
Cushingov syndróm a akromegália na podklade pikoadenómu hypofýzy
Cushing’s syndrome and acromegaly based on picoadenoma of the pituitary gland
Cushing’s syndrome (CS) is a relatively rare disease characterized by autonomous hypersecretion of cortisol. The incidence of CS is estimated to be equal to 2–3 cases per million inhabitants per year. The incidence of acromegaly is 3–4 patients per 1 000 000 per year. The disease is caused by hypersecretion of growth hormone which is mainly caused by benign tumour of the pituitary gland. In our case report we present a 41 - year old woman suffering from both Cushing’s syndrome and acromegaly. The patient was examined in National Institute of Endocrinology and Diabetology Ľubochňa for a centripetal type of obesity and hirsutism. Laboratory tests revealed high plasma cortisol levels without circulating variation, hypercortisoluria and elevated plasmatic levels of ACTH. A 2 mg dexamethasone blockade was performed without adequate cortisol suppression in serum and urine up to 8 mg blockade resulted in suppression of 24 hour urine free cortisol. A magnetic resonance imaging (MR) scan revealed suspect pikoadenoma of the pituitary gland (size 2mm). Subsequently trans-sphenoidal resection was performed. Histopathological and immunohistochemical examinations did not reveal the ACTH-producing pituitary adenoma. After surgery hypercortisolism persisted with newly revealed hypersomatotropism. Treatment with Ketoconazole at dose 200mg 1/ 2-0-1 and somatostatin analogues (Lanreotide) at dose 120mg every 42 days were initiated. Control magnetic resonance imaging of the sella demonstrated small tumour of pituary gland of size 3×5mm. Later 3 years after first surgery another trans-sphenoidal resection of residue was performed. Histological and immunohistochemical examinations did not confirm adenoma with ACTH and RH secretion. After second surgery, IGF-1 plasma levels were not normalized with persistence of hypercortisolism. The treatment with Lanreotide at the initial dose as well as Ketoconazole was reinitiated (with increased dose of Ketoconazole to 1-1-1 tbl per 200mg).
Keywords:
acromegaly – cortisol – Cushing’s syndrome – Growth hormone – IGF-1
Autoři:
Ivana Ságová 1; Daniela Kantárová 2; Dušan Pávai 1; Milan Dragula 2; Anton Vaňuga 1,3; Peter Vaňuga 1
Působiště autorů:
Endokrinologické oddelenie, Národný endokrinologický a diabetologický ústav Ľubochňa
1; Interná klinika UN a JLFUK, Martin
2; Alphamedical, s. r. o.
3
Vyšlo v časopise:
Vnitř Lék 2020; 66(2): 82-86
Kategorie:
Kazuistika
Souhrn
Cushingov syndróm (CS) je pomerne vzácne ochorenie charakterizované autonómnou hypersekréciou kortizolu. Ročná incidencia CS je 2–3/milión obyvateľov. Incidencia akromegálie je 3–4 pacienti na 1 000 000 za rok. Ochorenie je spôsobené hypersekréciou rastového hormónu (RH) v 99% na podklade adenómu hypofýzy. V našej kazuistike prezentujeme 41-ročnú ženu s kombináciou Cushingovho syndrómu a akromegálie. Pacientka bola vyšetrená v NEDU Ľubochňa pre centripetálny typ obezity a hirzutizmus. Laboratórne bola prítomná hyperkortizolémia bez cirkadiánnej variácie, hyperkortizolúria s elevovanou hladinou adrenokortikotropného hormónu (ACTH). Realizovaná 2mg dexametazónová blokáda bez adekvátnej supresie kortizolu v sére a moči až v 8mg blokáde došlo k supresii kortizolúrie. Magnetická rezonancia (MR) s nálezom v.s. pikoadenómu hypofýzy veľkosti 2mm. Následne bola realizovaná transsfenoidálna resekcia pikoadenómu hypofýzy. Histopatologické a imunohistochemické nálezy neodhalili ACTH produkujúci adenóm hypofýzy. Pozákrokovo pretrvával hyperkortizolizmus s novozachyteným hypersomatotropizmom. Do liečby bol pridaný Ketokonazol 200mg tbl 1/2-0-1 a Lanreotid v dávke 120mg každých 42 dní. Kontrolná MR hypofýzy preukázala drobnú ložiskovú štruktúru s rozmermi 3×4mm. Po súhlase pacientky s odstupom 3 rokov bola vykonaná endoskopická revízia rezidua. Histologické a imunohistochemické vyšetrenia bez potvrdenia adenómu s ACTH a RH sekréciou. Pozákrokovo opäť nedošlo k znormalizovaniu plazmatických hladín IGF-1 s pretrvávaním hyperkortizolizmu. Bola opätovne začatá liečba Lanreotidom v pôvodnej dávke ako aj Ketokonazolom s navýšením dávky 200mg na 3-krát 1 tbl.
Klíčová slova:
akromegália – Cushingov syndróm – IGF-1 – kortizol – rastový hormón
Úvod
Cushingov syndróm (CS) je ochorenie charakterizované nadbytkom glukokortikoidov, ktoré môžu byť exogénneho, alebo endogénneho pôvodu (1). Endogénny CS je spôsobený autonómnou nadprodukciou ACTH, alebo samotného kortizolu; autonómna nadprodukcia CRH je veľmi vzácna. V klinickom obraze dominuje mesiačikovitá tvár, centripetálna distribúcia tuku, ukladanie tuku na krku, pletora, červenofialové strie na koži, adynamia, úbytok svalovej hmoty najmä v oblasti koreňových svalov, osteoporóza, amenorea a hypofunkcia gonád. Z kardiovaskulárnych prejavov býva prítomná artériová hypertenzia. Diabetes mellitus sa u CS vyskytuje ako dôsledok inzulín-rezistentného stavu spolu so zhoršenou sekréciou inzulínu pri nadbytku glukokortikoidov. V prípade výrazne zvýšenej glukokortikoidnej nadprodukcie (ektopický ACTH syndróm, adrenokortikálny karcinóm) môže byť prítomná ťažká hypokalémia a hypokalemická alkalóza. Diagnostika sa zakladá na laboratórnom vyšetrení voľného močového kortizolu, sérového kortizolu v diurnálnom profile a ACTH v sére. V ďalšej diferenciálnej diagnostike CS využívame supresívne dexametazónové testy. Zo zobrazovacích vyšetrení v diagnostike CS má dominantné postavenie MR hypofýzy a CT nadobličiek. Terapia CS je najmä chirurgická (transfenoidálna adenomektómia, adrenalektómia, odstránenie nádoru produkujúceho ACTH-like peptid). Ďalšími terapeutickými metódami sú rádioterapia (lineárny urýchľovač, Leksellov gamma nôž) a medikamentózna liečba (Pasireotid, adrenolytiká, adrenostatiká).
Akromegália je zriedkavé ochorenie vznikajúce v 99% na podklade adenómu hypofýzy, ktoré je charakterizované nadmernou sekréciou IGF-1 vyvolanou nadprodukciou rastového hormónu (2). Medzi najčastejšie príznaky a symptómy akromegálie patrí zvýšené potenie, parestézie, dysmorfia, artralgie, cefalea, slabosť, syndróm karpálneho tunela . Pacienti mávajú často sprievodné ochorenia ako artériovú hypertenziu, porušenú glukózovú toleranciu prípadne diabetes mellitus, syndróm spánkového apnoe, polypózu hrubého čreva (2). Zo samotného rastu tumoru môže byť prítomná bolesť hlavy, poruchy zorného poľa. Diagnostika sa zakladá na potvrdení vysokej hladiny rastového hormónu, najmä jeho nesupresibilite v orálnom glukózovom tolerančnom teste ako aj vysokej hladine inzulínu podobného rastového faktora 1 (IGF-1). Zo zobrazovacích vyšetrení má dominantné postavenie MR hypofýzy a celej selárnej oblasti. Transsfenoidálna chirurgia predstavuje prioritnú liečbu u pacientov s akromegáliou. Ak sa operačným výkonom nedosiahne biochemická kontrola ochorenia pristupuje sa k stereotaktickej rádioneurochirurgii (lineárny urýchľovač, Leksellov gama nôž). Z medikamentóznej liečby sú analógy somatostatínu prvou líniou liečby. Antagonisty receptora pre rastový hormón predstavujú alternatívu pri zlyhaní somatostatínovej liečby. Agonisty dopamínu majú doplnkový účinok najmä pri súčasnej sekrécií prolaktínu.
Kazuistika
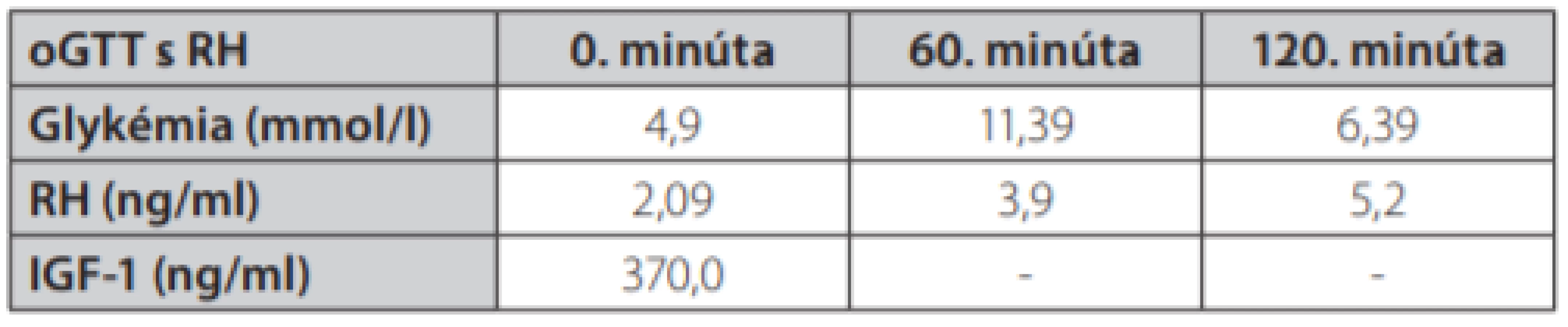
Uvádzame prípad 41-ročnej ženy (narodená v roku 1977) s kombináciou Cushingovho syndrómu a akromegálie. Pacientka bola prijatá na oddelenie endokrinológie Národného endokrinologického a diabetologického ústavu v Ľubochni pre centripetálny typ obezity (TV: 167 cm, TH: 85 kg, BMI: 30,48, OP: 112 cm) s váhovým prírastkom za posledný rok 9 kg s purpurovými striami v abdominálnej oblasti a hirzutizmus (zvýšené ochlpenie na tvári, brade, chrbte a končatinách). Menštruačný cyklus bol pravidelný. Cestou spádového gynekológa bolo realizované USG ovárií bez patologického nálezu. Anamnesticky sa pacientka liečila 2 roky na artériovú hypertenziu, 1 rok na depresívnu poruchu. Zo subjektívnych ťažkostí dominovala zvýšená únava. Laboratórne testy odhalili zvýšenú hladinu plazmatického a močového kortizolu, bez diurnálnej variácie plazmatickej kortizolémie, s mierne elevovanou hladinou ACTH, s mierne zvýšenou bazálnou hladinou androstendiónu a dehydroepiandrosterón-sulfátu (DHEA-S) (Tab. 1.). Bola realizovaná 2mg dexametázonová blokáda bez adekvátnej supresie kortizolu v sére a moči (Tab. 2.). V 8mg dexametazónovej blokáde došlo k supresii kortizolúrie (Tab. 2.). Doplnený perimeter bez výpadku v zornom poli. Následne bolo realizované MR hypofýzy s veľkosťou v kraniokaudálnej rovine 7–8mm,vo ventrodorzálnej rovine 12mm. Po i. v. aplikácií kontrastnej látky sa paramediálne vľavo aj vpravo zobrazuje hyposignálna lézia veľkosti 2mm v.s. pikoadenóm. Stopka hypofýzy je v stredovej rovine, optická chiazma, supraselárne a paraselárne priestory sú bez infiltrácie. Na základe uvedeného bola stanovená diagnóza Cushingovej choroby na podklade pikoadenómu hypofýzy. Následne bola vykonaná transsfenoidálna resekcia pikoadenómu hypofýzy. Histopatologické a imunohistochemické nálezy potvrdili tkanivo hypofýzy, so zachovanou acinárnou architektúrou, expresiou všetkých hypofýzových hormónov, Ki 67 prakticky 0, P53 negatívna, zachovaná retikulínová kostra bez atypií. Pozákrokovo u pacientky pretrvávala hypersekrécia kortizolu (zvýšená plazmatická kortizolémia bez diurnálnej variácie, zvýšená kortizolúria, elevovaná hladina ACTH) s novozachytenou zvýšenou plazmatickou hladinou inzulínu podobného rastového faktora 1, bazálna hladina rastového hormónu bola v norme (Tab. 1.). Doplnený orálno-glukózový tolerančný test s RH bez adekvátnej supresibility (Tab. 3.). Bola začatá liečba somatostatínovými analógmi (lanreotid) v dávke 120mg každých 42 dní s poklesom hladiny IGF-1 (Graf 1.). Doplnené kontrolne MR hypofýzy s odstupom 6 mesiacov pooperačne, kde v porovnaní s predoperačným MR došlo k regresii veľkosti hypofýzy na maximálny rozmer asi 6mm. Na laterálnom okraji adenohypofýzy vľavo v kontakte s ACI perzistuje drobná ložisková štruktúra s rozmermi 3×4mm. Ložisko je v T2W sekvencií diskrétne hyperintenzívne, na postkontrastných sekvenciách sa javí ako relatívne hypointenzívne. Supreselárna oblasť je bez expanzie, optochiazmatické štruktúry sú bez kompresie. Opakovane konzultovaný chirurg, ktorý odporúča reoperáciu, s ktorou pacientka v tom čase nesúhlasila. Pre pretrvávanie hyperkortizolizmu bol pridaný do liečby Ketokonazol v dávke 200mg tbl 1/2-0-1, avšak bez významného efektu na klinické prejavy ochorenia ako aj bez supresie hyperkortizolizmu. Liečba Pasireotidom nebola iniciovaná pre nesúhlas pacientky. Realizované denzitometrické vyšetrenie s kostnou hustotou v norme pre daný vek a pohlavie. S odstupom 3 rokov po 1. operácií bola po súhlase pacientky realizovaná endoskopická revízia rezidua. Histologicky a imunohistochemicky potvrdené nenádorové tkanivo adenohypofýzy so zachovanou acinárnou architektúrou (retikulín a kolagén IV), Ki 67 prakticky nulové, expresia všetkých hypofýzarných hormónov, niekoľko málo je expandovaných - mohlo by ísť o nodulárnu hyperpláziu tkaniva adenohypofýzy. Pozákrokovo ale nedošlo k znormalizovaniu plazmatických hladín IGF-1 s pretrvávaním hyperkortizolizmu. Bola opätovne začatá liečba somatostatínovými analógmi (lanreotid) v dávke 120mg každých 42 dní ako aj Ketokonazolom s navýšením dávky 200mg na 3-krát 1 tbl. Na uvedenej liečbe došlo v priebehu 6 mesiacov k zlepšeniu klinických ťažkostí pacientky (redukcia hmotnosti -5 kg, ústup slabosti, pokles TK s redukciou antihypertenzívnej liečby) s poklesom kortizolémie a kortizolúrie a normalizáciou hladiny IGF-1.



Diskusia
Súčasný výskyt akromegálie a Cushingovho syndrómu je vzácny. V našej kazuistike popisujeme zriedkavý prípad funkčného mikroadenómu hypofýzy s ACTH-RH sekréciou, ktorých výskyt bol doposiaľ popísaný v literatúre v 20 prípadoch (3). Plurihormonálne adenómy (PHA) predstavujú 10–15% všetkých funkčných adenómov hypofýzy. Niektoré štúdie však naznačujú prevalenciu až 31–36% u chirurgicky odstránených nádorov (4). Histologicky sú PHA monomorfné (rôzne hormóny sú exprimované z jediného morfologického bunkového typu) alebo plurimorfné (rôzne hormóny sú exprimované morfologicky divergentnými bunkami) (5). Najčastejšie hormonálne asociácie sú s prolaktínom a rastovým hormónom. Z 20 popísaných prípadov ACTH-RH PHA sa u 11 pacientov klinicky prezentovali akromegalické príznaky, 2 pacienti mali Cushingovu chorobu, u 3 bola prítomná pituárna apoplexia, zatiaľ čo len 4 pacienti boli s klinickými príznakmi akromegálie a Cushingovej choroby (3). Histologicky boli u 19 pacientov hypofyzárne adenómy pozostávajúce z dvoch odlišných bunkových populácií, iba v 1 prípade bol dôkaz jednej bunky produkujúcej ACTH aj RH. Klinické prejavy PHA vyplývajú jednak zo samotného rastu tumoru a sú to bolesť hlavy, poruchy zrakových nervov, pri supraselárnom raste s následnými výpadkami zorného poľa. Na druhej strane sú prejavy vyplývajúce z hypersekrécie konkrétnych hypofýzových hormónov. V prípade našej pacientky išlo o ACTH-RH sekréciu a dominovali prejavy Cushingovej choroby. Pre CS je charakteristický vzostup telesnej hmotnosti s centripetálnou distribúciou tuku, ukladanie tuku na krku (tzv. býčia šija), v oblasti tváre (tzv. mesiačikovitá tvár). Môže byť prítomné aj ukladanie tuku v mediastine, alebo epidurálne v miešnom kanáli (s možnými neurologickými prejavmi). Súčasne často dochádza k úbytku svalovej hmoty najmä v oblasti koreňových svalov. Pri CS môžu byť kožné zmeny ako purpurové strie, pletora tváre, známky hirzutizmu, akné, alopécia (u žien), zhoršené hojenie rán. Z kardiovaskulárnych prejavov býva prítomná hypertenzia, ktorá vzniká ako následok spoluúčasti nadbytku glukokortikoidov na pôsobenie mineralokortikoidov a katecholamínov. Z ďalších príznakov CS môže byť v klinickom obraze amenorea, hypofunkcia gonád, osteoporóza, psychické zmeny (emočná labilita, depresívne ladenie, afektívne poruchy, celková únavnosť, výpadky pamäti, poruchy koncentrácie), poruchy