
Evolutionary Mirages: Selection on Binding Site Composition Creates the Illusion of Conserved Grammars in Enhancers
The clustering of transcription factor binding sites in developmental enhancers and the apparent preferential conservation of clustered sites have been widely interpreted as proof that spatially constrained physical interactions between transcription factors are required for regulatory function. However, we show here that selection on the composition of enhancers alone, and not their internal structure, leads to the accumulation of clustered sites with evolutionary dynamics that suggest they are preferentially conserved. We simulated the evolution of idealized enhancers from Drosophila melanogaster constrained to contain only a minimum number of binding sites for one or more factors. Under this constraint, mutations that destroy an existing binding site are tolerated only if a compensating site has emerged elsewhere in the enhancer. Overlapping sites, such as those frequently observed for the activator Bicoid and repressor Krüppel, had significantly longer evolutionary half-lives than isolated sites for the same factors. This leads to a substantially higher density of overlapping sites than expected by chance and the appearance that such sites are preferentially conserved. Because D. melanogaster (like many other species) has a bias for deletions over insertions, sites tended to become closer together over time, leading to an overall clustering of sites in the absence of any selection for clustered sites. Since this effect is strongest for the oldest sites, clustered sites also incorrectly appear to be preferentially conserved. Following speciation, sites tend to be closer together in all descendent species than in their common ancestors, violating the common assumption that shared features of species' genomes reflect their ancestral state. Finally, we show that selection on binding site composition alone recapitulates the observed number of overlapping and closely neighboring sites in real D. melanogaster enhancers. Thus, this study calls into question the common practice of inferring “cis-regulatory grammars” from the organization and evolutionary dynamics of developmental enhancers.
Published in the journal:
. PLoS Genet 6(1): e32767. doi:10.1371/journal.pgen.1000829
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1000829
Summary
The clustering of transcription factor binding sites in developmental enhancers and the apparent preferential conservation of clustered sites have been widely interpreted as proof that spatially constrained physical interactions between transcription factors are required for regulatory function. However, we show here that selection on the composition of enhancers alone, and not their internal structure, leads to the accumulation of clustered sites with evolutionary dynamics that suggest they are preferentially conserved. We simulated the evolution of idealized enhancers from Drosophila melanogaster constrained to contain only a minimum number of binding sites for one or more factors. Under this constraint, mutations that destroy an existing binding site are tolerated only if a compensating site has emerged elsewhere in the enhancer. Overlapping sites, such as those frequently observed for the activator Bicoid and repressor Krüppel, had significantly longer evolutionary half-lives than isolated sites for the same factors. This leads to a substantially higher density of overlapping sites than expected by chance and the appearance that such sites are preferentially conserved. Because D. melanogaster (like many other species) has a bias for deletions over insertions, sites tended to become closer together over time, leading to an overall clustering of sites in the absence of any selection for clustered sites. Since this effect is strongest for the oldest sites, clustered sites also incorrectly appear to be preferentially conserved. Following speciation, sites tend to be closer together in all descendent species than in their common ancestors, violating the common assumption that shared features of species' genomes reflect their ancestral state. Finally, we show that selection on binding site composition alone recapitulates the observed number of overlapping and closely neighboring sites in real D. melanogaster enhancers. Thus, this study calls into question the common practice of inferring “cis-regulatory grammars” from the organization and evolutionary dynamics of developmental enhancers.
Introduction
The transcriptional output of developmental enhancers is affected by the spatial organization of the transcription factor binding sites they contain. The relative positioning of sites is known from individual cases to modulate direct competition between factors for the same site [1],[2], cooperative and repressive interactions between transcription factors [3],[4], and the formation of higher-order regulatory complexes [5]–[7]. However, we have a precise understanding of the relationship between binding site organization and function for few, if any, developmental enhancers.
In the absence of efficient experimental protocols for dissecting enhancer function, recent efforts have attempted to infer functional constraints on binding site organization from the distribution and evolution of binding sites in enhancers of interest. We recently examined developmental enhancers in species distantly related to D. melanogaster and found a strong preferential conservation of overlapping and proximal sites [8], a result which was confirmed by a recent survey of enhancer evolution across the twelve sequenced Drosophila genomes [9]. Others have focused on the density of overlapping and proximal sites, finding that both are significantly enriched [10],[11]. All of these studies, including ours, reached a similar conclusion: the evolutionary dynamics of binding sites in developmental enhancers suggest that clustered and/or overlapping sites are common functional necessities for enhancer activity.
This shared conclusion was premised on the idea that the observed non-random arrangement of sites must be a result of selection on the relative positioning of sites within enhancers. However, alternative explanations for these phenomena, especially the possibility that such arrangements might arise as a byproduct of other mutational and selective pressures [12], have not been explored.
Here we simulate the evolution of real and synthetic D. melanogaster enhancers constrained only to maintain their binding site composition and investigate the spatial organization of binding sites within enhancers evolving with no direct selection on the arrangement of sites within them. We show that this simple global constraint on enhancer composition is sufficient to produce many of the organizational and evolutionary features observed in real enhancers, including enrichment and apparent conservation of overlapping and clustered sites.
Results
Simulating enhancer evolution
We used simulations to explore the properties of enhancers evolving under selection on binding site composition. We subjected synthetic enhancers, in which a predefined number of binding sites for one or more transcription factors were randomly positioned in randomly generated sequence with the same composition as D. melanogaster non-coding DNA, to mutations sampled from the distribution of substitutions, insertions and deletions observed in D. melanogaster [13]. We applied a strong selective constraint to these mutated sequences. If the number of sites in the enhancer fell below a specified threshold, we rejected the new sequence. Otherwise, it was carried through to the next mutational step (Figure 1).

Because such a strict cutoff might not be realistic, we compared the results of these simulations to those involving a large population of enhancers in which suboptimal sequences were assigned a fitness penalty rather than immediately removed. None of the measures of binding site distribution and evolution discussed below differed appreciably between these models (see Text S1). Since these population simulations required significantly greater computational resources, we present only the results of the simpler model below.
Binding site turnover
The most basic property of our model of enhancer evolution is that most mutations that destroy a binding site will be rejected, as they bring the number of sites present in the enhancer below the specified fitness threshold (Figure 1B). However, the small size of most binding sites means that they are generated de novo by random mutation at an appreciable rate. And, once new sites are generated, mutations that destroy existing sites will be tolerated (Figure 1C), leading to non-homologous site conservation, or “binding site turnover” [14]–[17].
The rate at which mutations destroy existing sites for a given factor and create new ones depends on the size and degeneracy of the site recognized by the factor. To examine how specificity affects these rates, we simulated the evolution of enhancers constrained only to have a single site matching real, or randomly generated, transcription factor specificities. The rate of turnover varied considerably, depending on the size of the recognition site, its base composition and degeneracy (Figure 2), with the variance primarily explained by the rate at which new binding sites are generated from random DNA. Longer, less degenerate and GC-rich sites are generated from random sequence at a lower rate and thus turn over more slowly.

The expected half-life (measured in mutational distance) of binding sites for the typical D. melanogaster transcription factor was between one and two substitutions per site, or around 50 to 100 million years. This is consistent with previous studies of turnover rates for functional sites in real enhancers that have estimated that there have been around one to two turnover events per site per hundred million years [17],[18].
Selection on binding site composition alone leads to conserved structure in enhancers
Some transcription factors, such as the D. melanogaster proteins Bicoid (BCD) and Krüppel (KR), overlap in their binding specificities, so that the same bases can be part of binding sites for multiple factors [19]–[21]. In specific cases competition between BCD and KR for overlapping sites plays an important role in producing specific expression patterns [22],[23]. The high frequency of overlapping BCD and KR sites in other embryonic enhancers has been used as evidence for the generality of this mechanism [10].
However, when we simulated the evolution of synthetic 1,000 bp enhancers constrained to contain five BCD and five KR sites, we find an almost twofold elevation in the frequency of overlapping BCD and KR sites compared to the random expectation (Figure 3A). Thus selection acting to preserve enhancer composition alone indirectly leads to “higher order” structure in enhancers. This phenomenon is not specific to BCD and KR: rather it is a general property of factors with overlapping binding specificities (data not shown).

The increase in the density of overlapping sites is almost entirely due to their increased half-life relative to isolated sites. In the BCD/KR simulations described above, which had no explicit selection to maintain overlapping sites, overlapping sites persisted 1.5 to 2.0 times longer (depending on the specific choice of matrix) than isolated sites (Figure 3B, Figure S1, and Figure S2).
This difference in half-life between overlapping and isolated sites not only increases the density of overlapping sites; it also significantly alters how they are classified in comparative genomic analyses. Their longer half-life means that overlapping sites are more likely to be found at orthologous positions in related species. In particular, at evolutionary distances in the range typically used for comparative analyses (around one substitution per site) the likelihood of finding an orthologous overlapping pair of BCD and KR sites is two times larger than the likelihood of finding an orthologous singleton site (Figure 3B, Figure S1, and Figure S2). Thus, our simulations show that selection to maintain enhancer composition not only leads to an increase in the density of overlapping sites, it also makes it appear that selection is acting to specifically preserve them.
A deletion bias induces conserved binding site clustering
Binding sites in real enhancers are clustered, with an excess of short inter-binding-site distances at the expense of long ones [10],[11]. This clustering has been interpreted as evidence that long-range interactions between transcription factors or between transcription factors and nucleosomes are required for proper gene regulation [10],[11].
However, in our simulations, we also observed an increase in the proportion of small spacers (Figure 4A). This induced binding site clustering occurred whenever the mutation model included a bias for deletions over insertions, a known property of Drosophila species [24]. When simulations were run with only point mutations, or with balanced insertions and deletions, no increase in short spacers was observed.

Unlike point mutations, deletions can disrupt multiple non-overlapping binding sites. In our simulations, deletions affecting two or more sites were less than half as likely to be accepted as were deletions affecting single sites (10.5% compared to 23.2% of the time). Thus it is possible that the induced binding site clustering arises from the protective effect proximal sites have against each other's deletion (Figure 4B). Indeed, in simulations that exclusively involved deletions, tightly spaced but non-overlapping sites showed a substantial increase in half-life (Figure S3). However, in simulations with a realistic balance of mutations and indels this effect was minimal (Figure S4), as the frequency of multi-site deletions was low relative to single site deletions and point mutations.
Instead, the induced binding site clustering appears to be driven simply by the deletion of spacer DNA between sites. Since, in our simulations, deletions between sites occur more frequently than sites are lost, sites get closer together over time, distorting the distribution of inter-site distances. A corollary of this phenomenon is that sites that are observed to be close together tend to be older, and therefore more likely to be labeled as conserved, than isolated sites (Figure 4C). Thus, both binding site clustering and an apparent preferential conservation of clustered sites are expected to occur even in the absence of any selection on enhancer organization.
A deletion bias distorts evolutionary inference
Sequence features present in multiple related species are generally considered to reflect those found in the shared ancestor, whether through selection or common descent. However, the deletion bias-induced tendency for sites to get closer together over time distorts this relationship. To illustrate this, we placed two sites at a fixed distance and monitored the distance between them over time in a large number of independent simulations. With indels, but no bias towards deletions either in frequency or in average length, the intersite spacing quickly diverges between simulations (Figure 5A). However, with the observed Drosophila deletion bias, the spacing between sites in the different simulations is strongly correlated (Figure 5B). Thus, with a deletion bias, the spacing between sites after speciation will appear conserved and yet reflect neither selection nor the ancestral state.

To examine how this relationship between inter-site spacing and age might affect evolutionary inference, we simulated the divergence after speciation of regulatory sequences containing pairs of binding sites separated by varying distances. We then compared, at different times after divergence, the inter-site spacing in orthologous evolved sequences. Roughly following practice in the field, we considered the spacing to be “conserved” if the sites were within 30 bp in both species. Even where the starting spacing was 30 bp, the probability that it remained within 30 bp in both species in the absence of a deletion bias is small at evolutionary distances beyond one substitution per site (Figure 5C). But with a deletion bias, the probability is substantially higher, and is appreciable even for starting spacings of 50 or 100 bp (Figure 5C–5E). Thus, comparison of binding site spacing in multiple species with deletion biases will often lead to the incorrect inference that selection has acted to preserve close spacing of binding sites.
