tab. 4: Farmakoekvivalentní a maximální doporučené dávky jednotlivých statinů.
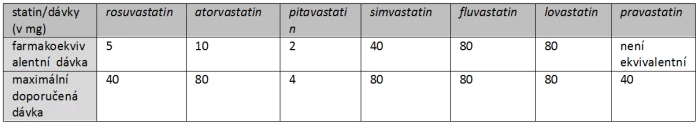
Farmakoekvivalentní dávka je odvozena od srovnatelného poklesu LDL-C. Z tabulky je patrno, že u rosuvastatinu či atorvastatinu můžeme základní dávku až osminásobně zvýšit. Pouze tyto dva statiny jsou schopny snížit LDL-C o více než 50 % a měla by jim být dána přednost. Hlavní místo fluvastatinu jakožto nejbezpečnějšího statinu je v léčbě dyslipidemie při špatné toleranci ostatních statinů.
Literatura u autora
Byl pro Vás kurz přínosný? Rádi byste se k němu vyjádřili? Napište nám − Vaše názory a postřehy nás zajímají. Zveřejňovat je nebudeme, ale rádi Vám na ně odpovíme.