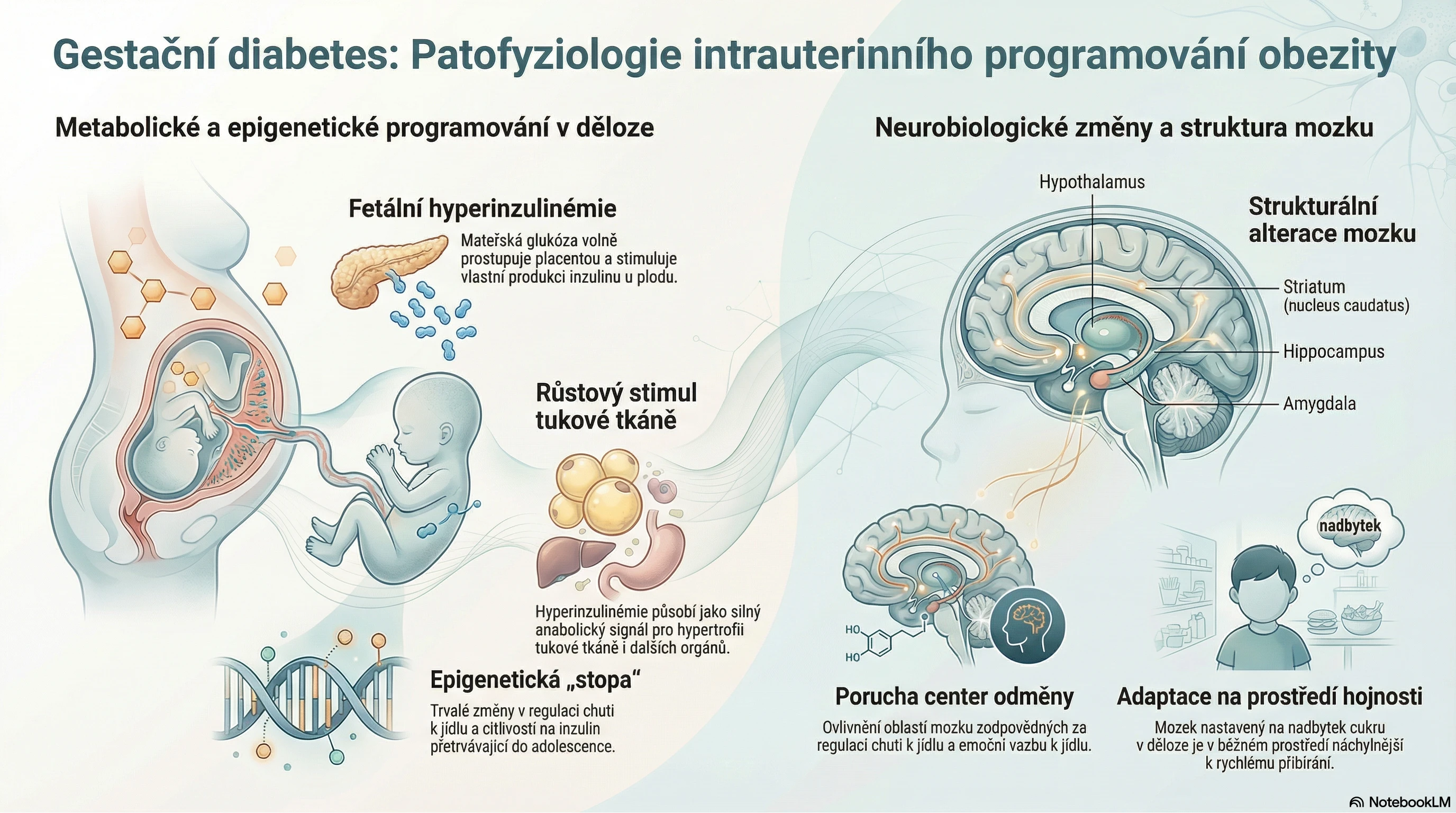
GM2 gangliosidózy, způsobené deficitem β-hexosaminidázy (Hex) A (Tayova−Sachsova choroba) nebo B (Sandhoffova choroba), patří mezi střádavá lyzosomová onemocnění. V lyzosomech se při nich akumuluje gangliosid GM2, což je glykosfingolipid membrány neuronů. Různé mutace v genech pro HexA/B vedou k různému stupni snížení enzymové aktivity, přičemž zbytková aktivita určuje tíži onemocnění a spektrum klinických příznaků.1
Prevalence přenašečů Tayovy−Sachsovy choroby v běžné populaci činí asi 1 : 250–300, což odpovídá odhadované incidenci autosomálně recesivního onemocnění cca 0,3/100 000 živě narozených. V rizikových populacích (aškenázští Židé, francouzští Kanaďani aj.) je však výskyt přenašečů onemocnění až 100× vyšší.2 Prevalence Sandhoffovy choroby v běžné populaci není známa, odhady se pohybují v rozmezí 0,07–0,2/100 000.3
Deficit HexA/B se manifestuje primárně v neuronech a vyznačuje se progresivním neurologickým postižením s poruchami motoriky, slabostí, hypotonií, snížením responzivity, poruchami vidění a křečemi. Akutní formy s časným nástupem vedou u obou onemocnění k rychlé deterioraci a úmrtí pacienta obvykle do 3 let věku. Neurologické příznaky u subakutních juvenilních forem zpravidla nastupují mezi 2. a 5. rokem života, kdy dochází ke zpomalení či regresi dosud normálního psychomotorického vývoje a rozvoji neurologických příznaků (abnormální vzorec chůze, dysartrie, kognitivní úpadek). Pacienti obvykle umírají ve 2. dekádě života, často po aspiraci.2, 3
V případě Tayovy−Sachsovy choroby s pozdním nástupem (LOTS) se pomalu progredující neurologické a/nebo psychiatrické příznaky začínají objevovat během dospívání či v časné dospělosti. Jedná se o široké spektrum symptomů, jež mohou napodobovat jiná neurodegenerativní onemocnění. Mezi časné příznaky patří neurogenní slabost dolních končetin s atrofií kvadricepsu, dysartrie, poruchy rovnováhy, tremor, mírná spasticita či dystonie. Až 40 % nemocných zažívá psychiatrické projevy, časté bývají úzkosti a depresivní poruchy, může se vyskytnout i akutní psychóza s mánií. Atrofie mozečku bývá patrná již od časného stadia onemocnění téměř u všech pacientů. U některých nemocných se postupně rozvíjí pokles kognitivních funkcí a paměti.2
Sandhoffova choroba s pozdním nástupem (LOSD) se vyznačuje progresivní neuropatií dolních končetin se slabostí a atrofií (zejména extenzory kolene a flexory kyčle), poruchami rovnováhy, třesem nebo ataxií. Obvyklé jsou distální senzorické neuropatie, progresivní dysartrie a neurokognitivní úpadek. Na zobrazovacím vyšetření může být přítomná cerebelární, míšní či mírná kortikální atrofie.3
Deficit HexA/B se stanovuje vyšetřením enzymové aktivity v séru, periferních leukocytech nebo jiných tkáních. Stanovení lze provést i testem ze suché krevní kapky (DBS). Diagnózu potvrdí nález patogenní varianty genu HEXA (Tayova−Sachsova choroba) či HEXB (Sandhoffova choroba).2, 3
V diferenciální diagnostice je třeba zohlednit další onemocnění, a to jak dědičná (například amyotrofickou laterální sklerózu, spinální muskulární atrofii, Friedreichovu či spinocerebelární ataxii aj.), tak i získaná neurologická, včetně otrav (olovem a jinými těžkými kovy) nebo infekčních onemocnění.2, 3
S diferenciálně diagnostickou rozvahou může pomoci validovaný on-line nástroj accelRare, který na základě dotazníku, kam lékař vyplní příznaky a anamnézu, navrhne seznam potenciálních vzácných onemocnění odpovídajících profilu pacienta. Platforma lékaři poskytne přehled typických symptomů těchto onemocnění, doporučená další vyšetření pro upřesnění diagnózy i seznam pracovišť, kam je možno pacienta s podezřením na vzácné onemocnění odeslat.4
GM2 gangliosidózy s pozdním nástupem jsou velmi vzácné dědičné metabolické poruchy s primárně neurologickou manifestací. Měli bychom na ně pomýšlet zejména u pacientů s neurogenní slabostí dolních končetin, dysartrií a cerebelárními příznaky, jež se rozvinou v dospívání či časné dospělosti. U velké části nemocných jsou přítomné také psychiatrické problémy, a to včetně akutních psychóz. Ačkoliv chorobu modifikující léčba dosud není k dispozici, včasné stanovení diagnózy je nezbytné pro zavedení optimální podpůrné péče.5 S předběžnou diagnostikou může pomoci accelRare − validovaný on-line nástroj pro časný záchyt vzácných onemocnění.4
(este)
Zdroje:
1. Picache J. A., Zheng W., Chen C. Z. Therapeutic strategies for Tay-Sachs disease. Front Pharmacol 2022; 13: 906647, doi: 10.3389/fphar.2022.906647.
2. Toro C., Shirvan L., Tifft C. HEXA disorders. GeneReviews, University of Washington, Seattle, 2020 Oct 1.
3. Xiao C., Tifft C., Toro C. Sandhoff disease. GeneReviews, University of Washington, Seattle, 2022 Apr 14.
4. Frequently asked questions. accelRare, 2026. Dostupné na: www.accelrare.fr/faq-accelrare
5. González-Sánchez M., Ramírez-Expósito M. J., Martínez-Martos J. M. Advances in diagnosis, pathological mechanisms, clinical impact, and future therapeutic perspectives in Tay−Sachs disease. Neurol Int 2025 Jun 25; 17 (7): 98, doi: 10.3390/neurolint17070098.