
Plicní onemocnění a autoimunitní hemolytická anémie asociovaná s IgG4
Lung diseases and autoimmune hemolytic anemia associted with IgG4 disease
IgG4 related disease (IgG4-RD) is a rare and relatively new group of systemic inflammatory diseases characterized by inflammatory, fibrotic or sclerotic involvement of one or more organs accompanied by increased IgG4plasma cells tissue infiltration andusually elevated serum IgG4(IgG4 > 1.35g/l, normal range 0.08–1.40 g/l) level. Histopathological findings are crucial for the diagnostics of this disease. The authors present a case report of a patient with IgG4 associated disease manifested by a rare combination of autoimmune hemolytic anemia and pulmonary involvement.
Keywords:
autoimmune haemolytic anemia – IgG4 associated disease – interstitial lung disease
Autoři:
Martina Doubková 1; Radoslav Matěj 2; Zita Chovancová 3; Michael Doubek 4,5
Působiště autorů:
Klinika nemocí plicních a tuberkulózy LF MU a FN Brno, pracoviště Bohunice
1; Ústav patologie a molekulární medicíny 3. LF UK a Thomayerovy nemocnice, Praha
2; Ústav klinické imunologie a alergologie LF MU a FN u sv. Anny Brno
3; Interní hematologická a onkologická klinika LF MU a FN Brno pracoviště Bohunice
4; Středoevropský technologický institut MU Brno
5
Vyšlo v časopise:
Vnitř Lék 2020; 66(4): 47-52
Kategorie:
Přehledové články
Souhrn
IgG4 asociovaná onemocnění představují relativně novou a poměrně vzácnou skupinu systémových zánětlivých onemocnění, která je charakterizována zánětlivým, fibrotizujícím či sklerotizujícím postižením jednoho nebo více orgánů doprovázené lymfoplazmocelulární infiltrací tkání s výrazným zastoupením IgG4 plazmatických buněk a většinou zvýšenou koncentrací sérových IgG4 imunoglobulinů (dle definice > 1,35 g/l; referenční rozmezí 0,08–1,40 g/l). Pro stanovení diagnózy je klíčový histopatologický nález. Autoři prezentují kazuistiku pacienta s IgG4 asociovaným systémovým onemocněním projevujícím se vzácnou kombinací autoimunitní hemolytické anémie a plicního postižení napodobujícího radiologicky metastatický proces.
Klíčová slova:
autoimunitní hemolytická anémie – IgG4 asociované onemocnění – intersticiální plicní proces
Úvod
S imunoglobulinem IgG4 asociovaná onemocnění (z anglického IgG4-related disease; IgG4-RD) tvoří relativně vzácnou a v současnosti zatím málo probádanou heterogenní skupinu systémových zánětlivých onemocnění. Jsou charakterizována přítomností fibrotizujícího nebo sklerotizujícího postižení jednoho nebo více orgánů, pro které je histologicky typická přítomnost lymfoplazmocelulární infiltrace bohaté na IgG4 plazmatické buňky (1). Toto onemocnění bývá doprovázeno také zvýšenou sérovou koncentrací IgG4 podtřídy imunoglobulinů (dle definice > 1,35 g/l; referenční rozmezí 0,08–1,40 g/l) (2), přičemž až u jedné třetiny pacientů s IgG4-RD může být koncentrace IgG4 v séru v normě (3, 4). Choroby asociované s IgG4 mohou postihnout celou řadu orgánů (např. žlučové cesty, slinné žlázy, periorbitální tkáň, ledviny, plíce, mízní uzliny, meningy, cévy, měkké tkáně nebo prostatu). Postižení tkání může vést až k orgánovému selhání, navíc často imituje maligní nádorové onemocnění.
V našem sdělení prezentujeme vzácný případ pacienta s IgG4 asociovaným systémovým onemocněním projevujícím se vzácnou kombinací autoimunitní hemolytické anémie (AIHA) a plicního postižení napodobujícího radiologicky metastastatickou diseminaci nádorového procesu.
Kazuistika
Šedesátisedmiletý pacient, nekuřák, byl přijat na hematologicko-onkologickou kliniku pro nově zjištěnou autoimunitní hemolytickou anémii s tepelnými protilátkami. Pacient trpěl únavou, úbytkem hmotnosti, bolestmi svalů, bez teplot. Nestěžoval si na dušnost či kašel. Léčil se jen s arteriální hypertenzí. V pracovní anamnéze nebylo pozoruhodností, dříve pracoval jako počítačový technik, nyní důchodce zpívající v opeře. Kromě antihypertenziv neužíval pravidelně žádné léky. Biochemické vyšetření bylo v normě. Byla zjištěna imunitní hemolytické anémie s tepelnými protilátkami (koncentrace hemoglobinu 64g/l). Vyšetřením kostní dřeně nebyla zjištěna infiltrace patologickou buněčnou populací. Vzhledem k těžké anémii byla zahájena terapie intravenózními kortikoidy a pro známky anemického syndromu bylo nutné podat transfuzi erytrocytů. V rámci základního diagnostického algoritmu bylo pátráno po příčinách anémie. Na zadopředním snímku hrudníku byly četné difuzní nodulární zastínění (Obr. 1). Na HRCT hrudníku byla přítomna mnohočetná ložiska a hladké, okrouhlé noduly, některé i neostré s naznačenou spikulací do okolí a hraniční mediastinální a hilová lymfadenopatie (Obr. 2). Plicní funkční vyšetření neprokázalo ventilační poruchu, poruchu plicní difuze ani hypoxemii. Bronchoskopie s bronchoalveolární laváží (BAL) ukázala neutrofilní alveolitidu (79 % neutrofilních granulocytů v BAL; norma < 5 %), ale výsledný diferenciální rozpočet byl limitován regresivními změnami a mikrobiologické vyšetření prokázalo méně než 104 CFU/ml β -hemolytického streptokoka. Transbronchiální plicní biopsie zastihla drobnou část plicního parenchymu, která byla histologicky bez patologie. Autoprotilátky byly negativní (antinukleární protilátky, protilátky proti extrahovatelným nukleárním antigenům, dvouvláknové DNA, cyklickým citrulinovaným peptidům, cytoplazmě neutrofilů a revmatoidní faktor). Základní imunologické vyšetření prokázalo zvýšené sérové hladiny celkových IgG a IgE imunoglobulinů (IgG 18,19 g/l (referenční rozmezí 7,51–15,6g/l); IgM 1,64g/l (0,4–2,3g/l); IgA < 0,05g/l (0,7–4,0g/l); IgE 684 U/l (0–90 U/l)). Pro podezření na metastazující plicní proces bylo u pacienta indikováno provedení plicní biopsie pomocí videoasistované hrudní torakoskopie (VATS). Dle histopatologického vyšetření se jednalo o fibrotizující plicní proces nejasné etiologie, diferenciálně diagnosticky bylo pomýšleno na silikotický uzlík. Vzhledem k tomu, že pacient nebyl exponován křemičitému prachu a nepracoval v rizikovém prašném prostředí, byla diagnóza pneumokoniózy nepravděpodobná. Během léčby kortikoidy v úvodní dávce 2mg/kg/den, kterou pacient podstoupil na hematologické klinice, došlo k postupnému ústupu anémie. Patologická zastínění na zadopředním snímku hrudníku však i nadále přetrvávala. Vzhledem ke stále nejasné diagnóze byl histopatologický vzorek zaslán ke konziliárnímu vyšetření. Diagnóza silikózy byla vyloučena a nález byl uzavřen jako diagnóza IgG4 plicního asociovaného onemocnění. Pro tuto diagnózu byla splněna všechna histopatologická kritéria (hustý lymfocytární -plazmocelulární infiltrát s vyšším zastoupením IgG4 plazmocytů a eozinofilních granulocytů, výrazná fibróza uspořádaná do vírovitých vzorců a obliterující flebitida) (Obr. 3 a 4). Imunologické laboratorní vyšetření ukázalo hraniční elevaci IgG4 podtřídy imunoglobulinů (IgG4 1,43g/l), ostatní imunologické parametry humorální i buněčné imunity byly v normě. Pacient tak splňoval všechna kritéria s IgG4 asociovaného onemocnění. V současnosti je pacient stabilní, nemá žádné klinické potíže, užívá malou udržovací dávku kortikoidů z indikace hematologické. Na kontrolním HRCT hrudníku byl po 8 měsících léčby kortikoidy zatím stacionární nález. Vzhledem k tomu, že pacient nemá žádné potíže, nyní nepřidáváme další imunosupresivní léčbu.




Diskuze
IgG4 asociovaná onemocnění představují skupinu chronických relabujících zánětlivých onemocnění, pro jejichž diagnostiku je zásadní imunohistochemický průkaz denzní lymfoplazmocelulární proliferace s přítomností zvýšené koncentrace IgG4 plazmatických buněk, různě akcentovaná fibróza až skleróza, obliterující flebitida a mírná eozinofilie se znaky fibrózy, obliterující flebitida a mírné eozinofilie (2).
Zpočátku byly tyto změny popisovány v různých orgánech a považovány za samostatná onemocnění (Tab. 1), nicméně později se zjistilo, že se jedná o stejný patogenetický projev v různých orgánech. Proto byla tato onemocnění sloučena pod soubornou diagnózu tzv. IgG4-asociovaných onemocnění (IgG4-RD) (4, 5). První mezinárodní kongres věnovaný této problematice se konal v roce 2011 v Bostonu a první diagnostická kritéria tohoto onemocnění byla publikována v roce 2012 (Tab. 2) (4–6). Navrhovaná diagnostická kritéria z roku 2011 pro IgG4-RD s plicním postižením shrnují Tab. 3 a 4 (7).




Přesná epidemiologická data nejsou známa a prevalence nemocí asociovaných s IgG4 je pravděpodobně podhodnocená, a to zejména v Evropě a Jižní Americe. V těchto zemích je povědomí o tomto onemocnění nižší než v Asijských zemích (zejména Koreji a Japonsku), kde byla nemoc prvně popsána (8, 9).
Přesný patofyziologický mechanismus a charakteristika vyvolávajících příčin rozvoje IgG4 asociovaných onemocnění není plně objasněna. K rozvoji fibroinflamatorního procesu však zřejmě přispívá více imunopatologických mechanismů, přičemž by se na něm mohly podílet genetické faktory, prodělané infekce nebo mechanismy autoimunitní (4, 5, 10, 11). Hlavní úlohu v patogenezi onemocnění hrají pravděpodobně T -lymfocyty (12–14). Naivní Th -lymfocyty se pod vlivem cytokinů diferencují na pomocné (Th1 a Th2), regulační (Treg) a Th17 T -lymfocyty. IgG4 asociovaná onemocnění jsou charakterizována převahou Th2 imunitní odpovědí s expresí odpovídajících cytokinů (zejména IL4, IL5, IL10 a IL13), viz Obr. 5 (15). Tyto interleukiny (zejména IL5) jsou zodpovědné také za eozinofilii. Na vzniku nemoci mají podíl rovněž Treg lymfocyty, které jsou zdrojem hlavního profibrotického cytokinu transformujícího růstového faktoru β (TGFβ), který se zásadně podílí na vzniku fibrózy. Produkce IgG4 je závislá na Th2 cytokinech (IL5, IL13) a Treg cytokinech (IL10) (13, 14). Koncentrace celkových IgG imunoglobulinů v séru je dána součtem koncentrace 4 jeho podtříd (IgG1–IgG4), IgG4 tvoří méně než 5 % celkového IgG u zdravých osob a je zároveň nejméně zastoupenou podtřídou IgG imunoglobulinů. U nemocných s IgG4-RD může být tvorba IgG4 zvýšená. IgG4 je považován za nezánětlivý nebo spíše protizánětlivý imunoglobulin, protože prakticky neváže C1q složku komplementu a nespouští klasickou dráhu aktivace komplementu a má nízkou afinitu k Fc receptorům (16). Nicméně u některých onemocnění se na jejich patogenezi podílí (např. pemphigus vulgaris, idiopatická membranózní glomerulonefritida nebo myasthenia gravis) (17–19). Původně se myslelo, že přítomnost zvýšené koncentrace IgG4 podtřídy imunoglobulinů v séru je u pacientů s IgG4-RD pouze epifenoménem bez patogenetického významu, nicméně v pokusech na myších se ukázalo, že při podání IgG imunoglobulinů pacientů s IgG4-RD myším, u nich dochází ke stejnému orgánovému postižení jako u pacientů s IgG4-RD (20). To by mohlo ukazovat na možnou úlohu IgG4 v rámci patogeneze tohoto onemocnění (Obr. 4 – imunologická podstata onemocnění s histopatologickými nálezy).
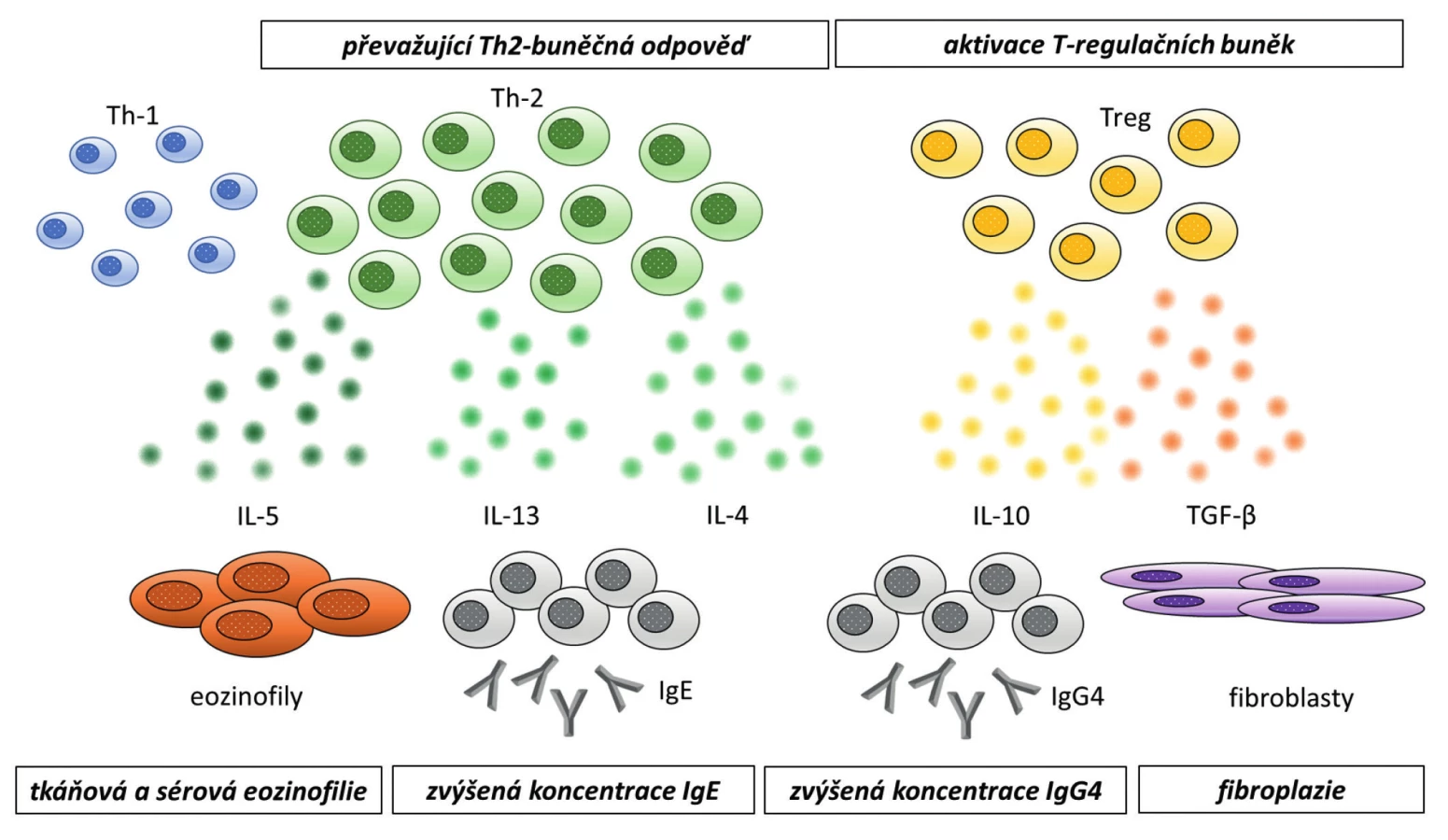
Vzhledem k vzácnému výskytu a heterogenitě této skupiny onemocnění chybí dosud široce používané a obecně akceptované standardy pro diagnostiku a léčbu (1). Zvýšená koncentrace IgG4 podtřídy imunoglobulinů (> 1,35 g/l) není pro IgG4-RD specifická a může se vyskytovat i u jiných onemocnění, např. některých malignit nebo infekcí (6).
IgG4-RD se řadí mezi autoimunitní onemocnění, protože obvykle dobře odpovídají na léčbu systémovými kortikoidy (1). Pacienty s IgG4-RD můžeme dle typu a rozsahu jejich onemocnění buď jen sledovat (tzv. strategie „watch and wait“) nebo léčit imunosupresivní terapií. Pouhé sledování je možné např. u pacientů s asymptomatickou lymfadenopatií nebo mírným zvětšením podčelistních žláz v případě histopatologicky ověřené diagnózy (21). Nicméně musíme mít na paměti, že i subklinické asymptomatické formy onemocnění mohou postupně vést k nevratnému a závažnému poškození postižených orgánů. Naopak fibrotické léze mohou být projevem nevratných fibrotických změn, tzv. „vyhoření“ („burnoutu“) a nemusí reagovat na terapii. Na druhou stranu nasazení imunosupresivní léčby vyžadují všichni symptomatičtí pacienti s aktivní IgG4-RD. Podobně jako u jiných systémových zánětlivých onemocnění i zde dochází k relapsům či recidivám onemocnění. Základním lékem první linie jsou systémové kortikoidy v dávkách 30–40mg/denně po dobu 2–4 týdnů s postupným snižováním každé 1–2 týdny na co nejmenší účinnou dávku. Délka terapie se různí od 3–6 měsíců až po dobu 3 let (8, 21–24). Dalším lékem v případě kortikorezistence nebo recidiv jsou jiná imunosupresiva (azathioprin, mykofenolát mofetil, methotrexát, takrolimus, cyklofosfamid) (21, 25). Slibnou možností léčby je u kortikorezistentních pacientů s IgG4-RD terapie pomocí anti -CD20 monoklonálních protilátek (21, 25, 26). Rituximab je chimérickou myší monoklonální protilátkou namířenou proti molekule CD20 na povrchu B -lymfocytům. Pacienti s IgG4-RD mají zvýšený počet cirkulujících plazmablastů. Marker CD20 není přítomen na povrchu plazmablastů a plazmatických buněk, proto rituximab přímo tyto buňky neovlivňuje, ale přes depleci celkových B -lymfocytů vede k depleci také cirkulujících IgG4 plazmocytů a cirkulujících plazmablastů (25).
V naší kazuistice prezentujeme případ pacienta s autoimunitní hemolytickou anémií a plicním postižením, imitujícím dle zobrazovacích metod metastatický rozsev nádorového onemocnění, které bylo nakonec diagnostikováno jako IgG4-RD. Koincidence tohoto onemocnění s autoimunitní hemolytickou anémií je vzácná (27–29). Patogenetickým podkladem rozvoje AIHA je tvorba autoprotilátek proti povrchovým antigenům červených krvinek. Vzniklý imunokomplex může aktivovat komplement klasickou cestou, což vede k intravaskulární hemolýze. Druhým mechanismem destrukce erytrocytů je vazba Fc receptoru fagocytů na Fc fragmenty autoprotilátek nebo receptoru pro komplement na povrchu fagocytů na opsonizační C3b složku komplementu, což vede k extravaskukární hemolýze ve slezině či játrech. Fc receptory fagocytů rozpoznávají navázané autoprotilátky podtřídy IgG1 a IgG3, které jsou zároveň nejpotentnějšími aktivátory komplementového systému. Patogeneze koincidence AIHA a IgG4-RD zatím zůstává neobjasněna, nicméně se předpokládá účast cytokinového prostředí Th2 typu imunitní odpovědi. To se pravděpodobně významných způsobem podílí jednak na rozvoji plicní fibrózy, ale také na produkci autoprotilátek způsobujících AIHA (30). V tkáních pacientů s IgG4-RD byla sice prokázána zvýšená exprese Th2 cytokinů (IL4, IL5 a IL13) a regulátorů jejich produkce (IL10 a TGFβ) (12), což by mohlo svědčit na propojení mezi IgG4-RD a AIHA skrz Th2 cytokinový profil, přesto se ale nepředpokládá, že by se IgG4 imunoglobuliny účastnily patogeneze AIHA, protože špatně aktivují komplementový systém a mechanismy fagocytózy (31).
Závěr
gG4 asociovaná onemocnění představují relativně vzácnou skupinu chronických relabujících zánětlivých onemocnění postihujících různé orgány lidského těla. Rozmanitý klinický obraz odvíjející se od typu postižených orgánů stěžuje diagnostiku tohoto onemocnění. Hlavním klíčem pro diagnózu IgG4-RD je histopatologické vyšetření, ale to vyžaduje, aby v rámci diferenciální diagnostiky bylo na toto onemocnění primárně pomýšleno. Kazuistika našeho pacienta ukazuje možnost vzácné koincidence IgG4 - RD a AIHA, nicméně vzájemný vztah obou nemocí není plně objasněn.
KORESPONDENČNÍ ADRESA AUTORA:
MUDr. Martina Doubková, Ph.D.,
Klinika nemocí plicních a tuberkulózy LF MU a FN Brno,
pracoviště Bohunice,
Jihlavská 20,
625 00 Brno
Cit. zkr: Vnitř Lék 2020; 66(4): e22–e27
Článek přijat redakcí: 18. 8. 2019
Článek přijat k publikaci: 24. 9. 2019
Zdroje
1. Laccarino L, Talarico R, Scirè CA, et al. IgG4-related diseases: state of the art on clinical practice guidelines. RMD Open 2019; 4: (Suppl. 1): e000787.
2. Umehara H, Okazaki K, Nakamura T, et al. Current approach to the diagnosis of IgG4-related disease -Combination of comprehensive diagnostic and organspecific criteria. Mod Rheumatol 2017; 27 : 381–391.
3. Bozzala Cassione E, Stone JH. IgG4-related disease. Curr Opin Rheumatol 2017; 29 : 223–227.
4. Stone JH, Zen Y, Deshpande V IgG4-related disease. N Engl J Med. 2012; 366 : 539–551.
5. Stone JH, Khosroshahi A, Deshpande V, et al. Recommendations for the nomenclature of IgG4-related disease and its individual organ system manifestations. Arthritis Rheum 2012; 64 : 3061–3067.
6. Umehara H, Okazaki K, Masaki Y, et al. Comprehensive diagnostic criteria for IgG4-related disease (IgG4-RD), 2011. Mod Rheumatol 2012; 22 : 21–30.
7. Matusi S, Yamamoto H, Minamoto S, et al. Proposed diagnostic criteria for IgG4-related respiratory disease. Respir Investig 2016; 54 : 130–132.
8. Kamisawa T, Funata N, Hayashi Y, et al. A new clinicopathological entity of IgG4-related autoimmune disease. J Gastroenterol 2003; 38 : 982–984.
9. Masaki Y, Dong L, Kurose N, et al. Proposal for a new clinical entity, IgG4-positive multiorgan lymphoproliferative syndrome. Analysis of 64 cases of IgG4-realted disorders. Ann Rheum Dis 2009; 68 : 1310–1315.
10. Mikulová Š, Jílek D, Richter J. Nemoc asociovaná s IgG4. Úvod, patogeneze, diagnostika. 1. část. Alergie 2015; 17 : 16–24.
11. Mikulová Š, Jílek D, Richter J. Nemoc asociovaná s IgG4. Klinický obraz, orgánová postižení a terapie. 2. Část. Alergie 2015; 17 : 91–99.
12. Zen Y, Fujii T, Harada K, et al. Th2 and regulatory immune reactions are incresased in immunoglobulin G4-related sclerosing pancreatiits and cholangiitis. Hepatology 2007; 45 : 1538–1546.
13. Aalberse RC, Stapel SO, Schuurman J et al. Immunoglobulin G4: an odd antibody. Clin Exp Allergy 2009; 39 : 469–477.
14. Satoguina JS, Weyand E, Larbi J, et al. T regulatory-1 cells induce IgG4 production by B cells: role of IL-10. J Immunol 2005; 174 : 4718–4726.
15. Desphande V, Khorohahi A. Diagnostic guidelines for IgG4-related disease with a focus on histophatological criteria. Diagnostic histopathology 2013; 19 : 119–127.
16. Nirula A, Glaser SM, Kalled SL, et al. What is IgG4? A review of the biology of a unique immunoglobulin subtype. Curr Opin Rheumatol 2011; 23 : 119–124.
17. Anhalt GJ, Labib RS, Voorhees JJ et al. Induction of pemphigus in neonatal mice by passive transfer of IgG from patients with the disease. N Engl J Med 1982; 306 : 1189–1196.
18. Beck jr. LH, Salant DJ. Membranous nephropathy: recent travels and new roads ahead. Kidney Int 2010; 77 : 765–770.
19. Plomp JJ, Hujbers MG, van der Maarel SM, et al. Pathogenic IgG4 subclass autoantibodies in MuSK myasthenia gravis. Ann NY Acad Sci 2012; 1275 : 114–122.
20. Shiokawa M, Kodama Y, Kuriayma K, et al. Pathogenicity of IgG in patients with IgG in patients with IgG4-related disease. Gut 2016; 65 : 1322–1332.
21. Khosroshahi A, Wallace ZS, Crowe JL, et al. International consensus guidance statement on the management and treatment of IgG4-related disease. Arthiritis Rheumatol 2015; 67 : 1688–1699.
22. Ghazale A, Chari ST, Zhang L, et al. Immunoglobulin G4-associated cholangitis: clinical profile and response to therapy. Gastroenterology 2008; 134 : 706–715.
23. Raina A, Yadav D, Krasinskas AM, et al. Evaluation and management of autoimmune pancreatitis: experience at al large US center. Am J Gastroenterol 2009; 104 : 2295–306.
24. Losse S, Žurková M. Plicní projevy nemoci asociované s IgG4. Postgrad Med 2017; 19: (Suppl. 2): 50–54.
25. Khoroshahi A, Bloch DB, Desphande V, et al. Rituximab therapy leads to rapid decline of serum IgG4 levels and prompt clinical improvement in IgG4related systemic disease. Arthritis Rheum 2010; 62 : 1755–1762.
26. Adam Z, Chovancová Z, Nová M, et al. Remise „the disease associated/related with immunoglobulin IgG4“ provázeného mnohočetnou lymfadenopatií po léčbě rituximabem a dexametazonem: kazuistika. Vnitř Lék 2018; 64 : 290–299.
27. Hasegawa S, Mine S, Hagiwara S. IgG4-related disease combined with autoimmune hemolytic anemia and steroid -responsive transient hypercalcemia. Clin Med Insights Case Rep 2015; 8 : 51–55.
28. Wang KC, Liao HT, Tsai CY IgG4-related disease coexisting with autoimmune haemolytic anaemia. BMJ Case Rep 2018; pii: bcr-2018–224814.
29. Yoshida M, Marumo Y, Naitoh I, et al. Autoimmune hemolytic anemia obscured by the obstructive jaundice associated with IgG4-related sclerosing cholangitis in a patient with type 1 autoimmune pancreatitis: a case report and review of the literature. Intern Med 2018; 57 : 1725–1732.
30. Noguchi S, Yatera K, Jinbo M, et al. IgG4-related lung disease associated with autoimmune hemolytic anemia: a case report and a literature review. Intern Med 2016; 55 : 2469–2474.
31. Masutani H, Okuwaki K, Kida M, et al. First case of IgG4-related sclerosing cholangitis associated with autoimmune hemolytic anemia. World J Gastroenterol 2014; Doi: 10.3748/wjg.v20.i26.8740.
Štítky
Diabetologie Endokrinologie Interní lékařstvíČlánek vyšel v časopise
Vnitřní lékařství

2020 Číslo 4
- Inovace v hojení ran: krytí Zetuvit Plus Silicone Border pro optimální management exsudátu z ran
- Ivabradin zlepšuje kvalitu života starších pacientů se srdečním selháním
- Fixní kombinace kandesartan/amlodipin v terapii arteriální hypertenze
- Clopidogrel je v prevenci kardiovaskulárních příhod přínosnější než kyselina acetylsalicylová
Nejčtenější v tomto čísle
- Jaterní fibróza
- Hodnocení operačního rizika u pacientů s jaterní cirhózou
- Mikroskopická polyangiitída
- Hyperurikemie z perspektivy nefrologického pacienta
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy