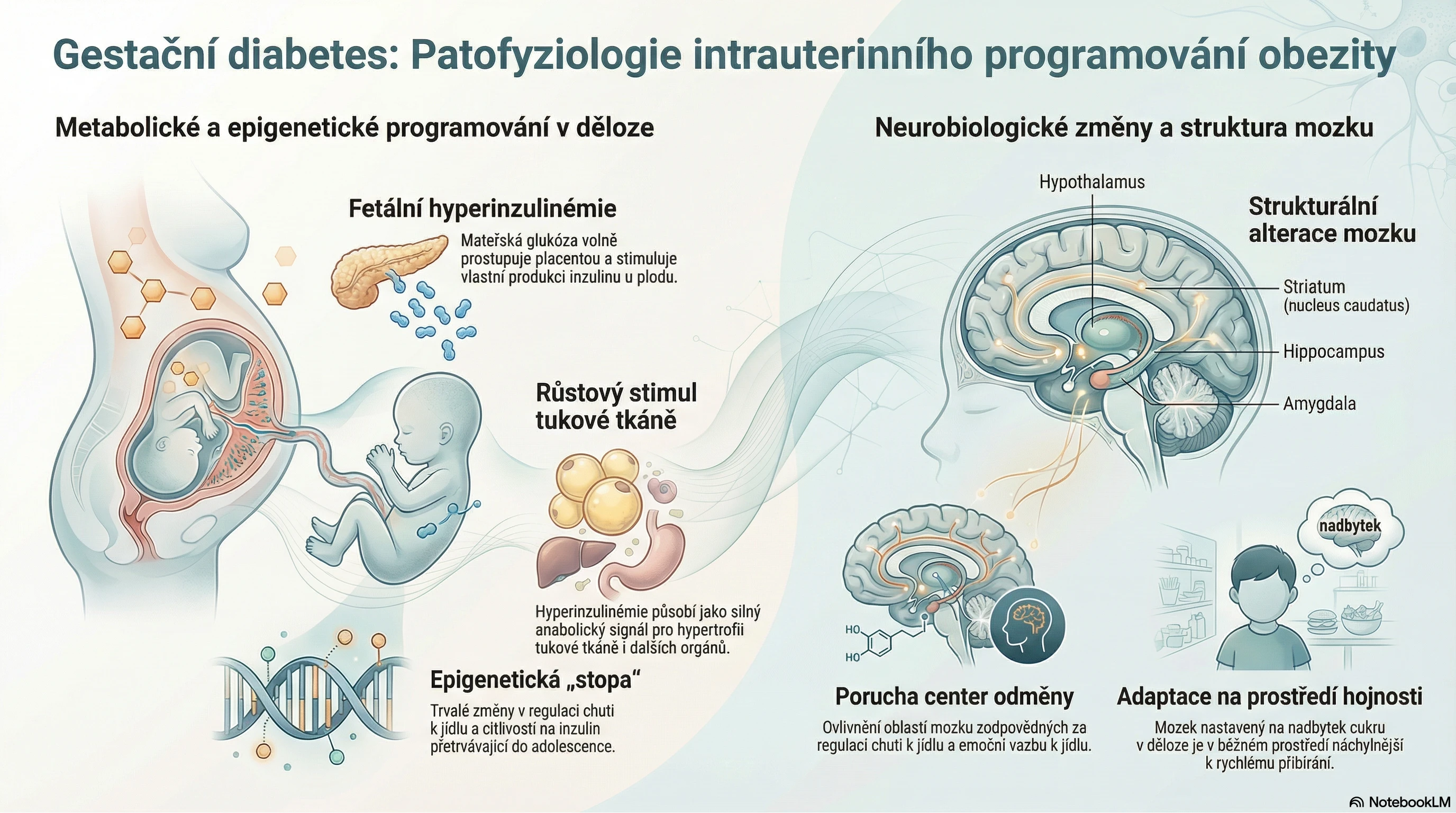
Deficit kyselé sfingomyelinázy je vrozené multisystémové onemocnění způsobené patogenními variantami genu SMPD1. Celosvětová prevalence se odhaduje přibližně na 0,1–1/100 000 narozených. V některých populacích (například u aškenázských Židů) je prevalence vyšší, udává se asi 1/40 000 porodů. I když se jedná o kontinuální spektrum klinické manifestace, konkrétní projevy, doba do rozvoje onemocnění i jeho tíže se u jednotlivých podtypů značně liší. Velkou variabilitu pozorujeme dokonce též v rodinách se stejnou mutací.1
Nejtěžším průběhem se vyznačuje rapidně progredující infantilní neuroviscerální ASMD, dříve označovaná jako Niemannova–Pickova choroba (NPD) typu A. Nejčastěji se projevuje hepatosplenomegalií, kterou lze obvykle pozorovat ve 2–4 měsících věku. Dalším typickým příznakem je třešňová skvrna na sítnici, která je u většiny dětí patrná nejpozději v 1 roce věku. Střádání sfingomyelinu v alveolárních septech, stěnách bronchů i pleuře vede k intersticiálnímu onemocnění plic s častými respiračními infekty. Neurologické příznaky se rozvíjejí přibližně od 7 měsíců, následuje zastavení psychomotorického vývoje okolo 10 měsíců věku. Neurodegenerace s hypotonií rychle progreduje a častou příčinou úmrtí těchto pacientů, kteří se obvykle nedožívají více než 3 let, bývá respirační selhání.1–3
Nejmírnější formou onemocnění je chronická viscerální ASMD (dříve NPD typu B), pro niž je typický pozdější nástup v dětství nebo i v dospělosti. Onemocnění se vyznačuje multisystémovými projevy s pomalejší progresí bez manifestací v centrální nervové soustavě (CNS). Typická je hepatosplenomegalie a postupné zhoršování plicních i jaterních funkcí.2 Nemocní s mírnou formou ASMD typu B mohou mít obvyklou délku dožití.3
U pacientů s chronickou neuroviscerální ASMD (typ A/B) se onemocnění může rozvinout v dětství či v dospělosti a k orgánovým projevům se přidává i neurodegenerace s pomalejší progresí oproti typu A.3 Neurologické symptomy mohou být relativně mírné (hypotonie, hyporeflexie), ale vyskytnout se může také těžká progresivní ataxie, spasticita nebo kognitivní úpadek.1
Kurativní terapie ASMD v současnosti není k dispozici, onemocnění je však léčitelné. Základem péče zůstává podpůrná symptomatická léčba. K dispozici je také enzymová substituční terapie rekombinantní kyselou sfingomyelinázou, která však vzhledem k neprostupnosti hematoencefalické bariéry pro léčivo není schopna ovlivnit neurologické manifestace. U některých pacientů může být vhodným postupem transplantace hematopoetických kmenových buněk (HSCT).1
Vzácnost a fenotypová variabilita ASMD často vedou k misdiagnostice, zejména u podtypů s pozdním nástupem. Mezi nejtypičtější příznaky patří hepatosplenomegalie, přičemž zvětšení sleziny může být masivní. Častá je zvýšená hladina jaterních enzymů (u velké části pacientů s ASMD typu B je přítomná fibróza jaterní tkáně). Většina nemocných jeví známky intersticiálního plicního onemocnění na snímcích hrudníku, relativně časté jsou také osteopenie či osteoporóza. Velká část pacientů s ASMD vykazuje aterogenní lipidový profil s vysokou hladinou triglyceridů i celkového a LDL cholesterolu a nízkou koncentrací HDL cholesterolu.1
Prvním krokem ke stanovení diagnózy obvykle bývá vyšetření biomarkerů testem ze suché krevní kapky (DBS), následuje stanovení aktivity kyselé sfingomyelinázy v periferních leukocytech a případně genetické vyšetření. Nejdůležitější ovšem je na ASMD u dospělých pacientů s nevysvětlenou hepatosplenomegalií vůbec pomyslet.
Při podezření na ASMD nebo jiné střádavé onemocnění může s předběžnou diferenciální diagnostikou pomoci nový nástroj accelRare.4 Lékař během 5–10 minut ve svém prohlížeči anonymně vyplní jednoduchý dotazník s anamnézou pacienta, na jehož základě accelRare vygeneruje seznam vzácných onemocnění, která nejlépe odpovídají symptomům daného pacienta. Navíc navrhne případná další vyšetření a seznam specializovaných pracovišť, kam je pacienta možno odeslat.
Časná diagnostika ASMD je klíčová pro prevenci progrese orgánového poškození. Pro léčbu tohoto vzácného multisystémového onemocnění máme k dispozici nedávno vyvinutou enzymovou substituční terapii. Klinické podezření na ASMD bychom měli pojmout zejména u pacientů s hepatosplenomegalií doprovázenou poruchami funkce jater, plicními potížemi nebo aterogenním lipidovým profilem.1 S diagnostickou rozvahou může pomoci on-line nástroj accelRare.4
(este)
Zdroje:
1. Geberhiwot T., Wasserstein M., Wanninayake S. et al. Consensus clinical management guidelines for acid sphingomyelinase deficiency (Niemann–Pick disease types A, B and A/B). Orphanet J Rare Dis 2023; 18 (1): 85, doi: 10.1186/s13023-023-02686-6.
2. Wasserstein M. P., Schuchman E. H. Acid sphingomyelinase deficiency. GeneReviews, University of Washington, Seattle, 2023 Apr 27.
3. Arslan N., Coker M., Gokcay G. F. et al. Expert opinion on patient journey, diagnosis and clinical monitoring in acid sphingomyelinase deficiency in Turkey: a pediatric metabolic disease specialist's perspective. Front Pediatr 2023; 11: 1113422, doi: 10.3389/fped.2023.1113422.
4. Frequently asked questions. accelRare, 2026. Dostupné na: www.accelrare.fr/faq-accelrare