A plausible evolutionary scenario explains positional information in a Drosophila enhancer
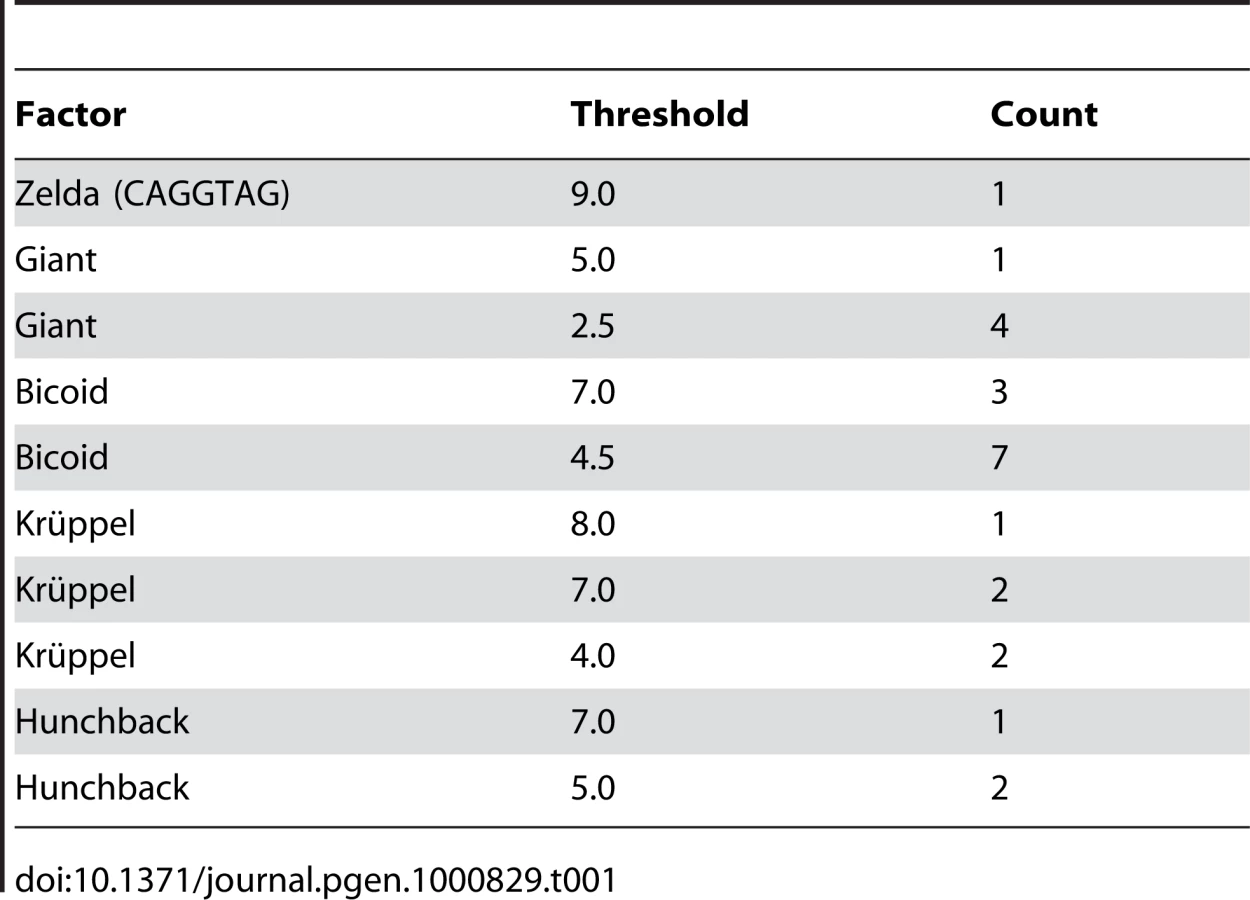
To assess whether the above-described effects could replicate the degree of binding site overlap and clustering that is observed in extant enhancers, we simulated the evolution of the well-characterized eve stripe 2 enhancer [23], with compositional constraints derived from the extensive biochemical and genetic literature on this enhancer. In particular we required five Krüppel, ten Bicoid, three Hunchback, five Giant [25], and a single Zelda [26] binding site (see Table 1). We also required that a certain number of sites for each factor be predicted high-affinity sites (based on the number of high-affinity sites in the D. melanogaster enhancer).
We simulated 1,000 replicates of this enhancer to twenty substitutions per site, and found that both the number of overlapping BCD and KR sites, and the number of sites in close proximity to others, in the real enhancer were well within the range typically generated by this architecture-free evolutionary model (Figure 6A and 6B).

Discussion
New molecular methods and ever more sophisticated computational approaches have made significant progress towards understanding the mechanisms of gene regulation. Sequence affinities and binding sites for many transcription factors in many organisms are known, and increasing attention is now being paid to the ‘grammar’ that may link them together [27]–[29].
A common strategy in our work and that of many of our colleagues has been to infer functional constraints on enhancer activity from the apparent conservation of aspects of the organization of transcription factor binding sites within enhancers. However, the results of the simulations presented here show that many of our conclusions were based on naïve assumptions about the expected distribution of binding sites in enhancers evolving with no constraints on their organization.
The value of simulations
In retrospect, the properties we observed are straightforward consequences of coupling selection on binding site composition with a deletion-biased mutational process. One does not need simulations to see why overlapping sites will clearly turn over less frequently than isolated sites, that a deletion bias will drive sites closer together over time, and how both phenomena distort comparative analyses.
But as self-evident as these results may appear, they have never been noted before, despite more than a decade of intense comparative genomic analysis of enhancer structure and function in Drosophila. Indeed, prior to performing these simulations we did not consider that the clustering of binding sites in Drosophila enhancers might arise from a deletion bias. We simply attempted to have our simulations accurately reflect the underlying mutational process in our simulations, with the consequences evident only in the results. This highlights the value of simulations of simple evolutionary processes in uncovering unappreciated consequences of our models and assumptions.
Furthermore, although the general effects of selection on binding site composition and of a deletion bias can be intuited, specific quantitative aspects of the model are difficult to work out analytically. For example, while we have developed a mathematical model for the effect on half-life of overlapping sites in enhancers (see Text S2), it is difficult to extend this model to enhancers with multiple sites. Simulations can answer these questions simply and effectively.
Generality
The simulations we performed here used non-coding DNA, transcription factor binding sites, and mutation patterns from D. melanogaster. Interspecies differences in the composition of non-coding DNA, specificity of transcription factors, and base substitution patterns will have minimal effect on our conclusions. However, differences in the indel rate and the balance of insertions and deletions could significantly alter the existence or magnitude of the induced binding site clustering. Although the deletion biased mutation process we used in our model is often thought of as a Drosophila-specific phenomena, there is increasing evidence that short indels are deletion biased in all species [30]–[35]. Thus, we expect this effect to be general, although the magnitude will differ depending on the indel rate and bias (see Text S3).
Conclusions
Lynch has eloquently argued that biologists are often too quick to assume that organismal and genomic complexity must arise from selection for complex structures and too slow to adopt non-adaptive hypotheses [12]. Our results lend additional support to this view, and extend it to show that indirect and non-adaptive forces can not only produce structure, but also create an illusion that this structure is being conserved.
We do not doubt that many aspects of transcriptional regulation constrain the location of transcription factor binding sites within enhancers. Indeed a large body of experimental evidence supports this notion, and we remain committed to identifying and characterizing these constraints. But if this process is to be fueled by comparative sequence analysis, as we believe it must be, it is essential that we give careful consideration to the neutral and indirect forces that we now know can produce evolutionary mirages of structure and function.
Methods
Simulation of enhancer evolution
Starting sequences 1,000 basepairs in length were generated randomly to match the base composition of D. melanogaster non-coding DNA, and binding sites were added to bring the starting density of sites to the specified thresholds. Mutations were sampled randomly from point mutations, insertions, and deletions. 80% of mutations were point mutations generated from an HKY85 [36] model with GC content 40% and kappa two; 12% were deletions and 8% insertions with size distributions drawn from [13]. The deletion bias (60%), and proportion of all mutations that were indels (20%), were also according to [13]. Except where noted, simulations took place for 100,000 mutation/selection rounds. To compensate for the change in the size of the enhancer when insertions and deletions occurred, bases were removed or added from the nearest edge of the sequence. New base pairs added with a 40% GC content. The simulation software was written in Python and utilizes the Motility [37] binding site identification package.
Simulations using BCD and KR used matrices from in vitro footprinting [38], one-hybrid assays [39], and SELEX [40], with cutoff scores chosen to match expected numbers of their sites in the even-skipped stripe two enhancer: 5.5, 4.9, and 4.1 for BCD and 5.6, 4.1, and 0.0 for KR for the three sources of matrices. Unless noted otherwise, simulations used matrices from the footprinting data set.
In the simulations in presented in Figure 5, we sought only to examine the evolution of site spacing over time and not the conservation and/or turnover of individual binding sites. Thus, we preconditioned in each case that neither could binding sites be generated from random sequence nor could existing binding sites be disrupted. To this end, in these simulations, all mutations affecting positions contained within existing binding sites were considered precluded by selection and discarded, and, similarly, the sequence was not scored for new binding sites created by mutations. We generated Figure 5A and 5B by simulating 980,000 300 base pair sequences to 30 substitutions per site, and Figure 5C–5E by simulating 480,000 300 base pair sequences to ten substitutions per site. In the even indels case, the distribution of insertion lengths was set equal to the distribution of deletion lengths.
Properties of the simulations were computed following a lengthy (∼30 subs/site) burn-in period that allowed the randomly generated starting model to reach equilibrium. We tested several sets of neutral mutation and selective parameters to make sure this burn-in period was sufficient (Figure S5, Figure S6, and Figure S7).
Generation of randomized binding sites
We chose binding site lengths randomly between five and twelve. At each position, we chose a consensus nucleotide and assigned its frequency by sampling a Gaussian with mean 0.8 and standard deviation 0.2. Subsequent nucleotide frequencies were chosen similarly, each being given a frequency chosen from a Gaussian with a mean and standard deviation of 80% and 20% of the remaining probability mass, respectively. Weight matrices were constructed against a 40% GC bias and threshold scores were sampled from a uniform distribution spanning zero to the maximum scores of the sites. Information content was calculated by weighting all N-mers above the score threshold with the GC bias and subtracting the information in an N-mer of random sequence of equal length and GC bias.
Conditional probability of overlapped sites
To find the expected probability KR and BCD sites would overlap in random DNA, we sampled random ten-mers from a 40% GC background distribution. If this sequence contained a KR site, then we added flanking sequence of length N-1, where N is the length of a BCD site. If this sequence also contained a BCD site, then we considered it as an overlap. The probability of a BCD site generating a KR site was found in an analogous manner. The post-selection conditional probability was directly calculated by simulating an enhancer with five sites for each transcription factor as described above and counting observations of singleton and overlapped binding sites.
Half-lives of binding sites
We determined the half-lives of sets of binding sites by randomly sampling individual sites in our simulations and observing their degradation as the simulations progressed. Our data consisted of simulations of 1,000 enhancers, each run for 30,000 iterations. For each enhancer, after a burn-in period of 10,000 iterations, we took a ‘snapshot’ of the binding sites present every 3,000 iterations. In each subsequent iteration of the simulation, the presence or absence of each binding site in the snapshot was assessed: if it had been destroyed by a point mutation or indel in that iteration, then a site ‘death’ was recorded. This process was repeated for 2,000 post-snapshot iterations of the simulation.
Generation of the even-skipped stripe two enhancer
We used one-hybrid binding sequences for Hunchback, Giant, Bicoid, and Krüppel from [39] and created weighted matrices as described. We used the same methods to generate a Zelda-consensus matrix from the sequences listed in [41]. Our enhancer sequence and matrices are available in Dataset S1. In order to determine the required number of sites for each matrix, we assessed the number of hits it had to the eve stripe 2 sequence at several score cutoffs. If the number of hits at a given score cutoff exceeded the number expected by chance, then this number/score cutoff pair was accepted as a requirement, provided that it did not substantially increase the total required number of sites for that factor beyond that described in [23]. The constraint on the enhancer is described in Table 1.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. StanojevicD
SmallS
LevineM
1991 Regulation of a segmentation stripe by overlapping activators and repressors in the Drosophila embryo. Science 254 1385 1387
2. NibuY
SengerK
LevineM
2003 CtBP-independent repression in the Drosophila embryo. Mol Cell Biol 23 3990 3999
3. KulkarniMM
ArnostiDN
2005 cis-regulatory logic of short-range transcriptional repression in Drosophila melanogaster. Mol Cell Biol 25 3411 3420
4. LebrechtD
FoehrM
SmithE
LopesFJ
Vanario-AlonsoCE
2005 Bicoid cooperative DNA binding is critical for embryonic patterning in Drosophila. Proc Natl Acad Sci U S A 102 13176 13181
5. KulkarniMM
ArnostiDN
2003 Information display by transcriptional enhancers. Development 130 6569 6575
6. ArnostiDN
KulkarniMM
2005 Transcriptional enhancers: Intelligent enhanceosomes or flexible billboards? J Cell Biochem 94 890 898
7. MerikaM
ThanosD
2001 Enhanceosomes. Curr Opin Genet Dev 11 205 208
8. HareEE
PetersonBK
IyerVN
MeierR
EisenMB
2008 Sepsid even-skipped enhancers are functionally conserved in Drosophila despite lack of sequence conservation. PLoS Genet 4 e1000106 doi:10.1371/journal.pgen.1000106
9. KimJ
HeX
SinhaS
2009 Evolution of regulatory sequences in 12 Drosophila species. PLoS Genet 5 e1000330 doi:10.1371/journal.pgen.1000330
10. MakeevVJ
LifanovAP
NazinaAG
PapatsenkoDA
2003 Distance preferences in the arrangement of binding motifs and hierarchical levels in organization of transcription regulatory information. Nucleic Acids Res 31 6016 6026
11. PapatsenkoD
GoltsevY
LevineM
2009 Organization of developmental enhancers in the Drosophila embryo. Nucleic Acids Res
12. LynchM
2007 The frailty of adaptive hypotheses for the origins of organismal complexity. Proc Natl Acad Sci U S A 104 Suppl 1 8597 8604
13. TanayA
SiggiaED
2008 Sequence context affects the rate of short insertions and deletions in flies and primates. Genome Biol 9 R37
14. MacArthurS
BrookfieldJF
2004 Expected rates and modes of evolution of enhancer sequences. Mol Biol Evol 21 1064 1073
15. HeX
LingX
SinhaS
2009 Alignment and prediction of cis-regulatory modules based on a probabilistic model of evolution. PLoS Comput Biol 5 e1000299 doi:10.1371/journal.pcbi.1000299
16. LudwigMZ
PatelNH
KreitmanM
1998 Functional analysis of eve stripe 2 enhancer evolution in Drosophila: rules governing conservation and change. Development 125 949 958
17. DermitzakisET
ClarkAG
2002 Evolution of transcription factor binding sites in Mammalian gene regulatory regions: conservation and turnover. Mol Biol Evol 19 1114 1121
18. MosesAM
PollardDA
NixDA
IyerVN
LiXY
2006 Large-scale turnover of functional transcription factor binding sites in Drosophila. PLoS Comput Biol 2 e130 doi:10.1371/journal.pcbi.0020130
19. DrieverW
Nusslein-VolhardC
1988 A gradient of bicoid protein in Drosophila embryos. Cell 54 83 93
20. StanojevicD
HoeyT
LevineM
1989 Sequence-specific DNA-binding activities of the gap proteins encoded by hunchback and Kruppel in Drosophila. Nature 341 331 335
21. TreismanJ
DesplanC
1989 The products of the Drosophila gap genes hunchback and Kruppel bind to the hunchback promoters. Nature 341 335 337
22. Rivera-PomarR
JackleH
1996 From gradients to stripes in Drosophila embryogenesis: filling in the gaps. Trends Genet 12 478 483
23. SmallS
KrautR
HoeyT
WarriorR
LevineM
1991 Transcriptional regulation of a pair-rule stripe in Drosophila. Genes Dev 5 827 839
24. PetrovDA
2002 DNA loss and evolution of genome size in Drosophila. Genetica 115 81 91
25. BergmanCM
CarlsonJW
CelnikerSE
2005 Drosophila DNase I footprint database: a systematic genome annotation of transcription factor binding sites in the fruitfly, Drosophila melanogaster. Bioinformatics 21 1747 1749
26. LiangHL
NienCY
LiuHY
MetzsteinMM
KirovN
2008 The zinc-finger protein Zelda is a key activator of the early zygotic genome in Drosophila. Nature 456 400 403
27. RastegarS
HessI
DickmeisT
NicodJC
ErtzerR
2008 The words of the regulatory code are arranged in a variable manner in highly conserved enhancers. Dev Biol 318 366 377
28. WonKJ
SandelinA
MarstrandTT
KroghA
2008 Modeling promoter grammars with evolving hidden Markov models. Bioinformatics 24 1669 1675
29. GertzJ
SiggiaED
CohenBA
2009 Analysis of combinatorial cis-regulation in synthetic and genomic promoters. Nature 457 215 218
30. NeafseyDE
PalumbiSR
2003 Genome size evolution in pufferfish: a comparative analysis of diodontid and tetraodontid pufferfish genomes. Genome Res 13 821 830
31. GraurD
ShualiY
LiWH
1989 Deletions in processed pseudogenes accumulate faster in rodents than in humans. J Mol Evol 28 279 285
32. PetrovDA
HartlDL
1998 High rate of DNA loss in the Drosophila melanogaster and Drosophila virilis species groups. Mol Biol Evol 15 293 302
33. PetrovDA
SangsterTA
JohnstonJS
HartlDL
ShawKL
2000 Evidence for DNA loss as a determinant of genome size. Science 287 1060 1062
34. RobertsonHM
2000 The large srh family of chemoreceptor genes in Caenorhabditis nematodes reveals processes of genome evolution involving large duplications and deletions and intron gains and losses. Genome Res 10 192 203
35. BensassonD
PetrovDA
ZhangDX
HartlDL
HewittGM
2001 Genomic gigantism: DNA loss is slow in mountain grasshoppers. Mol Biol Evol 18 246 253
36. HasegawaM
KishinoH
YanoT
1985 Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J Mol Evol 22 160 174
37. BrownCT
XieY
DavidsonEH
CameronRA
2005 Paircomp, FamilyRelationsII and Cartwheel: tools for interspecific sequence comparison. BMC Bioinformatics 6 70
38. DownTA
BergmanCM
SuJ
HubbardTJ
2007 Large-scale discovery of promoter motifs in Drosophila melanogaster. PLoS Comput Biol 3 e7 doi:10.1371/journal.pcbi.0030007
39. NoyesMB
MengX
WakabayashiA
SinhaS
BrodskyMH
2008 A systematic characterization of factors that regulate Drosophila segmentation via a bacterial one-hybrid system. Nucleic Acids Res 36 2547 2560
40. LiXY
MacArthurS
BourgonR
NixD
PollardDA
2008 Transcription factors bind thousands of active and inactive regions in the Drosophila blastoderm. PLoS Biol 6 e27 doi:10.1371/journal.pbio.0060027
41. De RenzisS
ElementoO
TavazoieS
WieschausEF
2007 Unmasking activation of the zygotic genome using chromosomal deletions in the Drosophila embryo. PLoS Biol 5 e117 doi:10.1371/journal.pbio.0050117
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2010 Číslo 1
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- A Major Role of the RecFOR Pathway in DNA Double-Strand-Break Repair through ESDSA in
- Kidney Development in the Absence of and Requires
- The Werner Syndrome Protein Functions Upstream of ATR and ATM in Response to DNA Replication Inhibition and Double-Strand DNA Breaks
- Alternative Epigenetic Chromatin States of Polycomb Target Genes
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy