
Genome Sequencing and Comparative Genomics of the Broad Host-Range Pathogen AG8
The fungus Rhizoctonia solani is divided into several sub-species which cause disease in a range of plant species that includes most major agriculture, forestry and bioenergy species. This study focuses on sub-species AG8 which causes disease of cereals, canola and legumes, and compares its genome to other R. solani sub-species and a wide range of fungal and non-fungal species. R. solani is unusual in that it can possess more than one nucleus per cell. The multiple nuclei and sequence mutations between them made assembly of its genome challenging, and required novel techniques. We observed signs that DNA sequences originating from multiple nuclei in AG8 exhibit a high frequency of single nucleotide polymorphisms (SNPs) and more SNP diversity than most fungal populations. These SNP mutations also have similarities to repeat-induced point mutations (RIP). Moreover in AG8, RIP-like SNPs are not restricted to intergenic regions but are also widely observed in gene-coding regions. This is novel as RIP has previously only been reported in repetitive DNA of distantly-related fungi that have only a single nucleus per cell. We generated a list of 308 genes with similar properties to known plant-disease proteins, in which we found higher rates of non-synonymous mutations than normal.
Published in the journal:
. PLoS Genet 10(5): e32767. doi:10.1371/journal.pgen.1004281
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1004281
Summary
The fungus Rhizoctonia solani is divided into several sub-species which cause disease in a range of plant species that includes most major agriculture, forestry and bioenergy species. This study focuses on sub-species AG8 which causes disease of cereals, canola and legumes, and compares its genome to other R. solani sub-species and a wide range of fungal and non-fungal species. R. solani is unusual in that it can possess more than one nucleus per cell. The multiple nuclei and sequence mutations between them made assembly of its genome challenging, and required novel techniques. We observed signs that DNA sequences originating from multiple nuclei in AG8 exhibit a high frequency of single nucleotide polymorphisms (SNPs) and more SNP diversity than most fungal populations. These SNP mutations also have similarities to repeat-induced point mutations (RIP). Moreover in AG8, RIP-like SNPs are not restricted to intergenic regions but are also widely observed in gene-coding regions. This is novel as RIP has previously only been reported in repetitive DNA of distantly-related fungi that have only a single nucleus per cell. We generated a list of 308 genes with similar properties to known plant-disease proteins, in which we found higher rates of non-synonymous mutations than normal.
Introduction
Rhizoctonia solani (formerly, teleomorph: Thanetophorus cucumeris) is a globally-distributed, soil-borne fungal phytopathogen employing a necrotrophic lifestyle. Collectively, the host-range of the R. solani species spans numerous plant species vital to the agriculture, forestry and bioenergy industries, including but not limited to: wheat, rice, barley, canola, soybean, corn, potato and sugar beet [1]. Chemical control methods may not be feasible nor economical for the control of many soil-borne pathogens [2]. Hence, agronomic controls such as crop-rotation are heavily relied upon to fight this disease, though the polyphagous habit of some isolates can include commonly rotated crop species. For example, cereal and legume rotations are susceptible to AG8 [1], [3]; and corn, canola and soybean rotations are susceptible to AG1 and AG2 [4]–[5]. Susceptible crop species possess at best, low to moderate levels of genetic resistance which are of limited use to conventional breeding strategies [6]–[8]. The impact of R. solani has been observed to increase in incidence and severity with increased adoption of conservation (no-till) farming techniques [2].The combinations of these factors places R. solani as a significant threat to global food security and other agro-forestry industries.
The R. solani species complex is comprised of fourteen anastomosis groups (AGs), most of which are reproductively incompatible with each other and are numbered AG-1 through AG-13. The ‘bridging isolate’ AG-BI is the exception, being compatible with multiple AGs [1], [9]. Despite an apparently low level of phylogenetic divergence between AGs [10] they exhibit diverse phenotypic variation, particularly with respect to the host-ranges of phytopathogenic AGs (Supporting Table S1A). Less frequently, certain AGs have been observed to have a predominantly saprophytic or mycorrhizal life-cycle.
Our study presents a comprehensive genome assembly and functional analysis of R. solani AG8, causative agent of bare patch of wheat, barley and legume species [3], [11]–[12]. Of the AGs that infect wheat, AG8 is the most damaging. In Australia, the impact of R. solani on wheat and barley production is estimated upwards of $77 million per annum and bare patch also remains a major problem for the production of wheat and other crops in the US [13]. The host-range of the sequenced isolate WAC10335 (zymogram group ZG1-1 [14]) also extends to legume species of agricultural and scientific importance: Lupinus spp. (lupin) [15] and Medicago truncatula (barrel medic) [16], but not to the non-legume Arabidopsis [17]. As a basidiomycete, the plant pathogens most closely related to R. solani with genome sequences available are the biotrophic smuts [18]–[20], rusts [21]–[22] and the tree-pathogenic Moniliophthora spp. [23], which possess vastly different lifestyles. Thus, the information gained from R. solani is expected to be of importance in filling gaps in our knowledge of plant pathogen biology, which apart from rusts and smuts, is skewed towards the ascomycetes.
Significant genomic resources for other AGs of R. solani have also recently become publicly available, formerly being limited to EST libraries of AG1-IA [24] and AG4 [25]. The recent generation of whole genome sequences of R. solani AGs presents new opportunities for comparative genomics between R. solani anastomosis groups. The most comprehensive whole-genome study to date has been that of the rice pathogen AG1-IA [26] [GenBank: AFRT00000000]. The genome assembly of the closely related AG1-IB was published recently [27] [GenBank: CAOJ00000000], however full scaffold sequences were not in the public domain at the time of writing and thus AG1-IB data has not been used for synteny comparisons in this study. The mitochondrial genome sequence of the potato pathogen AG3 strain Rhs1AP and its comparison to that of AG1-IB has been published recently [28]. A draft nuclear genome for AG3 is also available (http://www.rsolani.org with kind permission from Cubeta et al.), however a nuclear gene dataset and genome survey have not yet been published [29].
R. solani AG8 exists as a multi-nuclear heterokaryon in which individual R. solani cells may carry multiple nuclei and copy number can vary between cells. An average of 8 nuclei per cell has previously been observed in AG8, but numbers commonly ranged from 6 to 15 [1]. While reduction of nuclear complexity via protoplast isolation has been reported for R. solani [30]–[32], we chose to assemble a representative haploid assembly of all AG8 nuclei in an agriculturally-relevant isolate and investigate mechanisms and type of sequence variations between nuclei in this largely asexual isolate. We report evidence of SNP-level diversity between heterokaryotic nuclei of a complex fungal genome, which has not previously featured extensively in genome studies of fungal phytopathogens. The heterozygosity between nuclei of AG8 compounded the complexity of its de novo genome assembly [available from GenBank: AVOZ00000000] and we also describe novel bioinformatic approaches used to overcome these challenges. This study also compares whole-genome synteny between R. solani anastomosis groups (AG8, AG1-1A and AG3) and uses comparative genomics techniques to highlight genes and functions unique to AG8 and AG1-1A. Predicted properties of AG8 proteins have been leveraged to generate a list of 308 ‘effector-like’ genes that may be related to plant-pathogenicity. These collective resources will be important for further experimentation in this pathosystem.
Results & Discussion
The haploid consensus genome assembly of R. solani AG8
The heterokaryotic nature of the R. solani genome posed considerable challenges for genome assembly. To overcome these challenges we developed a novel genome assembly pipeline (Figure 1). The assembly process, including software and parameters, is described in the Materials and Methods section with additional information in Supporting Text S1. Preliminary de novo assemblies exhibited high levels of sequence redundancy and heterozygosity across gene-encoding regions. We confirmed that multiple nuclei were present in variable numbers within cells of the sequenced isolate (Figure 2A). In order to reduce sequence redundancies caused by the assembly of heterozygous homeologs, the process used to assemble the AG8 genome included a step to merge haplotype contigs prior to scaffolding. This step was followed by generation of a haploid ‘majority consensus’ sequence from alignments of genomic sequence reads to merged scaffolds. However prior to this study, the extent of sequence variation between homeologous chromosomes originating from different nuclei was unknown. Alignment of genomic deep-sequencing reads to the genome assembly indicated an abundance of heterozygous SNP mutations throughout the AG8 assembly (Figure 2B). As many as 74% of heterozygous SNP alleles were transition mutations between cytosine and thymine (or their complementary bases guanine and adenine) (Figure 2C, Supporting Table S2A). Nucleotides flanking these C→T ‘hypermutations’ exhibited a moderate bias of approximately 40% for a G at the 3′ base (i.e. CpG→TpG) (Supporting Table S2B). These cytosine and CpG hypermutations were widespread across the AG8 genome and occurred within protein-coding genes and repetitive DNA regions at similar levels (Figure 2D), with only a slight reduction in CpG frequency in genes relative to repeats. One of the consequences of C→T mutation is the introduction of stop codons into protein-coding open-reading frames (ORFs) [33]. We reason that it is possible for ORFs to be inactivated by nonsense mutations in the majority of nuclei, yet still produce functionally active, full length proteins from a low number of non-mutated nuclei in R. solani AG8. Thus the assembly process also included a step which reverted heterozygous mutations between C and T to cytosine, regardless of allele frequencies. The final R. solani AG8 draft assembly comprises 861 scaffolds, has a total length of 39.8 Mbp which is consistent with previous haploid cytogenetic estimates of 37 to 46 Mbp [34], an N50 of 65 and an N50 length of 160.5 kbp (Table 1).


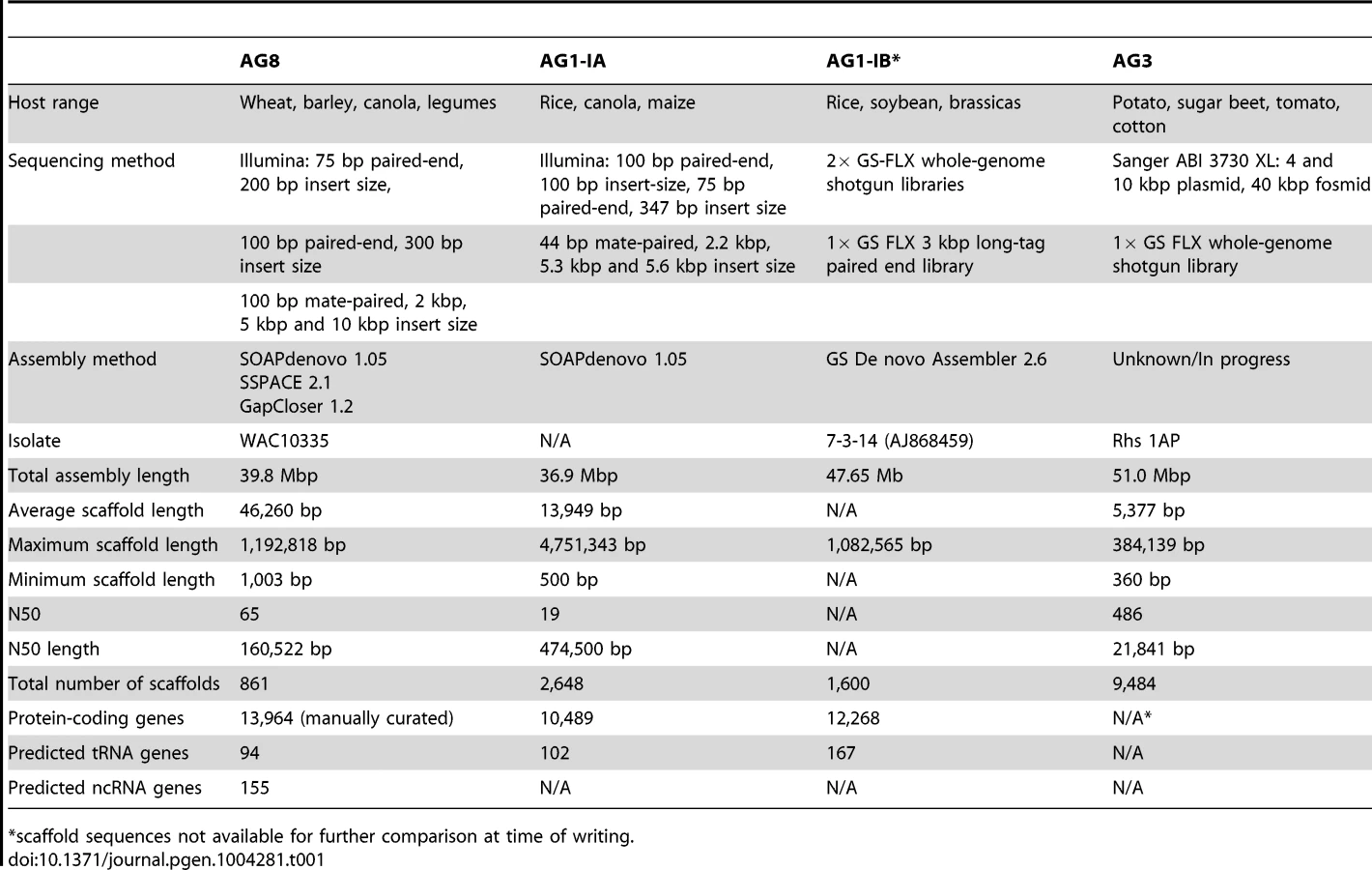
The AG8 genome assembly statistics compared favorably with those of other R. solani isolates AG1-1A, AG1-1B and AG3 as shown in Table 1. Sequence comparisons between the whole genome assemblies of R. solani AG8, AG1-1A and AG3 exhibited widespread co-linearity or macrosynteny [35] (Figure 3, Supporting Table S3). No conclusive evidence for dispensable chromosomes, as reported for F. oxysporum [36], was observed.

A single scaffold (Scaffold_77) of ∼140 kbp in length was predicted to represent the mitochondrial genome. The ends of the mitochondrial scaffold sequence were confirmed to be physically joined in a circular configuration by PCR (Supporting Text S2). The mitochondrial scaffold contained the expected set of fungal mitochondrial genes (atp6, cytb, cox1-3, nad1-5 & nad4L, rps5, rns & rnl) and was abundant with LAGLIDADG and GIY-YIG intronic endonucleases. This is consistent with recent reports for the mitochondrial genomes of AG3 and AG1-IB, which are of similarly large sizes (235.8 kbp and 162.8 kbp respectively) and possess high abundances of endonucleases [28].
Within the nuclear genome, repetitive DNA sequences (Supporting Table S4A) represented just over 10% of its total length. Gypsy retrotransposons were the most abundant repeat type and represented 4% of the nuclear genome. Protein-coding gene-based tri-nucleotide simple sequence repeats, WD40-like and tetratrichopeptide repeats, represented approximately 1%. Comparing the repetitive content of AG8 with available repeat data for AG1-1A, we observed more repetitive DNA in the assembly of AG8 (10.03% of the assembly) compared to that of AG1-1A (5.27%) [26]. It should be noted that critical differences in assembly, de novo repeat prediction and repeat classification methods may limit the comparability of these two datasets, however the proportions of the most dominant repetitive elements was strikingly similar. The most dominant transposable elements in both AG8 and AG1-1A were LTR retrotransposons: the most common being the Gypsy/Dirs1 family at 3.98% and 3.43% respectively; followed by the Ty1/Copia family at 0.14% and 0.60% respectively. This pattern of Gypsy being more numerous than Copia retroelements, appears to be typical of most fungal genomes [37]. Non-coding RNA (ncRNA) genes were predicted in silico (Supporting Table S5A), which overall made up less than 0.007% (26.5 kbp) of the total genome length.
Manual curation of 13,964 R. solani AG8 protein-coding genes with RNA-seq support, enables prediction of proteome and secretome properties
To enable discovery and accurate annotation of protein-coding genes present in the AG8 assembly, particularly those expressed in the presence of plant tissues, three high-coverage Illumina RNA-seq libraries were aligned to the genome to delineate gene exon boundaries. To obtain transcript data for as many genes as possible, the libraries included one library of AG8 undergoing vegetative growth in culture and two “infection-mimicking” libraries. These libraries were derived from AG8 grown on water agar containing wheat (Triticum aestivum) or Medicago truncatula seedlings separated by a permeable nitrocellulose membrane. This enabled collection of fungal tissue whilst reducing plant tissue contamination to negligible amounts. Alignment of RNA-seq data and proteins from related fungal species and pathogenicity gene databases were combined with in silico gene predictions to automatically predict gene structure annotations, which were then manually curated.
The density of gene-coding regions was relatively even throughout the assembled genomic scaffolds (Figure 2Eii), with reduced density at some scaffold termini with high levels of repeats (Figure 2Eiii). A total of 13,964 protein-coding AG8 genes that can serve as a reference for R. solani comparative genomics were predicted after RNA-seq-assisted manual gene annotation. Of these, 8,449 proteins had a BLASTP match to the NCBI NR protein database (Supporting Figure S1, Supporting Table S6). The taxonomic distribution of lowest-common ancestor taxa for these BLASTP matches indicated wide conservation of 83% (7016/8449) of R. solani AG8 with fungal proteins, 52.5% (4436/8449) specifically conserved within the Basidiomycota (Supporting Figure S1) and 17.9% conserved within the class Agaricomycetes. The extracellular secreted component of these proteins was predicted using a combination of SignalP [38], WolfPsort [39] and Phobius [40] (Figure 4). A total of 1,959 proteins (14.0% of all proteins) were predicted to be secreted by one or more methods and 608 (4.4%) were predicted to be secreted by all three methods. For comparative purposes, SignalP predictions were applied to R. solani AG8 and across 86 fungal species (Supporting Table S7). There were 911 secreted proteins predicted by SignalP for AG8, which was similar to the numbers predicted for closely-related plant-pathogenic species of the class Agaricomycetes. The secretome counts across biotrophic Basidiomycetes of other classes were relatively variable, e.g. Puccinia striformis (1,264), P. graminis f. sp. tritici (2,012) and Ustilago maydis (595). However AG8 was within a similar range to the average predicted secretome count across all fungi (1,052), which was predominantly comprised of necrotrophs.

To surmise the biological processes important to R. solani AG8 in the infection process, we predicted the functions of its 13,964 genes by comparison to the CAZy (Carbohydrate-Active enZyme) and Pfam (Protein family) databases. In total, we assigned CAZy annotations to 1,137 genes (Supporting Table S8B,C) and Pfam annotations to 6,099 genes (44.5%) (Supporting Table S9A).
Analysis of CAZymes present in the R. solani AG8 genome (Figure 5) revealed a dual bias for the degradation of the structures of plant cells and modification of the fungal cell wall for growth or protection from host-defences (Supporting Table S8C). The most abundant CAZy families are described here. The most prevalent glycoside hydrolase (GH) CAZyme class (GH18) represented chitinases, followed in frequency by classes representing cellulases (GH5), polygalacturonases (GH28) and beta-glucanases (GH16), which degrade major components of plant cell walls. The most abundant glycosyltransferase (GT) classes were strongly geared towards cellulose (GT2, GT41), hemicellulose (GT77, GT4, GT34) and chitin (GT2) degradation. The most common carbohydrate esterase (CE) class contained choline esterases (CE10). Polysaccharide lyase (PL) CAZymes were strongly biased towards pectin-degradation, with the two most dominant classes (PL1 and PL3) both representing pectate lyases. The three most abundant carbohydrate-binding (CBM) class CAZymes were lectin-like proteins. Two of these (CBM13 and CBM57) are predicted to bind cellulose and hemicelluloses and include ricinB-like lectins. The third (CBM18) contains sialic-acid-binding lectins, which may play a role in protection from plant host-defenses by ‘shielding’ sugars protruding from the fungal cell wall [41]. The fourth most frequent CBM class (CBM1) binds chitin and cellulose and appears to be conserved exclusively within fungal species.

Pfam domains in R. solani AG8 were compared to Pfam annotations assigned to a panel of 50 pathogenic and non-pathogenic fungal species (obtained from the JGI Integrated Microbial Genomes database) (Supporting Figure S2, Supporting Table S9A) [42]. R. solani AG8 exhibited high abundance of tyrosine protein kinase signalling, membrane transport, protein-protein binding, reduction-oxidation, DNA methylation and a bias among cell-wall degrading enzymes towards pectin and peptidase degradation. Pfam domains with protein-protein binding functions were dominated by various classes of tetratrichopeptide repeats, but also included other domains involved in protein binding interactions: (WD40-like) PD40 beta-propeller [Pfam: PF07676]; Ankyrin [Pfam: PF13606] and leucine-rich repeats [Pfam: PF00560]. The most abundant peptidase domain was the CHAT (Caspsase HetF-Associated with TPRs) domain [Pfam: PF12770] which may be involved in programmed cell death. In summary, R. solani AG8 possesses a number of gene families whose members have a broad range of potential biological roles, for example those encoding caspases or protein-binding functions. Further study would be required to determine their relevance to plant pathogenicity or other lifestyle characteristics. These findings do however indicate that R. solani AG8 possesses a large number of carbohydrate-binding lectins of unknown function as well as a battery of CAZymes suitable for consumption of carbohydrates commonly found in cereal hosts, but also is geared towards the degradation of pectin.
Comparison of functional annotations between AG8 and AG1-IA suggest molecular functions of importance in host-specific plant-pathogen interactions
Publicly-available protein data for AG1-IA [26] was also used to generate functional annotations for AG1-IA. Statistical comparisons between functions predicted in AG8 and AG1-IA were performed using Fisher's exact test (p≤0.05) (Supporting Table S10A). R. solani AG8 and AG1-IA primarily infect two different hosts - wheat and rice respectively. Differences between them in their relative abundances of functionally-annotated genes may reveal important differences in their infection strategies. Overall, fewer Pfam domains were found to be significantly higher in AG1-IA than in AG8. In AG1-IA (Supporting Table S10B), the Pfams that were significantly more abundant and may be related to pathogenicity included several types of transmembrane transporter domain and formin-like proteins that may be involved in cytokinesis. Many more functions were found to be increased in AG8 relative to AG1-IA (Supporting Table S10C), however most of these were of too broad or unknown function to infer their biological roles. Nevertheless, several functions stood out as potentially important for plant pathogenicity in AG8, including CAZymes, peptidases, membrane transporters, transcription factors and toxin-like proteins. Peptidases abundant in AG8 included the CHAT and C14 domain caspases as well as fungalysin-like peptidases. The CAZyme functions that were significantly more numerous in AG8 were predominantly glycosyl-hydrolases (polygalacturonases, β-galactosidases), pectate lyases and carbohydrate binding proteins (ricin-like and jacalin lectins and fungal-specific CBM1 proteins). Fungal pathogens of dicots generally possess higher numbers of pectin-degrading enzymes than monocot pathogens [43]. Though an important pathogen of monocot cereals, most notably wheat, the sequenced isolate of R. solani AG8 was isolated from the dicot lupin and is also an important pathogen of other leguminous dicots. The abundance of pectate lyases in AG8 relative to AG1-IA is likely to reflect the broad host range of the sequenced AG8 isolate.
Interestingly, AG8 had more members of two Pfams similar to ricinB lectins [44] and delta endotoxins [45], highly toxic proteins commonly associated with defence against insect predators which have been prioritised for further study. In contrast to AG1-IA which had none, AG8 possessed 3 delta-endotoxin-like proteins (RSAG8_06697, RSAG8_07821 and RSAG8_07820) with the Pfam domain Bac_thur_toxin [Pfam: PF01338]. This domain was originally defined based on the insecticidal delta endotoxins of Bacillus thuringiensis. Pfam matches and orthology analysis suggested the presence of orthologous delta endotoxin-like proteins in other phytopathogenic species including Fusarium graminearum (Fusarium head blight of wheat and barley) and the bacteria Dickeya dadantii (syn. Erwinia chrysanthemi, soft-rot, wilt and blight on a range of plant hosts and septicaemia of pea aphid) [46] (Supporting Table S9A, Supporting Table S11). Whether these ricinB and delta-endotoxin homologs confer an advantage against competitors or predators or may instead be toxic to the plant host remains to be determined.
Prediction of 308 R. solani AG8 plant-pathogenicity gene candidates
Effector proteins have been observed to be secreted by several microbial pathogens [47] and cause disease on their respective hosts. A set of characteristics common to plant pathogenicity effectors from fungi that would allow reliable bioinformatic predictions has not yet been accurately defined. However experimentally validated effectors tend to be low molecular weight, secreted, cysteine-rich proteins which may contain certain conserved amino-acid motifs near the N-terminus [47]–[48] (Supporting Table S12). Effector-like proteins were predicted in AG8, requiring: complete annotation from translation start to stop with <3 consecutive unknown (‘X’) amino acids; predicted molecular weight ≤30 kDa; predicted as secreted with 0–1 predicted transmembrane domains; and with ≥4 cysteine residues. A total of 308 AG8 proteins matched all of these criteria. These candidates were searched for known motifs previously associated with plant pathogenicity, however the occurrence of these motif matches was not significant relative to the complete protein dataset. As an example the RxLR-like motif (Kale et al., 2011), though found in 73% of the predicted effector candidates, was also found in 77% of the whole R. solani AG8 proteome (Supporting Table S13) indicating this permissive motif may not be useful for effector candidate prediction in R. solani AG8. We were also unable to identify any novel N-terminal-associated motifs that were highly conserved among these 308 proteins (Supporting Text S3). However, we observed the ratio of non-synonymous to synonymous mutations (dN/dS) within these 308 candidate genes to be 0.97 compared to 0.77 across all genes. Our understanding of the roles of these 308 effector candidates will benefit from the addition of further experimental data, resulting in a more succinct list of candidates with a potential direct role in disease on one or more of the many plant hosts of R. solani AG8. Unfortunately, no method for the stable transformation of R. solani AG8 is presently available and thus functional testing of candidate pathogenicity genes will be challenging.
To gain further support for an association with pathogenicity, approximately 10% (29) of the 308 predicted ‘effector-like’ genes were randomly selected and their mRNA expression relative to a set of 7 constitutively expressed genes was compared between R. solani AG8 sampled at 7 days post-infection of wheat and 7 day-old AG8 mycelia grown on media. Of these 29 genes, 25 (85%) had a positive fold-change and 17 had a significantly higher relative expression in-planta (Student's t-test; p≤0.05, log2 fold change ≥1) (Supporting Table S14B). This dataset highlights several plant-pathogenicity candidates, but other genes also important for pathogenicity may not be changing in abundance during infection relative to in-vitro growth.
Widespread CpG-biased hypermutations may be similar to repeat-induced point mutations (RIP) observed in mononuclear species
Repeat-induced point mutations (RIP) are fungal-specific SNP mutations previously reported to be restricted to the filamentous Ascomycota (Pezizomycotina) [49]. RIP in the Pezizomycotina involves transition mutations converting cytosine to thymine, with a moderate bias for CpA dinucleotides [49]. Other features of RIP include targeted mutation of repetitive DNA, with single-copy DNA regions being largely unaffected. An important exception to this is where RIP mutations ‘leak’ into single-copy DNA regions from neighbouring repetitive DNA which occurs more frequently closer to repeats [50].
The small number of studies looking for RIP-like mutations in the Basidiomycota do not exhibit the characteristic CpA mutation bias observed in the Pezizomycotina [49], however two studies have reported a CpG dinucleotide bias between repetitive DNA sequences within the Basidiomycota and a TpCpG trinucleotide bias specific to the subphylum Pucciniomycotina [51]–[52]. As an Agaricomycete, we expect R. solani to exhibit a bias towards CpG but not TpCpG. However, it should also be noted that hypermutations of CpG may also be caused by widely conserved processes involving the methylation of cytosine to 5-methylcytosine (5mC) and subsequent deamination which converts 5mC to thymine [53]. Importantly, conversion of cytosine to thyimine via methylation and deamination does not actively target repetitive DNA or ‘leak’ in the same manner as RIP.
Analysis of nucleotides immediately flanking heterozygous C↔T SNP sites in AG8 exhibited a CpG dinucleotide bias consistent with previous observations of ‘RIP-like’ cytosine hypermutations in the Basidiomycota [52] (Figure 2D). The distribution of these RIP-like mutations in AG8 was observed to occur across repetitive and gene-encoding regions alike at a relatively constant ratio versus non-RIP-like mutations, where heterozygous C↔T alleles comprised ∼70–80% of all SNP mutations (Figure 2Eiv-v) and in turn CpG dinucleotides comprised ∼40–50% of heterozygous C↔T alleles. In mononuclear fungal genomes, RIP has previously only been observed to act upon repetitive DNA or to ‘leak’ into adjacent non-repetitive sequences [50]. Due to the novel genome assembly process for AG8 which involved merging of redundant haplotypes, a survey of SNP mutations in its annotated repetitive DNA would likely lead to incorrectly inflated counts of RIP-like mutations. Therefore we instead looked at the frequency of CpG↔TpG mutations versus their distance from the nearest repeat, which indicated that CpG mutations were more frequent closer to repeats (Figure 6). Furthermore, although the ratios of (C↔T/all SNPs) and (CpG↔TpG/C↔T) were relatively similar between genes and other regions of the genome, the frequency of mutations in gene regions were lower than in the genome as a whole, suggesting strong selection pressures to retain protein function. The ratio of CpG/CpH (where H = not G) was slightly lower in repeats (0.3) than in genes (0.4) (Table 2) and we speculate that this likely to be due to complete (i.e. homozygous) conversion of C→T occasionally occuring across all copies of a repeat, as they are under no selection pressure to retain their pre-RIP sequences. Thus there would be fewer sites that can be detected as heterozygous SNPs by aligning genomic reads to the genome assembly.

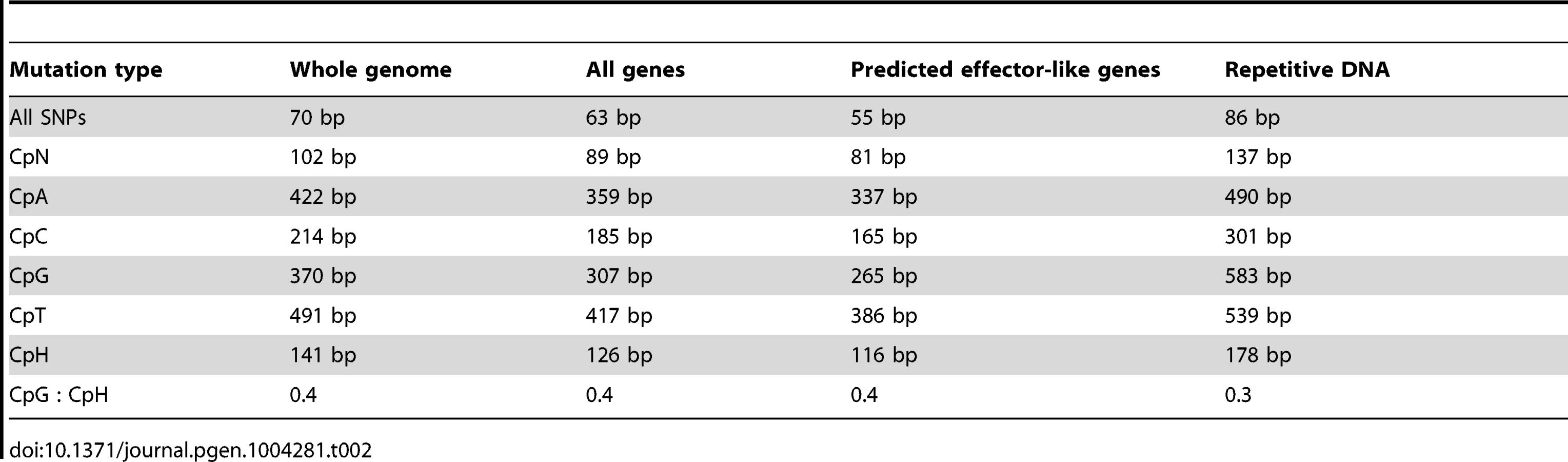
Regardless of whether the underlying process is similar to RIP or not, CpG-biased hypermutation is likely to play an important role in the evolution of the AG8 genome. RIP has been recently proposed to have the potential to randomly introduce nonsense mutations, converting longer secreted proteins into small, secreted proteins thus making them gradually more effector-like [54]. Stop-codon frequency across the 12,771 annotated AG8 genes possessing stop codons is highest for TGA (40%) compared to TAA (31%) and TAG (29%). As TGA stop codons would be the primary nonsense product of CpG-biased hypermutation, similar evolutionary processes may also occur in AG8. Furthermore, the presence of multiple nuclei in AG8 could potentially compensate for loss of gene function due to hypermutation in one or more nuclei, allowing for a higher tolerance for the accumulation of mutations in gene-coding regions. Analysis of total SNP, and CpN dinucleotide frequencies (expressed in Table 2 as average distance in bp between mutations), showed that a SNP mutation occurred on average every 70 bp, cytosine hypermutations occurred every 89 bp and that there was a 40% bias towards CpG mutations occurring every 307 bp. Within the 308 predicted ‘effector-like’ genes, SNP mutations occurred on average every 55 bp, cytosine hypermutations every 81 bp and CpG mutations occurred every 265 bp. However, the ratios of (C↔T/all SNPs) and (CpG↔TpG/C↔T) were not significantly different between the complete set of 13,964 AG8 genes and the 308 effector-like genes. Interestingly, despite apparently similar mutation ratios, the ratio of non-synonymous to synonymous SNP mutations (dN/dS) was 0.97 in ‘effector-like’ candidates compared to 0.77 across all genes. This may suggest that the increased mutation rate conferred by CpG-biased hypermutation is advantageous for accelerating the adaptation of pathogenicity genes which, if being actively counter-acted by plant defences, are likely to be under diversifying selection.
Comparison of SNP-level diversity within the R. solani AG8 whole-genome with diversity in other species
The density of heterozygous SNP mutations within AG8 was compared to SNP densities between the genome assemblies of AG8 and AG1-IA, AG1-IB and AG3 (Table 3). SNP density in AG8 was highest within intronic regions (19.6 SNPs/kbp), moderate in coding exons and genes (14.5–15.9 SNPs/kbp) and lowest in intergenic regions (11.5 SNPs/kbp). Comparisons of SNP mutations between AG8 and alternate AGs exhibited an approximately ten-fold increase in SNP density compared to the rate of heterozygous SNPs within AG8. The corresponding values within for comparisons between AG8 and AG1-IA ranged from 162.8–228.2 SNPs/kbp, AG1-IA from 141.6–200.3 SNPs/kbp and AG3 from 98.5–145.3 SNPs/kbp. We note however that in these comparisons between the genome assemblies of AG8 and other AGs, it was not possible to ascertain whether these SNPs (or homologous bases) were homozygous or heterozygous in the alternate AG. Nevertheless a higher SNP density between the AG8 genome and those of the other three AGs, relative to heterozygous AG8 SNPs, was consistent in all three comparisons.

Comparisons between individual genomes and fungal population genetics studies were also used to place the SNP diversity within R. solani AG8 into a wider context. Similar to AG8, the Basidiomycete stripe rust fungus Puccinia striformis is heterokaryotic but exhibits a lower SNP density within its genome assembly of 5.98 SNPs/kbp [21]. It may be significant that P. striformis is binucleate and therefore only possesses 2 nuclei per cell as opposed to the 6–15 nuclei that have been observed within cells of R. solani AG8 [1]. Similarly, SNP variation across a population of shiitake mushroom (Lentinula edodes) was reported to be 4.6 SNPs/kbp (186,0789 SNPs in 40.2 Mbp) [55]. In barley powdery mildew (Blumeria graminis), the SNP rate observed between pairs of isolates was lower at 1 SNP/kbp [56]. Across isolates of the multinucleate endomycorrhizal Glomeromycete Rhizophagus irregularis [57] and the beetle-symbiont Leptographium longiclavatum [58], even lower SNP densities of 0.2 SNPs/kbp (28,872 SNPs in 140.9 Mb) and 0.6 SNPs/kbp (17,266 in 28.9 Mbp) respectively, were observed. In contrast, a population study of the multinucleate human pathogens Coccidioides immitis and C. posadasii reported a rate of 23.7 SNPs/kbp relative to the C. immitus RS reference genome assembly (687,250 SNPs in 28.95 Mb) [59], which though slightly higher is within a similar range to R. solani AG8 (Table 3). In conclusion, the SNP diversity in R. solani AG8 appears to be higher than that observed thus far within individual isolates of binucleate rusts, between isolates of the same pathogenic species and across non-pathogen populations. Furthermore, diversity within R. solani AG8 is comparable to a population of another multinucleate pathogen (C. immitus) and much higher than that observed within a population of a multinucleate non-pathogen (R. irregularis). We speculate that the combination of multinuclearity and selection pressures relating to pathogenicity may be driving the accumulation of widespread heterozygous SNP diversity in R. solani AG8.
Conclusions
In this study, we present a novel bioinformatics pipeline for the accurate and comprehensive assembly of a complex fungal genome, the heterozygous and multinucleate pathogen Rhizoctonia solani AG8 (Figure 1). The combination of genome and transcriptome sequencing allowed for data-driven gene prediction and comparative genomics with other publically available genomes of alternate anastomosis groups and other fungal species. Using a combination of novel genome assembly methods, RNA-seq, manual gene curation and comparative genomic techniques, a list of 308 ‘effector-like’ plant-pathogenicity gene candidates has been predicted. Analysis of mRNA expression for a subset of candidate pathogenicity genes during infection of wheat has highlighted several candidates for further study. Additionally, comparisons to available data for alternate AGs of R. solani have highlighted important differences, which may be related to differing host ranges, host tissue preference and environmental stress tolerance. The resources presented here should provide powerful tools for the identification of host-specialised mechanisms for fungal-plant interactions and pathogenicity for this important group of fungal pathogens.
CpG-biased hypermutations were observed between nuclei of AG8, within genes and repeat sequences alike and have some similarities with repeat-induced point mutation (RIP). Previous observations of RIP in haploid fungal genomes have only reported its activity upon repetitive sequences [49], [52] or non-repetitive regions within a finite distance of a repeat [50]. Although we observed hypermutation within genes, intriguingly these mutations were more numerous with increasing proximity to repeats, suggesting that repeats are mutated more frequently than genes and that a process similar to ‘RIP-leakage’ may occur. Furthermore, the molecular mechanisms of RIP have not yet been fully characterised [60] and the consequences of combining RIP-like hypermutation and multinuclearity in R. solani are unknown. In the basidiomycete human pathogen Cryptococcus neoformans, increases in ploidy and the accumulation of mutations have been implicated as mechanisms for its adaptation to immune - and drug-related selection pressures [61]. Also of note is that across isolates of the human-pathogenic and multinucleate ascomycete Coccidiodies immitus, higher relative frequencies of repeat-associated CpG mutation have also been observed [59] (unusual for species of the Pezizomycotina which typically exhibit a bias towards mutation of CpA [49]). We speculate that RIP-like SNP mutations accumulating in multiple nuclei may similarly be a means by which R. solani is also able to rapidly generate allelic diversity despite being predominantly clonally propagated [1]. Loss-of-heterozygosity and copy-number variation analyses to confirm this hypothesis would require further study using a sequencing platform which can produce longer read lengths and higher base-call accuracies than those used in this study. However, if this is the case, this mechanism may be a factor contributing to the relatively mild effectiveness of fungicide treatment against this pathogen [62] and its adaptation to a broad range of plant hosts.
Materials and Methods
Isolation and sequencing of R. solani AG8
R. solani AG8 isolate WAC10335 was isolated from lupin and provided by the Department of Agriculture and Food of Western Australia (DAFWA). Anastomosis group was confirmed by ribosomal ITS sequences and host-range was confirmed by inoculation assays on wheat, lupin and Medicago truncatula [3]. R. solani does not readily produce sexual or asexual spores thus single spore isolation was not possible, therefore a single rapidly growing hyphal tip was excised from a colony growing on PDA and transferred to water agar containing 250 µg/ml cefotaxime. Pathogenicity of the resulting culture was confirmed as equivalent to the original. A pure in vitro culture of R. solani was produced by incubation in PDB at 25°C with gentle shaking for 7 days. Hyphae were filtered from the culture through sterile Miracloth and rinsed with sterile water. DNA was purified by grinding hyphal tissue in liquid nitrogen and suspension in DNA extraction buffer (2% (w/v) CTAB, 1.4 M NaCl, 0.2% (v/v) β-mercaptoethanol, 20 mM EDTA and 100 mM Tris-HCl) and mixing at 60°C. Following two rounds of chloroform/isoamylalcohol extraction, the aqueous supernatant was treated with RNase I (Invitrogen) at 20 µg/ml. The DNA was purified through an additional two rounds of chloroform/isoamylalcohol extraction and precipitated by adding 0.1 volumes of 3M NaOAc (pH 5.2) and 0.6 volumes isopropanol. The resulting DNA pellet was resuspended in 10 mM Tris-HCl (pH 8.0) buffer and quantitated by Qubit (Invitrogen) and BioAnalyser prior to sequencing.
Next-generation sequencing and pre-assembly data quality control
Two Illumina paired-end libraries of genomic DNA were sequenced, with 75 and 100 bp read lengths and median insert lengths of 250 bp and 300 bp respectively. Three Illumina genomic mate-pair libraries with insert lengths of 2 kbp, 5 kbp and 10 kbp were also obtained. Paired-end libraries were combined and trimmed for sequencing adapter/primer sequences, low-quality (<Q30), and low-complexity sequences via CutAdapt v1.1 [63] filtered for adapter sequences from the Truseq RNA and DNA sample preparation kits versions 1 and 2. Pairs with one or more reads ≤50 bp after trimming were discarded. Where possible, overlapping 3′ ends between pairs were merged into long singleton reads via FLASH v 1.2.2 [64]. FLASH was also applied to the mate-paired libraries, to remove paired-end contamination of incorrect insert length and pair orientation which would complicate genome assembly (Supporting Text S1).
For the purpose of gene annotation, Illumina paired-end libraries of 100 bp read lengths were obtained from 3 mRNA libraries derived from AG8 grown under: vegetative conditions (7 days at 25°C in PDB with gentle shaking) (non-infection) and; Medicago or wheat infection-mimicking conditions. Under infection-mimicking conditions, AG8 was cultured on a film of nitrocellulose overlaid on water agar containing young sterilised Medicago truncatula or wheat seedlings. After seven days incubation at 25°C the film and hyphae were removed, ensuring negligible plant contamination in subsequent RNA extractions with TRIzol (Sigma-Aldrich, St. Louis, MO). Two sequencing libraries were generated per mRNA library, with 200 bp and 500 bp insert sizes. Transcript libraries were trimmed for contaminant sequences via Cutadapt v1.1 as per genomic reads.
Genome assembly
Complex genome structure caused by heterozygosity and multinuclearity prevented the use of commonly employed de novo assembly methods. To this end, a novel pipeline was developed for AG8 (Supporting Text S1). Paired-end libraries were assembled with SOAPdenovo v1.0.5 (k-mer length = 61) [65]. This assembly was scaffolded with SSPACE 2.0 using the parameters (end extension, min size 500 bp) [66] and subject to 5 rounds of Gapcloser2 [65] using paired-end and 3′ end merged single-end reads. Mate-paired reads were used for scaffolding but excluded from gap-closing to avoid introducing inversion errors (Supporting Text S1). Haplotype redundancy was reduced using HaploMerger v20111230 [67] (batchD: filterAli = 0; minlength = 10 bp; maxInternal = 10000000; mincoverage = 0). Tandem duplication assembly errors (common to polyploid assemblies) were corrected by a twofold approach (Supporting Text S1). The first method involved intra-scaffold re-assembly between rounds of scaffolding and gap-closing, where gaps were broken and tested for overlap via CAP3 [68]. The second method involved self-alignment via BLASTN [69], applied after scaffolding, gap-closing and N-breakage rounds had completed. Alignments occurring in tandem on the same sequence were identified, and the sequences of the second repeat plus the intermediate region were removed from the assembly if repeats were ≤500 bp apart or ≥30% polyN in intermediate region. Introduction of errors by these processes was corrected by re-alignment of raw genome reads with bowtie2 v 2.0.5 [70] followed by local-realignment at indels, variant-calling and variant-consensus generation via GATK v1.6.11 [71]. Variant Call Format (VCF v4.0) tables of SNP and indel variation between the paired-end, 3′-end merged (long single-end reads), 2 kbp mate-paired, 5 kbp mate-paired and 10 kbp mate-paired sequence libraries relative to the genome assembly sequences, were merged with VCFtools v0.1.6 [72] where variants agreed between at least 2 out of the 5 libraries. The most frequent alleles in the merged VCF were incorporated into the consensus sequence of the final assembly, with the exception of sites where cytosine (C) to thymine (T) (reverse complement: G to A) polymorphisms were observed at which the assembly was reverted to the C (or G) allele regardless of allelic frequency. The genomic distribution of SNP mutations was calculated using BEDTools v0.1.7 [73].
Genome assembly sequences of AG8, AG1-IA and AG3 were compared using MUMmer 3.0 [74] using both nucmer and promer (parameters: –maxmatch). Summary statistics were derived from coordinate outputs.
Repetitive DNA prediction, annotation and curation
Repetitive sequences were predicted via RepeatScout v1.0.5 [75], requiring consensus sequences ≥50 bp and ≥5 copies. Full-length repeats were reconstructed from RepeatScout outputs with CAP3 (v10.15.7, -h100 -p80 -z1) [68], manually curated and mapped to the genome assembly via RepeatMasker v3.2.9 (parameters: -e crossmatch -s) [76]. Repeat types were characterised using a combination of BLASTn vs NCBI Nucleotide, BLASTx vs NCBI Protein [69], CENSOR vs REPBASE v17.11 [77] and TEClass [78]. Repeat regions were also predicted with TransposonPSI v08222010 [79] and RepeatMasker vs REPBASE v17.11 (species = “Eukaryota”) [80]. All repeat data, excluding repeats corresponding to protein-coding genes (Supporting Table S4B), were used as negative support for gene annotation.
Transcriptome alignment and assembly
Exon splice-junctions were determined by aligning six RNA-seq libraries to the AG8 assembly via TopHat 2.0.4 (minimum intron size 20 bp, maximum intron 5000 bp, no coverage search, 2 splice mismatches, microexon search enabled, very sensitive, 20 read mismatches, 3 segment mismatches, max insertion length 12 bp, max deletion length 12 bp, report discordant and secondary alignments) [81]. Transcriptome de novo assembly was performed via Trinity r2012-03-17 (k-mer trimming with JellyFish 1.1.4, jaccard clipping, minimum contig length 150 bp, min k-mer coverage 5×, minimum glue 5, minimum percent read mapping 70%) [82]–[83]. Combined and individual library-specific assemblies were used for manual gene annotation.
Gene prediction, annotation and curation
The library-specific and the combined transcriptome assemblies were used to determine exon structure using PASA r2012-06-25 (minimum percent aligned 75%, maximum intron length 5000 bp). The output of PASA was passed to the EvidenceModeller r2012-06-25 auto-annotation pipeline [84], which also incorporated the following supporting data: splice-junctions determined from RNA-seq alignment to the genome assembly via Tophat 2.0.5; in silico gene prediction via GeneMark-ES v3.2.3 [85]; ESTs and proteins of previously sequenced fungi and PHIbase v3.4 [86] aligned to the genome assembly with AAT r03052011 [87]; predicted repetitive DNA (see above) and; non-coding locus predictions via tRNAscan-SE v1.23 (genomic, COVE only) [88] and Infernal v1.0 [89]. Gene annotations were evaluated by EvidenceModeller, visualised in Apollo v1.11.6 [90] and manually curated. Predicted protein translations were compared to the NCBI NR Protein database by BLASTP [69] (BLAST v2.2.26, e-value≤1e−3, top 20 hits) and the taxonomic distribution of their corresponding lowest-common ancestor taxa was summarised with MEGAN5 (LCA parameters: minimum support 1, minimum score 40, max expected 1e−3, top percentage 100) [91].
Functional annotation
Conserved protein domains in AG8 and AG1-IA were predicted with HMMER 3.0 [92] versus Pfam(A) v26.0 [88] with gathering cutoffs. Carbohydrate-active enzymes (CAZymes) were predicted with CAT v1.8 [93]. Multiple alignments of the most abundant CAZyme families were generated with MAFFT L-INS-i v7.130 [94] (Supporting Table S15). Comparison of Pfams were performed between R. solani AG8 and other sequenced fungi from JGI IMG v4 [42]. Orthology comparisons between AG8 predicted proteins and protein datasets from 197 fungal, oomycete, prokaryotic, insect and nematode species and included a range of pathogens with different host ranges and non-pathogens (Supporting Table S11) was performed via ProteinOrtho v4.26 (BLASTP v2.2.26, E-value = 1e-05, alg.-conn. = 0.1, coverage = 0.5, percent_identity = 25, adaptive_similarity = 0.95, inc_pairs = 1, inc_singles = 1, selfblast = 1, unambiguous = 0) [95]. Predicted secretome comparisons were performed using SignalP 4.1 [38] between R. solani AG8 and 86 other fungi (Supporting Table S7).
Candidate ‘effector-like’ pathogenicity genes were classified by: complete annotation with translation start and stop codons and ≤3 consecutive unknown ‘X’ amino acids; predicted to be secreted by at least one method; 0–1 predicted transmembrane domains (single domains can be mis-predicted within secretion signal peptides); predicted molecular weight ≤30 kDa; and ≥4 cysteine amino acids. Molecular weights and amino-acid compositions were predicted with Bio::Tools::SeqStats (BioPerl) [96]. Sub-cellular localisation, secretion status and transmembrane domains were predicted with Phobius v1.01 [40], SignalP v4.1 [38] and WolfPSort v0.2 [39]. Matches to motifs previously associated with plant pathogenicity effectors (Supporting Table S13) were searched with PREG [97] (Supporting Table S13). We also attempted to identify high frequency novel motifs within the ‘effector-like’ candidates with MEME v4.9.1 (model = ANR, minsites = 2, maxsites = 300, nmotifs = 50, minwidth = 5, maxwidth = 50) [98] (Supporting Text S3).
Heterozygous SNP mutations derived from genomic read alignment to the final genome assembly, as described above, within all genes and predicted ‘effector-like’ genes were tested for: 1) stop-codon bias; 2) gene structure location bias with SNPeff [99]; 3) non-synonymous vs synonymous SNP ratio (dN/dS) via SNPeff [99]; 4) frequency and density via BEDtools coverageBed [73].
Validation of expression of predicted plant-pathogenicity genes during wheat infection
Gene expression of selected genes (Supporting Table S14A) was tested via quantitative polymerase chain reaction (qPCR) in wheat roots at 7 days post-infection and in 7 day old in vitro grown PDB culture. Wheat samples were inoculated with millet seeds pre-infect with WAC10335 and grown in pots of vermiculite for 7 days at 24°C. Wheat seeds were surface-sterilised and germinated on moist filter paper at 4°C for 4 days, then planted into pre-infected vermiculite pots and covered by a layer of fresh fine vermiculite. The pots were transferred to a growth room at 16°C and 12 hours light/day (150 µmol.m−2.s−1) for 7 days. Plants were harvested and root and above ground tissues separated. RNA was extracted from root tissue using Trizol reagent (Sigma) according to the manufacturer's instructions and cDNA produced using Superscript III (Invitrogen) following the manufacturer's instructions. Quantitative PCRs used SsoFast EvaGreen Supermix (BioRad).
A total of 29 out of 308 predicted ‘effector-like’ pathogenicity genes were selected for testing based on their assigned functional annotations. Seven control genes were also selected based on stable expression, averaging ≥70 FPKM and ≤0.1× fold change between libraries, across the three RNA-seq libraries discussed in this study and/or for putative functions suggesting stable expression patterns (e.g. actin and tubulin). Primer pairs were designed from coding-exon sequences (CDS) using primer3 [100] (opt. amplicon 200 bp, primer 18–25 bp, opt. Tm 60°C, max. ΔTm 1°C, min. GC clamp 2 bp, max. homopolymer 3 bp). In silico PCR screening via e-PCR [101] required ≤1 amplicon (10 bp to 10 kbp) versus genome assembly and CDS sequences. Quantitative PCR was performed with 2 technical replicates and 3 biological replicates. Log2 fold-changes between in-vitro and infection samples were calculated by the ΔΔCT method in accordance with Anderson et al. [102], relative to the mean of 7 controls. A two-tailed Student's T-test was applied to relative abundances between in planta and in vitro samples (equal variance, p-value≤0.05).
Supporting Information
{kind=link}
{kind=link}
Zdroje
1. Sneh B, Burpee L, Ogoshi A, editors(1991) Identification of Rhizoctonia species. St. Paul, Minnesota, USA: APS Press.
2. PaulitzTC (2006) Low input no-till cereal production in the Pacific Northwest of the US: The challenges of root diseases. Eur J Plant Pathol 115 : 271–281.
3. AndersonJP, SinghKB (2011) Interactions of Arabidopsis and M. truncatula with the same pathogens differ in dependence on ethylene and ethylene response factors. Plant Signal Behav 6 : 551–552.
4. BellDK, SumnerDR (1982) Virulence of Rhizoctonia solani Ag-2 Type-1 and Type-2 and Ag-4 from Peanut Seed on Corn, Sorghum, Lupine, Snapbean, Peanut and Soybean. Phytopathology 72 : 947–948.
5. SumnerDR, BellDK (1982) Crop-Rotation and Yield Loss in Corn in Soil Infested with Rhizoctonia solani Ag-2 and Ag-4. Phytopathology 72 : 361–362.
6. BradleyCA, HartmanGL, NelsonRL, MuellerDS, PedersenWL (2001) Response of ancestral soybean lines and commercial cultivars to Rhizoctonia root and hypocotyl rot. Plant Dis 85 : 1091–1095.
7. KluthC, BuhreC, VarrelmannM (2010) Susceptibility of intercrops to infection with Rhizoctonia solani AG 2-2 IIIB and influence on subsequently cultivated sugar beet. Plant Pathol 59 : 683–692.
8. KluthC, VarrelmannM (2010) Maize genotype susceptibility to Rhizoctonia solani and its effect on sugar beet crop rotations. Crop Prot 29 : 230–238.
9. GarciaVG, OncoMAP, SusanVR (2006) Review. Biology and systematics of the form genus Rhizoctonia. Span J Agric Res 4 : 55–79.
10. SharonM, KuninagaS, MyakumachiM, SnehB (2006) The advancing identification and classification of Rhizoctonia spp. using molecular and biotechnological methods compared with the classical anastomosis grouping. Mycoscience 47 : 299–316.
11. MurrayGM, BrennanJP (2009) Estimating disease losses to the Australian wheat industry. Australas Plant Pathol 38 : 558–570.
12. MurrayGM, BrennanJP (2010) Estimating disease losses to the Australian barley industry. Australas Plant Pathol 39 : 85–96.
13. SchroederKL, ShettyKK, PaulitzTC (2011) Survey of Rhizoctonia spp. from wheat soils in the US and determination of pathogenicity on wheat and barley. Phytopathology 101: S161–S161.
14. SweetinghamMW, CruickshankRH, WongDH (1986) Pectic zymograms and taxonomy and pathogenicity of the Ceratobasidiaceae. Mycol Res 86 : 305–311.
15. Sweetingham MW, MacNish GC, editors(1994) Rhizoctonia Isolation, Identification and Pathogenicity: A Laboratory Manual. 2nd ed. South Perth, WA, Australia: Department of Agriculture.
16. AndersonJP, LichtenzveigJ, GleasonC, OliverRP, SinghKB (2010) The B-3 ethylene response factor MtERF1-1 mediates resistance to a subset of root pathogens in Medicago truncatula without adversely affecting symbiosis with rhizobia. Plant Physiol 154 : 861–873.
17. FoleyRC, GleasonCA, AndersonJP, HamannT, SinghKB (2013) Genetic and genomic analysis of Rhizoctonia solani interactions with Arabidopsis; evidence of resistance mediated through NADPH oxidases. PLoS ONE 8: e56814.
18. KamperJ, KahmannR, BolkerM, MaLJ, BrefortT, et al. (2006) Insights from the genome of the biotrophic fungal plant pathogen Ustilago maydis. Nature 444 : 97–101.
19. LaurieJD, AliS, LinningR, MannhauptG, WongP, et al. (2012) Genome comparison of barley and maize smut fungi reveals targeted loss of RNA silencing components and species-specific presence of transposable elements. Plant Cell 24 : 1733–1745.
20. SchirawskiJ, MannhauptG, MunchK, BrefortT, SchipperK, et al. (2010) Pathogenicity determinants in smut fungi revealed by genome comparison. Science 330 : 1546–1548.
21. CantuD, SegoviaV, MacleanD, BaylesR, ChenX, et al. (2013) Genome analyses of the wheat yellow (stripe) rust pathogen Puccinia striiformis f. sp. tritici reveal polymorphic and haustorial expressed secreted proteins as candidate effectors. BMC Genomics 14 : 270.
22. DuplessisS, CuomoCA, LinYC, AertsA, TisserantE, et al. (2011) Obligate biotrophy features unraveled by the genomic analysis of rust fungi. Proc Natl Acad Sci U S A 108 : 9166–9171.
23. MondegoJM, CarazzolleMF, CostaGG, FormighieriEF, ParizziLP, et al. (2008) A genome survey of Moniliophthora perniciosa gives new insights into Witches' Broom Disease of cacao. BMC Genomics 9 : 548.
24. RiouxR, ManmathanH, SinghP, de los ReyesB, JiaYL, et al. (2011) Comparative analysis of putative pathogenesis-related gene expression in two Rhizoctonia solani pathosystems. Curr Genet 57 : 391–408.
25. LakshmanDK, AlkharoufN, RobertsDP, NatarajanSS, MitraA (2012) Gene expression profiling of the plant pathogenic basidiomycetous fungus Rhizoctonia solani AG 4 reveals putative virulence factors. Mycologia 104 : 1020–1035.
26. ZhengA, LinR, ZhangD, QinP, XuL, et al. (2013) The evolution and pathogenic mechanisms of the rice sheath blight pathogen. Nat Commun 4 : 1424.
27. WibbergD, JelonekL, RuppO, HennigM, EikmeyerF, et al. (2012) Establishment and interpretation of the genome sequence of the phytopathogenic fungus Rhizoctonia solani AG1-IB isolate 7/3/14. J Biotechnol 167 : 142–155.
28. Losada L, Pakala SB, Fedorova ND, Joardar V, Shabalina SA, et al.. (2014) Mobile elements and mitochondrial genome expansion in the soil fungus and potato pathogen Rhizoctonia solani AG-3. FEMS Microbiol Lett In Press.
29. CubetaMA, DeanRA, ThomasE, BaymanP, JabajiS, et al. (2009) Rhizoctonia solani genome project: providing insight into a link between beneficial and plant pathogenic fungi. Phytopathology 99: S166.
30. LiuTH, LinMJ, KoWH (2010) Factors affecting protoplast formation by Rhizoctonia solani. N Biotechnol 27 : 64–69.
31. RobinsonHL, DeaconJW (2001) Protoplast preparation and transient transformation of Rhizoctonia solani. Mycol Res 105 : 1295–1303.
32. YangHA, SivasithamparamK, ObrienPA (1993) Improved Method for Protoplast Regeneration of Rhizoctonia solani. Soil Biol Biochem 25 : 633–636.
33. HaneJK, OliverRP (2010) In silico reversal of repeat-induced point mutation (RIP) identifies the origins of repeat families and uncovers obscured duplicated genes. BMC Genomics 11 : 655.
34. KeijerJ, HoutermanPM, DullemansAM, KorsmanMG (1996) Heterogeneity in electrophoretic karyotype within and between anastomosis groups of Rhizoctonia solani. Mycol Res 100 : 789–797.
35. HaneJK, RouxelT, HowlettBJ, KemaGH, GoodwinSB, et al. (2011) A novel mode of chromosomal evolution peculiar to filamentous Ascomycete fungi. Genome Biol 12: R45.
36. MaLJ, van der DoesHC, BorkovichKA, ColemanJJ, DaboussiMJ, et al. (2010) Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium. Nature 464 : 367–373.
37. MuszewskaA, Hoffman-SommerM, GrynbergM (2011) LTR Retrotransposons in Fungi. PLoS ONE 6: e29425.
38. PetersenTN, BrunakS, von HeijneG, NielsenH (2011) SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods 8 : 785–786.
39. HortonP, ParkKJ, ObayashiT, FujitaN, HaradaH, et al. (2007) WoLF PSORT: protein localization predictor. Nucleic Acids Res 35: W585–W587.
40. KallL, KroghA, SonnhammerELL (2004) A combined transmembrane topology and signal peptide prediction method. J Mol Biol 338 : 1027–1036.
41. AlvianoCS, TravassosLR, SchauerR (1999) Sialic acids in fungi: A minireview. Glycoconj J 16 : 545–554.
42. MavromatisK, ChuK, IvanovaN, HooperSD, MarkowitzVM, et al. (2009) Gene Context Analysis in the Integrated Microbial Genomes (IMG) Data Management System. PLoS ONE 4: e7979.
43. ZhaoZ, LiuH, WangC, XuJR (2013) Comparative analysis of fungal genomes reveals different plant cell wall degrading capacity in fungi. BMC Genomics 14 : 274.
44. CandyL, PeumansWJ, Menu-BouaouicheL, AstoulCH, Van DammeJ, et al. (2001) The Gal/GalNAc-specific lectin from the plant pathogenic basidiomycete Rhizoctonia solani is a member of the ricin-B family. Biochem Biophys Res Commun 282 : 655–661.
45. VachonV, LapradeR, SchwartzJL (2012) Current models of the mode of action of Bacillus thuringiensis insecticidal crystal proteins: a critical review. J Invertebr Pathol 111 : 1–12.
46. GrenierAM, DuportG, PagesS, CondemineG, RahbeY (2006) The phytopathogen Dickeya dadantii (Erwinia chrysanthemi 3937) is a pathogen of the pea aphid. Appl Environ Microbiol 72 : 1956–1965.
47. OlivaR, WinJ, RaffaeleS, BoutemyL, BozkurtTO, et al. (2010) Recent developments in effector biology of filamentous plant pathogens. Cell Microbiol 12 : 705–715.
48. KaleSD (2012) Oomycete and fungal effector entry, a microbial Trojan horse. New Phytol 193 : 874–881.
49. ClutterbuckAJ (2011) Genomic evidence of repeat-induced point mutation (RIP) in filamentous ascomycetes. Fungal Genet Biol 48 : 306–326.
50. Van de WouwAP, CozijnsenAJ, HaneJK, BrunnerPC, McDonaldBA, et al. (2010) Evolution of linked avirulence effectors in Leptosphaeria maculans is affected by genomic environment and exposure to resistance genes in host plants. PLoS Pathog 6: e1001180.
51. HoodME, KatawczikM, GiraudT (2005) Repeat-induced point mutation and the population structure of transposable elements in Microbotryum violaceum. Genetics 170 : 1081–1089.
52. HornsF, PetitE, YocktengR, HoodME (2012) Patterns of repeat-induced point mutation in transposable elements of basidiomycete fungi. Genome Biol Evol 4 : 240–247.
53. NabelCS, ManningSA, KohliRM (2012) The curious chemical biology of cytosine: deamination, methylation, and oxidation as modulators of genomic potential. ACS Chem Biol 7 : 20–30.
54. MeerupatiT, AnderssonKM, FrimanE, KumarD, TunlidA, et al. (2013) Genomic mechanisms accounting for the adaptation to parasitism in nematode-trapping fungi. PLoS Genet 9: e1003909.
55. AuCH, CheungMK, WongMC, ChuAK, LawPT, et al. (2013) Rapid genotyping by low-coverage resequencing to construct genetic linkage maps of fungi: a case study in Lentinula edodes. BMC Res Notes 6 : 307.
56. HacquardS, KracherB, MaekawaT, VernaldiS, Schulze-LefertP, et al. (2013) Mosaic genome structure of the barley powdery mildew pathogen and conservation of transcriptional programs in divergent hosts. Proc Natl Acad Sci U S A 110: E2219–2228.
57. LinK, LimpensE, ZhangZ, IvanovS, SaundersDG, et al. (2014) Single Nucleus Genome Sequencing Reveals High Similarity among Nuclei of an Endomycorrhizal Fungus. PLoS Genet 10: e1004078.
58. OjedaDI, DhillonB, TsuiCK, HamelinRC (2013) Single-nucleotide polymorphism discovery in Leptographium longiclavatum, a mountain pine beetle-associated symbiotic fungus, using whole-genome resequencing. Mol Ecol Resour 14 : 401–410.
59. NeafseyDE, BarkerBM, SharptonTJ, StajichJE, ParkDJ, et al. (2010) Population genomic sequencing of Coccidioides fungi reveals recent hybridization and transposon control. Genome Res 20 : 938–946.
60. FreitagM, WilliamsRL, KotheGO, SelkerEU (2002) A cytosine methyltransferase homologue is essential for repeat-induced point mutation in Neurospora crassa. Proc Natl Acad Sci U S A 99 : 8802–8807.
61. MorrowCA, FraserJA (2013) Ploidy variation as an adaptive mechanism in human pathogenic fungi. Semin Cell Dev Biol 24 : 339–346.
62. KatariaHR, VermaPR, GisiU (1991) Variability in the Sensitivity of Rhizoctonia solani Anastomosis Groups to Fungicides. J Phytopathol 133 : 121–133.
63. MartinM (2011) Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnetjournal 17 : 10–12.
64. MagocT, SalzbergSL (2011) FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27 : 2957–2963.
65. LiRQ, ZhuHM, RuanJ, QianWB, FangXD, et al. (2010) De novo assembly of human genomes with massively parallel short read sequencing. Genome Res 20 : 265–272.
66. BoetzerM, HenkelCV, JansenHJ, ButlerD, PirovanoW (2011) Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 27 : 578–579.
67. HuangSF, ChenZL, HuangGR, YuT, YangP, et al. (2012) HaploMerger: Reconstructing allelic relationships for polymorphic diploid genome assemblies. Genome Res 22 : 1581–1588.
68. HuangXQ, MadanA (1999) CAP3: A DNA sequence assembly program. Genome Res 9 : 868–877.
69. AltschulSF, MaddenTL, SchafferAA, ZhangJH, ZhangZ, et al. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25 : 3389–3402.
70. LangmeadB, SalzbergSL (2012) Fast gapped-read alignment with Bowtie 2. Nat Methods 9 : 357–359.
71. McKennaA, HannaM, BanksE, SivachenkoA, CibulskisK, et al. (2010) The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20 : 1297–1303.
72. DanecekP, AutonA, AbecasisG, AlbersCA, BanksE, et al. (2011) The variant call format and VCFtools. Bioinformatics 27 : 2156–2158.
73. QuinlanAR, HallIM (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26 : 841–842.
74. KurtzS, PhillippyA, DelcherAL, SmootM, ShumwayM, et al. (2004) Versatile and open software for comparing large genomes. Genome Biol 5: R12.
75. PriceAL, JonesNC, PevznerPA (2005) De novo identification of repeat families in large genomes. Bioinformatics 21: I351–I358.
76. Smit AFA, Hubley R, Green P (1996–2010) RepeatMasker Open-3.0. http://www.repeatmasker.org.
77. KohanyO, GentlesAJ, HankusL, JurkaJ (2006) Annotation, submission and screening of repetitive elements in Repbase: RepbaseSubmitter and Censor. BMC Bioinformatics 7.
78. AbrusanG, GrundmannN, DeMesterL, MakalowskiW (2009) TEclass-a tool for automated classification of unknown eukaryotic transposable elements. Bioinformatics 25 : 1329–1330.
79. Haas BJ (2007–2011) TransposonPSI. http://transposonpsi.sourceforge.net.
80. JurkaJ, KapitonovVV, PavlicekA, KlonowskiP, KohanyO, et al. (2005) Repbase update, a database of eukaryotic repetitive elements. Cytogenet Genome Res 110 : 462–467.
81. TrapnellC, PachterL, SalzbergSL (2009) TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25 : 1105–1111.
82. GrabherrMG, HaasBJ, YassourM, LevinJZ, ThompsonDA, et al. (2011) Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29 : 644–652.
83. MarcaisG, KingsfordC (2011) A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27 : 764–770.
84. HaasBJ, SalzbergSL, ZhuW, PerteaM, AllenJE, et al. (2008) Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol 9: R7.
85. BorodovskyM, LomsadzeA (2011) Eukaryotic gene prediction using GeneMark.hmm-E and GeneMark-ES. Curr Protoc Bioinformatics Chapter 4 Unit 4.6.1-10.
86. WinnenburgR, BaldwinTK, UrbanM, RawlingsC, KohlerJ, et al. (2006) PHI-base: a new database for pathogen host interactions. Nucleic Acids Res 34: D459–D464.
87. HuangXQ, AdamsMD, ZhouH, KerlavageAR (1997) A tool for analyzing and annotating genomic sequences. Genomics 46 : 37–45.
88. LoweTM, EddySR (1997) tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 25 : 955–964.
89. NawrockiEP, KolbeDL, EddySR (2009) Infernal 1.0: inference of RNA alignments. Bioinformatics 25 : 1335–1337.
90. LewisSE, SearleSM, HarrisN, GibsonM, LyerV, et al. (2002) Apollo: a sequence annotation editor. Genome Biol 3 research0082-0082.0014.
91. HusonDH, MitraS, RuscheweyhHJ, WeberN, SchusterSC (2011) Integrative analysis of environmental sequences using MEGAN4. Genome Res 21 : 1552–1560.
92. FinnRD, ClementsJ, EddySR (2011) HMMER web server: interactive sequence similarity searching. Nucleic Acids Res 39: W29–W37.
93. ParkBH, KarpinetsTV, SyedMH, LeuzeMR, UberbacherEC (2010) CAZymes Analysis Toolkit (CAT): Web service for searching and analyzing carbohydrate-active enzymes in a newly sequenced organism using CAZy database. Glycobiology 20 : 1574–1584.
94. KatohK, StandleyDM (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30 : 772–780.
95. LechnerM, FindeissS, SteinerL, MarzM, StadlerPF, et al. (2011) Proteinortho: Detection of (co-)orthologs in large-scale analysis. BMC Bioinformatics 12 : 124.
96. StajichJE, BlockD, BoulezK, BrennerSE, ChervitzSA, et al. (2002) The Bioperl toolkit: Perl modules for the life sciences. Genome Res 12 : 1611–1618.
97. RiceP, LongdenI, BleasbyA (2000) EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet 16 : 276–277.
98. BaileyTL, BodenM, BuskeFA, FrithM, GrantCE, et al. (2009) MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res 37: W202–208.
99. CingolaniP, PlattsA, Wang leL, CoonM, NguyenT, et al. (2012) A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 6 : 80–92.
100. RozenS, SkaletskyH (2000) Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol 132 : 365–386.
101. SchulerGD (1998) Electronic PCR: bridging the gap between genome mapping and genome sequencing. Trends Biotechnol 16 : 456–459.
102. AndersonJP, BadruzsaufariE, SchenkPM, MannersJM, DesmondOJ, et al. (2004) Antagonistic interaction between abscisic acid and jasmonate-ethylene signaling pathways modulates defense gene expression and disease resistance in Arabidopsis. Plant Cell 16 : 3460–3479.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2014 Číslo 5
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- PINK1-Parkin Pathway Activity Is Regulated by Degradation of PINK1 in the Mitochondrial Matrix
- Null Mutation in PGAP1 Impairing Gpi-Anchor Maturation in Patients with Intellectual Disability and Encephalopathy
- Phosphorylation of a WRKY Transcription Factor by MAPKs Is Required for Pollen Development and Function in
- p53 Requires the Stress Sensor USF1 to Direct Appropriate Cell Fate Decision
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy