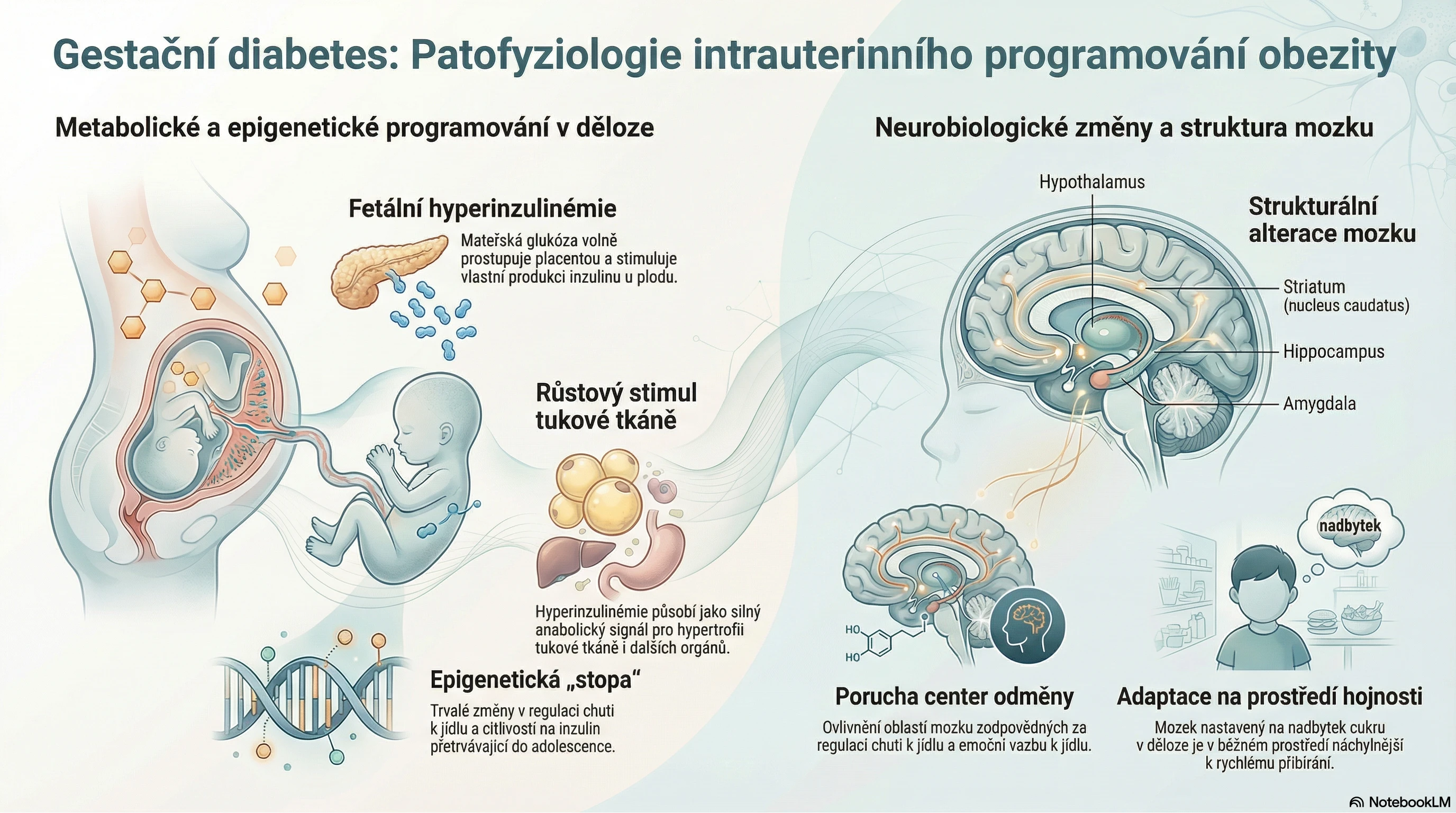
Intersticiální plicní procesy (IPP) představují skupinu chorob plicního parenchymu idiopatické či známé etiologie, které jsou charakterizované různou mírou zánětlivého a/nebo fibrotického poškození plicní tkáně. Nejrozsáhleji prostudovaným typem je idiopatická plicní fibróza (IPF) charakterizovaná progredující fibrotizací plicní tkáně.
Progresivní fibrózou s podobným průběhem jako IPF se projevuje i několik dalších onemocnění. Skupina takzvaných progredujících fibrotizujících plicních procesů zahrnuje intersticiální plicní procesy související s onemocněním pojivové tkáně (systémová sklerodermie, revmatoidní artritida, polymyozitida/dermatomyozitida), intersticiální plicní procesy související s chronickou sarkoidózou, chronická hypersenzitivní pneumonie, idiopatické nespecifikované intersticiální pneumonie a neklasifikovatelné intersticiální plicní procesy.
Některé typy IPP jsou považovány za primárně fibroproliferativní onemocnění, kdy dochází při poškození alveolů k proliferaci fibroblastů a fibrotizaci plicní tkáně. U jiných typů IPP je fibrotizace považována za následek primárně zánětlivých změn s přechodem do fibroproliferace za určitých podmínek. Progrese onemocnění je charakterizována postupným zhoršováním plicních funkcí, symptomů onemocnění a kvality života. Mezi základní klinické příznaky patří progredující námahová a poté i klidová dušnost, snadná unavitelnost, kašel a v pozdějších fázích cyanóza při hypoxémii. Mezi nejčastěji hodnocené parametry progrese patří měření usilovné vitální kapacity (FVC) a difuzní kapacity plic (DLCO). Klinický průběh jednotlivých onemocnění je heterogenní, avšak všechny spojuje progresivní fibrotizace.
Riziko progrese IPP související s onemocněním pojivové tkáně je nejvyšší v prvních několika letech od stanovení primární diagnózy. U části pacientů s chronickou hypersenzitivní pneumonií může dojít k částečnému zotavení, avšak současně může být u těchto pacientů pozorována rychlá progrese onemocnění, zejména pokud nelze identifikovat a odstranit vyvolávající antigen. O průběhu idiopatických nespecifikovaných intersticiálních pneumonií toho není mnoho známo, zdá se však, že oproti jiným fibrotizujícím IPP vykazují pomalejší progresi.
Onemocnění mohou být komplikována akutními exacerbacemi, které jsou také nejvíce prostudované v rámci IPF. Z dostupných studií však vyplývá, že se akutní exacerbace vyskytují i u dalších fibrotizujících IPP a velmi často jsou fatální. Progresivní fibrotizující intersticiální plicní procesy jsou asociovány s vysokou mortalitou, střední doba přežití se pohybuje v řádu jednotek let. Například u idiopatické plicní fibrózy je medián doby přežití od diagnózy 3–4 roky, podle recentní studie činí medián přežití u pacientů se systémovou sklerodermií 11,2 roku od prvního průkazu intersticiálního plicního procesu pomocí výpočetní tomografie o vysokém rozlišení (HRCT), u pacientů s revmatoidní artritidou byl podle retrospektivní analýzy stanoven medián přežití 5 let od první klinické návštěvy a 3,2 roku od průkazu obvyklé intersticiální pneumonie (UIP) na HRCT. Lepší prognóza je v této skupině pozorována u idiopatických nespecifikovaných intersticiálních pneumonií s 5letým přežitím přibližně 75 %.
U nemocných s progredujícími fibrotizujícími intersticiálními plicními procesy byla identifikována řada prognostických faktorů. Průběh nemoci u konkrétního pacienta však zatím není možné s jistotou předpovědět. Mezi prediktory špatné prognózy patří snížení FVC a DLCO, extenzivní rozsah fibrotických změn na HRCT nebo přítomnost voštiny a bronchiektázií nebo zvýšená sérová hladina mukoproteinu Krebs von den Lungen 6 (KL-6), která se rutinně používá v některých japonských centrech pro léčbu intersticiálních plicních procesů. Jako prediktory byly také vyvinuty kompozitní skórovací systémy (např. GAP) a identifikovány další biomarkery, některé genetické mutace a délka telomer.
Skupina progredujících fibrotizujících intersticiálních plicních procesů zahrnuje heterogenní skupinu chorob se společným rysem progredující fibroproliferace, která způsobuje zhoršování plicních funkcí, klinických symptomů a kvality života. Onemocnění jsou charakterizována nepříznivou prognózou s vysokou mortalitou, s mediánem přežití od diagnózy onemocnění v řádu několika let. V současnosti bylo identifikováno několik prognostických faktorů, průběh onemocnění však zatím nelze u konkrétního pacienta s jistotou předpovědět.
(holi)
Zdroj: Kolb M., Vašáková M. The natural history of progressive fibrosing interstitial lung diseases. Respir Res 2019; 20 (1): 57, doi: 10.1186/s12931-019-1022-1.