glukózovej tolerancie až diabetes mellitus (6). Diagnostika je založená na klinickom obraze, laboratórnych a zobrazovacích vyšetreniach. V laboratórnych parametroch nachádzame poruchu glukózovej tolerancie, polyglobúliu, leukocytózu, lymfopéniu, trombocytózu. Môže byť prítomná hypokalémia a alkalóza. V moči stanovujeme voľný močový kortizol za 24hod (vysoké hladiny u CS), sérový kortizol v diurnálnom profile (8 : 00, 16 : 00, 20 : 00, 24 : 00 hod), ktorý má narušený cirkadiánny rytmus u CS a bazálne hladiny ACTH v sére (7). V diferenciálnej diagnostike CS využívame supresívne dexametazónové testy (1mg, 2mg a 8mg dexametazónovú blokádu). V lokalizačnej diagnostike využívame MR hypofýzy a CT nadobličiek. U pacientov s adenómami vylučujúcimi ACTH je primárnou terapiou transsfenoidálna operácia. Chirurgická liečba Cushingovej choroby dosahuje 91% mieru remisie u mikroadenómov, ale u pacientov s makroadenómami klesá až na 65% (8). Približne 10–20% dospelých má po 10 rokoch rekurenciu (8). Rádioterapia je určená pacientom, ktorí sú po resekcii subtotálne resekovaní, alebo zostávajú nadmerným sekrétorom. Pooperačná stereotaktická rádiochirurgia dosiahla remisiu u približne 68% pacientov s CS (9). Pri čakaní na účinky rádioterapie môžu byť indikované inhibítory adrenálnej steroidogenézy (adrenostatiká – ketokonazol a metyrapon, adrenolytikum – mitotan). Ketokonazol je derivát imidazolu inhibujúci steroidogenézu, používaný ako antimykotikum. V indikácií CS začíname dávkou 400mg denne, účinné dávky sú väčšinou v rozmedzí 3krát 200–400mg denne. Časté nežiaduce účinky sú gastrointestinálne (nauzea, zvracanie) a kožné (rash, alopécia). Najzávažnejším je hepatotoxicita. U mužov môže podávanie ketokonazolu viesť k poklesu libida a k vzniku gynekomastie. U Cushingovej choroby je možnosťou liečby aj pasireotid, ktorý priamo pôsobí na kortikotrofové adenómové bunky a sekréciu ACTH (10). Pasireotid sa viaže na somatostatínové receptory okrem 4. podtypu. Efektom liečby pasireotidom je zníženie hladiny voľného močového kortizolu o viac ako 50% u 50% pacientov a jeho normalizácia u 25% pacientov (11). Ešte lepšie výsledky sú dosiahnuté pri súčasnej terapii s kabergolínom (normalizácia hladín voľného močového kortizolu až v 53%) a s ketokonazolom (normalizácia v 88%) (12). Najzávažnejším nežiaducim efektom liečby je hyperglykémia, poruchy glukózovej tolerancie a vznik alebo zhoršenie diabetes mellitus (až v 73% prípadov) (13). V prípade nedostupnosti liekov, alebo pri ich intolerancii sa môže navrhnúť bilaterálna adrenalektómia. Vzhľadom k včasnej diagnostike a následnej liečbe akromegálie u našej pacientky nedošlo k rozvoju klinickej symptomatológie ochorenia. U akromegálie sa klinický obraz rozvíja postupne v priebehu niekoľkých rokov až desaťročí. Medzi najčastejšie príznaky a symptómy ochorenia patrí zvýšené potenie, parestézie, dysmorfia, artralgie, cefalea, slabosť, syndróm karpálneho tunela a poruchy zraku (14). Diagnostika sa zakladá na potvrdení vysokej hladiny rastového hormónu, najmä jeho nesupresibilite v orálnom glukózovom tolerančnom teste ako aj vysokej hladine IGF-1. Zo zobrazovacích vyšetrení má dominantné postavenie MR hypofýzy. Pri liečbe adenómov vylučujúcich RH je transsfenoidálna chirurgia prvolíniovou terapiou. Efekt transfénoidálnej chirurgie u akromegalikov s mikroadenómami je 85–90% a 65% pri makroadenómoch hypofýzy (15). Na Slovensku a v Českej republike podľa údajov z registra sellárnych tumorov (RESET) sú výsledky horšie. Normalizácia hladín IGF-1 u 81% pacientov po chirurgickej liečbe po 3 mesiacoch bola prítomná u 54,5% mikroadenómov a 42,4% makroadenómov (16). Pooperačná rádioterapia (lineárny urýchľovač alebo Leksellov gamma nôž) sa vykonáva pre čiastočne resekované nádory alebo keď hladiny RH zostávajú pooperačne zvýšené. Nástup účinku tejto liečby môže trvať aj niekoľko rokov. Analógy somatostatínu (SSA) sú liečbou prvej línie pre pacientov s akromegáliou. Ich predoperačné podávanie môže zvýšiť úspešnosť samotného chirurgického zákroku (17). Dlhodobo pôsobiace formy oktreotidu a lanreotidu pôsobia najmä na somatostatínové receptory 2. typu a majú podobnú účinnosť (18, 19). Dlhodobo pôsobiaci oktreotid sa používa v dávkach 20–30mg i.m. à 4 týždne, lanreotid v dávke 60–120mg s. c. à 4 týždne. Efekt liečby SSA na normalizáciu hladín RH a IGF-1 sa značne líši medzi jednotlivými štúdiami v rozmedzí 20–70% (20). V nedávnej metaanalýze bol publikovaný účinok SSA na normalizáciu hladín RH u 55% liečených pacientov a na normalizáciu hladín IGF-1 u 56% liečených pacientov, bez významného rozdielu medzi jednotlivými typmi SSA (21). Účinnosť liečby lanreotidom pri normalizácií hladín RH u novodiagnostikovaných, neliečených pacientov bola iba 40% (22, 23). Zníženie objemu nádoru o viac ako 20% sa pozorovalo u 75% pacientov liečených s dlhodobo pôsobiacim oktreotidom a u 54,1% liečených lanreotidom, ale s rôznym trvaním sledovania (23, 24). Medzi najčastejšie nežiaduce účinky liečby SSA patria dyspeptické ťažkosti, najmä hnačky a cholecystolitiáza. S pokračujúcou liečbou väčšinou intenzita nežiaducich účinkov SSA klesá. Antagonista receptora pre rastový hormón pegvisomant sa podáva s. c. v dávke 10–40mg denne. V pilotných štúdiách bola dosiahnutá normalizácia hladín IGF-1 až u 95% pacientov užívajúcich pegvisomant v dávke 40mg denne (25, 26). Nedávno bola zverejnená štúdia zahŕňajúca 1 288 pacientov liečených pegvisomantom, u ktorých bola normalizácia hladín IGF-1 dosiahnutá u 63% pacientov (27). Rozdiely vo výsledkoch štúdií boli vysvetlené nedostatočnou titráciou dávky pegvisomantu ako aj komplikáciami v súvislosti s reálnym životom (27). Nežiaducim účinkom liečby je reverzibilný vzostup pečeňových transamináz a reakcie v mieste vpichu. Pegvisomant zlepšuje citlivosť na inzulín a dlhodobé sledovanie ukázalo významné zníženie hladiny glukózy nalačno. Rast tumoru pri liečbe pegvisomantom nie je obvyklý. V klinických štúdiách liečených pacientov s dostupnou MR bol výskyt nárastu veľkosti nádoru hypofýzy v 3,2% (27, 28). Na základe doporučení pre liečbu akromegálie z roku 2014 u pacientov s perzistujúcim ochorením pooperačne bola pri signifikantnom ochorení (t. j. so stredne ťažkými až závažnými príznakmi a symptómami nadbytku RH, bez známok lokálneho rastu tumoru) doporučená liečba SSA alebo pegvisomantom ako počiatočná adjuvantná liečebná terapia (29). U pacientov s miernym zvýšením hladín IGF-1 a miernymi príznakmi a symptómami nadbytku RH sa doporučuje liečba agonistami dopamínu, zvyčajne kabergolínom ako počiatočná adjuvantná liečebná terapia (29). Liečba SSA je doporučená ako primárna u pacientov s kontraindikáciou chirurgického výkonu (29). Pridanie pegvisomantu alebo kabergolínu sa odporúča u pacientov s nedostatočnou odpoveďou na SSA (29).
Záver
Súčasný výskyt Cushingovej choroby a akromegálie je raritný. Základom liečby akromegálie a Cushingovej choroby zostáva chirurgická resekcia hypofýzy skúseným chirurgom. Avšak významné percento pacientov má aj po operácii pretrvávajúce alebo rekurentné ochorenie s nutnosťou využitia iných terapeutických modalít (medikamentózna liečba a rádioterapia).
KORESPONDENČNÍ ADRESA AUTORA:
MUDr. Ivana Ságová, PhD.,
NEDU n.o.
Kollárová 282/3,
034 91 Ľubochňa
Cit. zkr: Vnitř Lék 2020; 66(E-2): e43–e47
Článek přijat redakcí: 1. 8. 2018
Článek přijat k publikaci: 16. 3. 2019
Zdroje
1. Ďurovcová V, Kršek M. Cushingův syndrom – charakteristika, diagnostika a léčba. Med Pro Praxi 2009; 6 : 295–299.
2. Capatina C, Wass JH. 60 Years Of Neuroendocrinology: Acromegaly. J Endocrinol 2015; 226 : 141–160.
3. Roca E, Mattogno PP, Porcelli T, et al. Plurihormonal ACTH-GH Pituitary Adenoma: Case Report and Systematic Literature Review. World neurosurgery 2018; 114 : 158–164.
4. Pawlikowski M, Kunert-Radek J, Radek M. Plurihormonality of pituitary adenomas in light of immunohistochemical studies. Endokrynol Pol 2010; 61 : 63–66.
5. Rasul FT, Jaunmuktane Z, Khan A, et al. Plurihormonal pituitary adenoma with concomitant adrenocorticotropic hormone (ACTH) and growth hormone (GH) secretion: a report of two cases and review of the literature. Acta Neurochir (Wien) 2014; 156 : 141–146.
6. Nieman LK. Cushing’s syndrome: update on signs, symptoms and biochemical screening. European Journal of Endocrinology 2015; 173 : 33–38.
7. Kiňová S. Endokrinné formy hypertenzie. Via pract 2011; 8 : 119–123.
8. Biller BMK, Grossman AB, Stewart PM, et al. Treatment of Adrenocorticotropin-Dependent Cushing’s Syndrome: A Consensus Statement. The Journal of Clinical Endocrinology & Metabolism 2008; 93 : 2454–2462.
9. Sheehan JP, et al. Gamma Knife surgery for pituitary adenomas: factors related to radiological and endocrine outcomes. Journal of Neurosurgery 2011; 114 : 303–309.
10. Fleseriu M. Medical treatment of Cushing disease: new targets, new hope. Endocrinology & Metabolism Clinics of North America 2015; 44 : 51–70.
11. Colao A, Petersenn S, Newell-Price J, et al. A 12-month phase 3 study of pasireotide in Cushing‘s disease. N Engl J Med 2012; 366 : 914–924.
12. Feelders RA, de Bruin C, Pereira AM, et al. Pasireotide alone or with cabergoline and ketoconazole in Cushing‘s disease. N Engl J Med 2010; 362 : 1846–1848.
13. Kršek M. Cushinguv syndrom a možnosti jehe řešení v roce 2012. Remedia. 2012; 6 : 386–392.
14. Biju Baby J, Veena SN, Jaishankar HP. Acromegaly – a case report. Journal of clinical and biomedical sciences 2012; 4 : 247–250.
15. John jr. AJ, Laws ER. Surgical Treatment of Pituitary Adenomas. Dostupné z https:// www.ncbi.nlm.nih.gov/books/NBK278983/.
16. Hána V, Švancara J, Bandúrová L, et al. Registry of sellar tumors – RESET: Diagnostic and therapy of acromegaly in Czech and Slovak republics in the 21st century. Diabetes, metabolizmus, endokrinologie a výživa 2013; 16 : 219–224.
17. Melmed S. New therapeutic agents for acromegaly. Nat Rev Endocrinol 2016; 12 : 90–98.
18. Colao A, Auriemma RS, Galdiero M, et al. Effects of initial therapy for five years with somatostatin analogs for acromegaly on growth hormone and insulin-like growth factor-I levels, tumor shrinkage, and cardiovascular disease: a prospective study. J Clin Endocrinol Metab 2009; 94 : 3746–3756.
19. Murray RD, Melmed S. A critical analysis of clinically available somatostatin analog formulations for therapy of acromegaly. J Clin Endocrinol Metab 2008; 93 : 2957–2968.
20. Zahr R, Fleseriu M. Updates in Diagnosis and Treatment of Acromegaly. European Endocrinology 2018; 10 : 57–61.
21. Carmichael JD, et al. Acromegaly clinical trial methodology impact on reported biochemical efficacy rates of somatostatin receptor ligand treatments: a meta-analysis. J Clin Endocrinol Metab 2014; 99 : 1825–1833.
22. Annamalai AK, et al. A comprehensive study of clinical, biochemical, radiological, vascular, cardiac, and sleep parameters in an unselected cohort of patients with acromegaly undergoing presurgical somatostatin receptor ligand therapy. J Clin Endocrinol Metab 2013; 98 : 1040–1050.
23. Caron PJ, et al. Tumor shrinkage with lanreotide Autogel 120 mg as primary therapy in acromegaly: results of a prospective multicenter clinical trial. J Clin Endocrinol Metab 2014; 99 : 1282–1290.
24. Mercado M, et al. A prospective, multicentre study to investigate the efficacy, safety and tolerability of octreotide LAR (long-acting repeatable octreotide) in the primary therapy of patients with acromegaly. Clin Endocrinol (Oxf) 2007; 66 : 859–868.
25. Trainer PJ, Drake WM, Katznelson L, et al. Treatment of acromegaly with the growth hormone-receptor antagonist pegvisomant. N Engl J Med 2000; 342 : 1171–1177.
26. van der Lely AJ, Hutson RK, Trainer PJ, et al. Long-term treatment of acromegaly with pegvisomant, a growth hormone receptor antagonist. Lancet 2001; 358 : 1754–1759.
27. van der Lely AJ, Biller BM, Brue T, et al. Long-term safety of pegvisomant in patients with acromegaly: comprehensive review of 1288 subjects in ACROSTUDY. J Clin Endocrinol Metab 2012; 97 : 1589–1597.
28. Tritos NA, et al. Effectiveness of first-line pegvisomant monotherapy in acromegaly: an ACROSTUDY analysis. Eur J Endocrinol 2017; 176 : 213–220.
29. Katznelson L, Laws EL, Melmed S, et al. Acromegaly: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab 2014; 99 : 3933–3951.
Štítky
Diabetologie Endokrinologie Interní lékařstvíČlánek vyšel v časopise
Vnitřní lékařství

2020 Číslo 2
- Diosmin a hesperidin: Co ukazuje farmakokinetika?
- Omeprazol a mechanismus hojení krvácení z peptického vředu
- Porovnání farmakologických vlastností mikronizovaného diosminu a hesperidinu v terapii chronické žilní insuficience a hemoroidů
- Srovnání vlivu omeprazolu a pantoprazolu na antiagregační účinek klopidogrelu
Nejčtenější v tomto čísle
- Diferenciální diagnostika hypoglykemie
- Hypoxemie/hypoxie a nové koncepty oxygenoterapie v intenzivní péči
- Thymom – možnosti diagnostiky
- Diosmin/hesperidin – spolupracující tandem nebo je diosmin klíčový a hesperidin jen neúčinnou příměsí?
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy