
Histiocytární choroby dospělých – pestrost jejich klinických projevů vyžaduje spolupráci lékařů mnoha oborů
Clinical presentations of the most common histiocytic disorders
Background: Histiocytic diseases are significantly rarer than diseases derived from lymphocytic, plasmacytic, or myeloid lineages, and thus are encountered infrequently in hematology and oncology clinics. The most common form is Langerhans cell histiocytosis, which in adults has an incidence of 1–2 cases per 1 million; the others are considerably rarer, with their occurrence reported only by the number of described cases rather than through incidence or prevalence. Their rarity leads to delays in establishing an accurate diagnosis. Objective: The group of histiocytic diseases includes seven clinical units: Langerhans cell histiocytosis, indeterminate dendritic cell histiocytosis, diseases from the juvenile xanthogranuloma group, Erdheim-Chester disease, Rosai-Dorfman disease, ALK-positive histiocytosis, and histiocytic sarcoma. Each of the described diseases has specific manifestations that distinguish it from the manifestations of other malignant blood disorders. The aim of this article is to remind the reader of these manifestations through images and text, thereby contributing to the timely recognition of these rare diseases. Conclusion: Treatment procedures are rapidly evolving, but the clinical presentations of these diseases remain unchanged. The disease profiles presented in this publication should aid in their early diagnosis and consequently in timely treatment.
Keywords:
histiocytosis
Autoři:
prof. MUDr. Adam Zdeněk, CSc. 1; MUDr. Eid Michal, Ph.D. 1,2; doc. MUDr. Řehák Zdeněk, Ph.D. 3; MUDr. Kubeš Václav, Ph.D. 4; doc. MUDr. Horváth Teodor, Ph.D. 5; doc. MUDr. Doubková Martina, Ph.D. 6; MUDr. Starý Karel 7; prof. MUDr. Šlampa Pavel, CSc. 8; MUDr. Čermák Aleš, Ph.D. 9; Doc. MUDr. Fadrus Pavel, Ph.D. 10; MUDr. Adamová Zuzana, Ph.D. 11; doc. MUDr. Vaníček Jiří, Ph.D. 12; MUDr. Nebeský Tomáš 13; MUDr. Kamarádová Kateřina, Ph.D. 14; MUDr. Boichuk Ivanna 1; prof. MUDr. Pour Luděk, Ph.D. 1
Působiště autorů:
Interní hematologická a onkologická klinika LF MU a FN Brno
1; Centrum precizní medicíny, FN Brno
2; Ústav nukleární medicíny, MOÚ Brno
3; Ústav patologie, LF MU a FN Brno
4; Chirurgická klinika LF MU a FN Brno
5; Klinika nemocí plicních a tuberkulózy LF MU a FN Brno
6; Interní gastroenterologická klinika LF MU a FN Brno
7; Klinika radiační onkologie, MOÚ Brno
8; Urologické klinika LF MU a FN Brno
9; Neurochirurgická klinika LF MU a FN Brno
10; Chirurgické oddělení, Moravskoslezská nemocnice Frýdek-Místek
11; Klinika zobrazovacích metod LF MU a FN u sv. Anny v Brně
12; Klinika radiologie a nukleární medicíny LF MU a Brno
13; Fingerlandův ústav patologie, LF UK v Hradci Králové
14
Vyšlo v časopise:
Klin Onkol 2026; 39(3): 150-168
Kategorie:
Přehled
doi:
https://doi.org/10.48095/ccko2026150
Souhrn
Východiska: Histiocytární choroby jsou podstatně vzácnější než nemoci odvozené z linie lymfocytární, plazmocytární či myeloidní, a proto se s nimi v hematologických a onkologických ambulancích setkáváme výjimečně. Nejčastější je histiocytóza z Langerhansových buněk, která u dospělých má incidenci 1–2 případy na 1 milion, ostatní jsou podstatně vzácnější a jejich výskyt je udáván jen počtem popsaných případů, nikoliv incidencí či prevalencí. Jejich vzácnost způsobuje prodlení při stanovení správné diagnózy. Cíl: Do skupiny histiocytárních chorob je řazeno sedm klinických jednotek – histiocytóza z Langerhansových buněk, histiocytóza z indeterminovaných dendritických buněk, choroby ze skupiny juvenilního xantogranulomu, Erdheimova-Chesterova choroba, Rosaiova-Dorfmanova choroba, ALK-pozitivní histiocytózy a histiocytární sarkom. Každá z popsaných chorob má specifické projevy, které ji odlišují od projevů ostatních maligních krevních chorob. Cílem následujícího textu je tyto projevy připomenout obrázky i textem, a přispět tak k časnému rozpoznání těchto vzácných chorob. Závěr: Léčebné postupy se prudce vyvíjejí, ale klinické obrazy těchto nemocí zůstávají stejné. Obrázky chorob uvedené v této publikaci by měly pomoci k jejich včasné diagnostice, a tím pádem i a včasné léčbě.
Klíčová slova:
histiocytóza
Úvod
V roce 2024 vyšel v časopise Klinická onkologie článek věnující se klasifikaci histiocytárních a dendritických chorob [1]. V něm jsou uvedeny změny, které přinesla nová WHO klasifikace těchto chorob ve srovnání s klasifikací starší [2]. V citovaném článku je také uvedena klasifikace dle Histiocyte Society, protože obě jsou stále používány [3]. Klasifikace WHO a Histiocyte Society se od sebe mírné liší. Klasifikace Histiocyte Society obsahuje i reaktivní histiocytární choroby a pro kožní formy histiocytóz má podstatně více termínů [3]. Proto její termíny patologové stále často používají.
V tomto textu se chceme k tématu vrátit, navázat na citovanou publikaci a zaměřit se na klinické obrazy těchto chorob a stručný text doplnit velmi četnými vyobrazeními, která jsme na našem pracovišti za posledních 35 let nasbírali. Histiocytární choroby jsou podstatně vzácnější než choroby odvozené z lymfocytární či myeloidní linie. Epidemiologické údaje typu incidence či prevalence lze dohledat pouze pro nejčastější z nich, histiocytózu z Langerhansových buněk (Langerhans cell histiocytosis – LCH).
Ve Velké Británii byla v letech 2013–2019 incidence LCH u dětí < 15 let 4,46/1 milion dětí, zatímco u osob ≥ 15 let byla incidence 1,06/1 milion obyvatel.
Další analýza je z Řecka, kde stanovili incidenci u osob > 18 let na 1,58/1 milion obyvatel. Tato data korespondují s analýzami před rokem 2000, kdy Baugartner uváděl incidenci LCH u osob > 18 let mezi jedním až dvěma případy na 1 milion obyvatel [4,5].
Ostatní histiocytární choroby jsou podstatně vzácnější, u Erdheimovy-Chesterovy choroby (Erdheim-Chester disease – ECD) se nově uvádí incidence kolem 3 případů na 10 milionů obyvatel a u dalších jen počet popsaných případů.
Ze sporadického výskytu těchto chorob vyplývají problémy s jejich časným a správným rozpoznáním. Proto se v této kapitole zaměříme hlavně na jejich klinický obraz, který je neměnný, zatímco v jejich léčbě dochází k rychlému vývoji. Informace o projevech těchto chorob neztratí v průběhu času nic na své platnosti, a proto je tento text doplněn četnými obrázky, které by měly napomoci k jejich správnému a časnému rozpoznání. Léčba se však bude neustále vyvíjet, a tak aktuálnost léčby bude čtenář muset vždy zkontrolovat v nějakém aktuálním zdroji.
Obrazy histiocytárních chorob jsou odlišné od obrazů chorob lymfoidní a myeloidní řady a vyžadují spolupráci širší skupiny odborníků. Základem pracovní skupiny pro histiocytózy je vždy patolog, který má možnost doplnit své histomorfologické vyšetření analýzou přítomnosti léčebně využitelných mutací. Odběry vzorků jsou doménou spolupracujících chirurgů. V případech plicních forem těchto chorob je nutná spolupráce s hrudními chirurgy, provedení plicní biopsie. Do kompetence hrudních chirurgů pak spadá léčba spontánních pneumotoraxů pacientů s histiocytózami. Diagnózu plicních forem LCH však mohou stanovit i plicní odborníci metodou bronchoalveolární laváže s vyšetřením počtu CD1a pozitivních buněk. U histiocytóz postihujících centrální nervovou soustavu (CNS) a mozkové obaly je nutná spolupráce s neurochirurgy. V případech s infiltráty v retroperitoneu, což je typické pro diseminovanou xantogranulomatózu – ECD, ale také pro chorobu asociovanou s imunoglobulinem IgG4 (IgG4-related disease), je nutný urologický diagnostický výkon, ale i případná léčba neprůchodnosti ureterů. Pro stanovení rozsahu choroby je zapotřebí zobrazovacích specialistů a velkou výhodou je zobrazení pomocí pozitronové emisní tomografie (PET). Naše spolupracující pracoviště na Masarykově onkologickém ústavu vyvinulo a ověřilo novou metodu hodnocení aktivity plicní formy LCH pomocí FDG-PET/CT. Vývoj této formy choroby je jinými metodami stanovitelný jen nepřesně [6,7].
Nepostradatelným spolupracovníkem je vždy endokrinolog, protože alterace tvorby hypotalamo-hypofyzárních hormonů je u této skupiny nemocí častá, pokud není zřejmá v době stanovení diagnózy, tak se může vyvinout v průběhu času. A dalším nepostradatelným spolupracovníkem je radioterapeut obeznámený s možností radioterapie popsaných chorob.
Cílem tohoto textu, na němž se podíleli všichni lékaři, kteří se podílejí na diagnostice a léčbě těchto chorob, je předložit čtenáři stručnou charakteristiku diagnóz a informovat o léčbě dle mezinárodních doporučení zveřejněných do léta 2025.
Histiocytóza z Langerhansových buněk (LCH)
Morfologická podstata
LCH je odvozena od Langerhansových dendritických buněk. Pro nemoc jsou charakteristická granulomatózní zánětlivá ložiska obsahující eozinofily, lymfocyty a dendritické Langerhansovy buňky. Nemoc může postihnout kterýkoliv orgán či tkáň. Pro přítomnost četných eozinofilů v kostních ložiscích se dříve používal termín „eozinofilní kostní granulom“.
Diagnózu LCH lze stanovit pouze histologickým vyšetřením. Mezi charakteristické znaky patří přítomnost proteinu S-100 v dendritických buňkách, exprese CD1a antigenu. Dalším typickým znakem je langerin (CD207), což je charakteristický protein pro Langerhansovy buňky, podílející se na tvorbě Birbeckových granulí. Uvedené znaky představují vysoce specifické markery této nemoci.
Stanovení správné diagnózy mohou komplikovat změny probíhající v čase. Nejvíce patologických dendritických buněk je přítomno v době vzniku ložiska. V průběhu času dochází k fibrotizaci, jizvení a počet patologických dendritických buněk klesá, což komplikuje správné histologické zařazení [1–3].
Zkušenosti našeho pracoviště
V roce 2010 jsme popsali soubor 22 pacientů s touto nemocí, od té doby se počet pacientů s touto nemocí registrovaných na našem pracovišti zvýšil na 37. V pěti případech se jednalo o osoby s diagnózou zjištěnou a léčenou v dětském věku, tito pacienti nám byli předáni z pediatrického pracoviště. U jednoho z nich se rozvíjí pozdní komplikace – atrofie mozečku a bazálních ganglií s projevy ataxie a dysartrie [8–11].
Jednotlivé klinické projevy
Po stanovení histologické diagnózy je jako u každé onkologické choroby třeba stanovit rozsah nemoci. Pro správné stanovení rozsahu nemoci je však nutné znát pestré projevy této nemoci a cíleně po nich pátrat. Klasifikaci rozsahu této nemoci uvádí tab. 1.

Kostní projevy LCH
LCH v dospělosti postihuje dominantně skelet, vytváří osteolytická ložiska, podobná osteolýze při mnohočetném myelomu. Nejčastěji je uváděno postižení kalvy, následuje osteolytické postižení žeber. S menší frekvencí bývají postiženy další části skeletu. U některých pacientů jsou kostní ložiska různého stáří, hojící se místa mají sklerotický lem. Ne všechna musí bolet. Zduření tkání přiléhajících ke kosti signalizuje, že choroba prorůstá do okolí a mnohdy zduření nad kostí upozorní na ložisko. Klasickou metodou průkazu ložisek je rentgenový snímek. Kostní ložiska mohou, ale nemusí mít dostatečně zvýšenou osteoneogenezu, takže, podobně jako mnohočetný myelom, nemusí být detekovatelná scintigrafií skeletu pomocí technecium-pyrofosfátu, zato 18FDG-PET/CT je vykreslí vždy. Za zvláště riziková kostní ložiska jsou považovány kostní defekty v oblasti orbitální se supraorbitálními infiltráty a obecně postižení obličejových kostí. Uvedené postižení je spojené s vyšší pravděpodobností pozdějšího postižení CNS [12]. Postižení kalvy chorobou ilustruje obr. 1.

Kožní projevy LCH
Kožní manifestace jsou u LCH velmi časté a mohou být vůbec prvními zachytitelnými projevy nemoci. Infiltrace kůže buňkami LCH často postihuje intertriginózní oblasti (perianální oblast, vulva, třísla, pupek). Zvláště postižení vulvy dělá diferenciálně diagnostické problémy, protože připadají v úvahu jiné kožní neoplazie či záněty. Tento typ postižení byl opakovaně v domácí literatuře popsán, naposledy Mlynčekem et al. [13].
Typická kožní morfa tvořená buňkami této nemoci je hnědorůžová papula velikosti 1–5 mm s tendencí ke splývání. Zvláště v oblasti kůže kštice se objevuje i tvorba šupinek (šupení). Popisovány jsou i vezikuly a pustuly. Papulózní projevy jsou často hodnoceny jako nespecifické či ekzémové. Šupící se plošky jsou zaměňovány za seboroickou dermatitidu, zvláště u kojenců a malých dětí při postižení vlasaté části hlavy.
Kožní forma LCH může mít také klinický obraz nehojícího se atopického ekzému. To, že jde o kožní projev LCH, nelze rozpoznat při makroskopickém pohledu, pro rozpoznání je vždy potřeba provést excizi a histologické hodnocení vzorku, jak popisují Mecherová et al. [14].
Kožní projevy se mohou sdružovat s kostním či viscerálním postižením, může však jít také o izolovanou morfu, která často spontánně regreduje [15] (obr. 2, 3).


buněk
Ale i jiné histiocytární choroby mohou postihovat kůži, např. multicentrická retikulohistiocytóza, tedy histiocytární onemocnění, které patologové pojmenovali termínem z klasifikace Histiocyte Society (obr. 4).

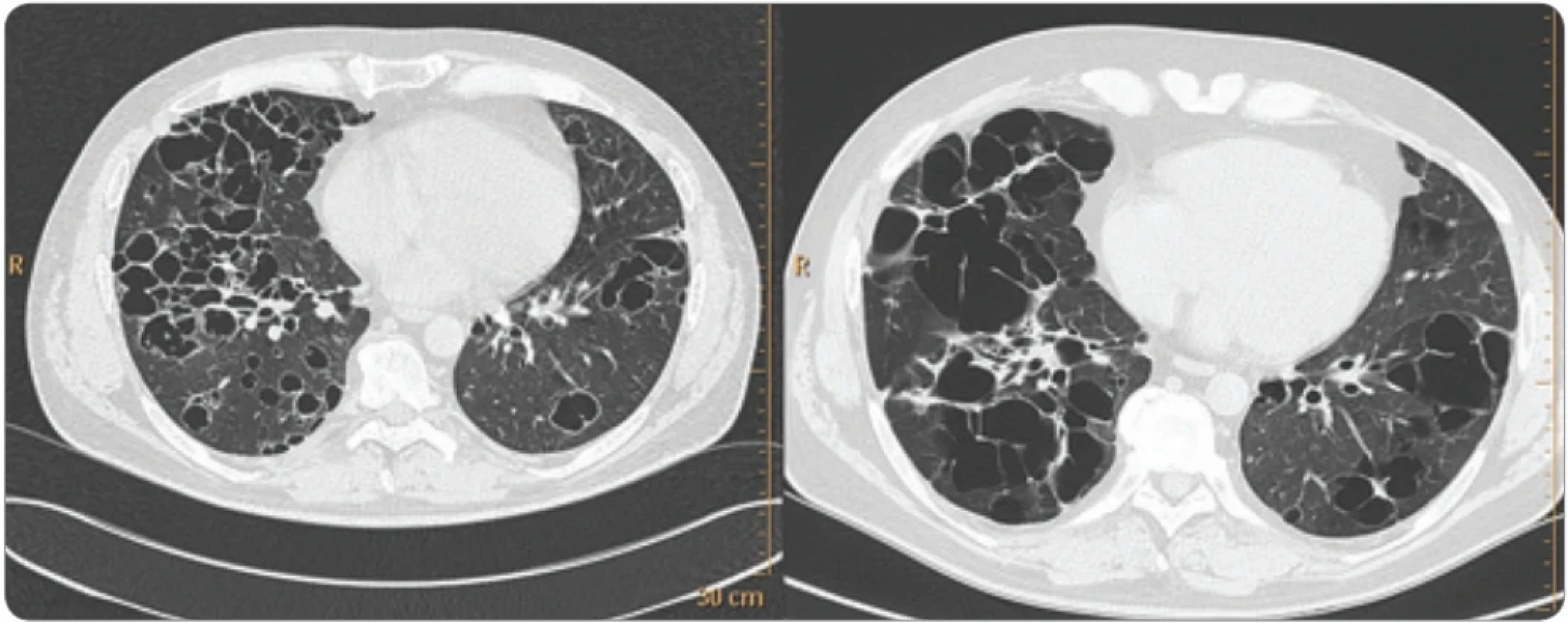
Plicní projevy LCH
Klinické projevy a příznaky LCH se s výjimkou plicních komplikací vyvíjejí pozvolna, pouze pneumotorax je akutním problémem. Respirační cesty jsou postiženy častěji u dospělých než u dětí, 60–100 % pacientů s plicní formou jsou kuřáci. Incidence plicního postižení mezi všemi pacienty s LCH se udává kolem 20 %. Pacienti přicházejí s dušností a bolestí na hrudníku, neproduktivním kašlem, někdy udávají teploty a úbytek hmotnosti, jak v domácí literatuře opakovaně popisují Doubková et al. [10].
Infiltrace plic vyvolává restriktivní změny, které předcházejí rentgenologickým změnám. Radiografický nález je tvořen cystami a intersticiálními nodulárními opacitami, uloženými obvykle blíže hilům. Optimálním prostředkem pro diagnostiku plicní formy histiocytózy je HRCT (high-resolution computed tomography), které velmi dobře zobrazí tenkostěnné cysty, mikronoduly, opacity a zesílení intersticiálních prostorů. Cysty jsou častěji v horních lalocích, méně jich je ve středních lalocích a vynechávají kostodiafragmatický úhel. Zesílení intersticia je při HRCT vyšetření zřetelné hlavně bazálně. Velmi podrobné informace o plicní formě LCH zveřejnila nedávno francouzská pracovní skupina vedená prof. Tazi, která se touto chorobou dlouhodobě a systematicky zabývá [16]. Prof. Tazi je i spoluautorem posledního léčebného doporučení pro LCH.
Prasknutí cyst a jejich komunikace s pleurální dutinou způsobují spontánní pneumotorax. Cysty představují problém i pro cestování letadlem, protože při sníženém tlaku mohou prasknout a způsobit pneumotorax [10,17,18].
Plicní formu LCH znázorňují obr. 5, 6. Plicní formu lze diagnostikovat nejen histologicky po odběru vzorku torakoskopicky, ale také metodou bronchoalveolární laváže s flowcytometrickou detekcí patologických buněk exprimujících znaky CD1a, ale i protein S-100.

buněk
Postižení endokrinního systému a CNS u LCH
LCH, podobně jako jiné histiocytózy, má afinitu k hypotalamu a hypofýze, které často postihuje. Diabetes insipidus je nejznámější endokrinologický projev LCH. Zvláště v dětském věku však deficit nepostihuje pouze sekreci adiuretinu, ale občas dochází v důsledku nemoci k deficitu i dalších hypofyzárních hormonů, vč. poškození tvorby somatotropinu. Defekt tvorby somatotropinu vzniklý v dětství má za následek nejen malý vzrůst, ale nedostatek somatotropinu může být příčinou i neúspěchu in vitro fertilizace, protože přiměřená tvorba somatotropinu či jeho substituce jsou podmínkou úspěšného dokončení gravidity [8,19,20]. Typické změny v oblasti hypotalamu a hypofýzy ilustruje obr. 7.

Po mnohaletém průběhu LCH se může dostavit atrofie cerebella a bazálních ganglií způsobující ataxii a další poruchy. Zde se předpokládá imunitní mechanizmus tohoto procesu a zatím není definována léčba, která by měla potenciál tyto změny zastavit či zvrátit [1–3].
Lymfadenopatie
Histiocytóza obvykle nezpůsobuje výraznou lymfadenopatii. Pokud ano, jde spíše o ložiskové než generalizované postižení [8,21]. U našich pacientů jsme se setkali jak s případem lokalizované lymfadenopatie, kterou vyléčil operační výkon, tak s případem generalizované lymfadenopatie, která měla stejný obraz při 18FDG-PET/CT zobrazení jako generalizovaný non-hodgkinský lymfom (obr. 8).

Uši a zevní sluchovod
Poruchy sluchu mohou nastat jak postižením zevního sluchového kanálu, tak poruchou středního či vnitřního ucha propagací choroby z processus mastoideus. Infiltrace je nebolestivá a postupně může vést k hluchotě. Časté jsou sekundární infekce, které jsou příčinou záměny za chronickou otitidu. Proto by u každé dlouhodobé afekce připomínající zánět zevního zvukovodu mělo být histologicky ověřeno, zda se nejedná o první projev LCH. Nemoc totiž dále progreduje do vnitřního ucha a bohužel je často diagnostikována až při operaci pro proces, který destruuje celé vnitřní ucho [22]. V domácí literatuře tyto projevy popsali Smilek et al. [23] (obr. 9).

Oči
Intraokulární postižení je vzácné, zatímco infiltrace orbitálního prostoru je relativně častá. Dětští lékaři se s ní setkávají u 20–30 % nemocných LCH. Projevuje se ptózou víčka, edémem papily a poruchou funkce VII. nervu. Může být poškozen i optický nerv, což si někdy kromě systémové léčby vynutí i akutní léčbu nitroložiskovou aplikací kortikosteroidů a radioterapii [24]. Sami jsme se setkali pouze s případy postižení zevní stěny orbity, které vyřešil operační výkon.
Játra a slezina
Játra i slezina mohou být touto chorobou také postiženy, což se projeví jejich zvětšením. Infiltrace jater může vyvolat příznaky jaterního selhání (pokles koncentrace albuminu, snížení aktivity koagulačních faktorů, žloutenka bez výrazného zvýšení jaterních enzymů). U chronických forem může vzniknout periportální fibrotizace s příznaky shodnými se sklerotizující cholangitidou a obstrukční biliární žloutenkou, kterou je nutno na základě biopsie odlišit od primární sklerotizující cholangitidy a adekvátně léčit [2,3].
Dutina ústní
Počínající infiltrace se v dutině ústní projevuje zduřením dásní a sliznice patra. Může dojít i k postižení kostí a uvolňování zubů či hypertrofii dásní. Progrese infiltrátů dásní vytváří ulcerace v ústech [25,26]. Proces je velmi často považován za paradentózu, od níž není bez histologie rozeznatelný a ke správnému stanovení diagnózy dojde až při značném poškození čelisti, jak popsali v domácí literatuře Fassmann et al. [27]. Diagnostické je histologické vyšetření. Změny v dutině ústní způsobené LCH ilustruje obr. 10.

Trávicí trakt
Sliznice střevního traktu je postižena jen zřídka. Prvními příznaky je celkové neprospívání a hubnutí. Klasické projevy malabsorpce se objevují až při rozsáhlejším postižení trávicího traktu. Naproti tomu často bývá postižen anální kanál a perianální oblast, infiltrace této oblasti někdy tvoří součást kožního postižení. Infiltrace kůže perianálně je makroskopicky nerozeznatelná od ekzému, pouze histologické vyšetření kůže může identifikovat LCH. Někdy má nemoc v této oblasti podobu verukovitých výrůstků podobných kondylomatům [28].
Léčba
Až v roce 2022 bylo konečně zveřejněno doporučení vytvořené mezinárodní skupinou expertů. Léčba se liší dle míry postižení organizmu. Toto doporučení analyzuje veškeré dosavadní zkušenosti a v letech 2023–2025 představuje nejpropracovanější instrukce pro léčbu, kterými bychom se měli řídit [29].
Radioterapie
Nejčastěji se její použití popisuje u unifokálního či multifokálního kostního postižení. Počty léčebných odpovědí se pohybují kolem 79–100 %. Není zcela jednota v doporučené dávce a je poměrně značný rozptyl publikovaných dávek (1,4–45 Gy). Obecně je za optimální dávku považováno 10–20 Gy pro dospělé při klasickém frakcionování, pro děti pak jen 10 Gy [29].
Léčba izolovaného postižení kůže
Kožní excize lze limitovat na solitární nevelká ložiska, ale neměla by se provádět v žádném případě mutilující operace. V případě multisystémového postižení reagují kožní morfy obvykle na systémovou léčbu.
V případě single system postižení omezeného na kůži lze použít fototerapii, psoralen a ultrafialové záření (PUVA) nebo ozáření úzkým pásmem ultrafialového světla (UVB).
Při postižení kůže lze s úspěchem použít i ozáření nízce energetickými elektrony (electron beam irradiation) [29].
Léčba multisystémového postižení
Lékem volby pro multisystémové postižení se stal kladribin. A i když se píše, že kladribin se aplikuje pacientům > 18 let, tak v mnoha z citovaných publikací byl použit pro léčbu dětských pacientů s LCH. Obvykle se podává v monoterapii v dávce 5 mg/m2 subkutánně 5 dní po sobě v počtu 4–6 cyklů [30]. Střední dávky cytosin-arabinosidu jsou další účinnou léčbou, ale jejich použití se popisuje častěji u dětských než u dospělých pacientů. Pro velmi agresivní formy LCH lze použít polychemoterapeutické režimy s etoposidem, jinak používané pro lymfomy. Na našem pracovišti jsme v této indikaci používali režim CHOEP (cyklofosfamid, doxorubicin, vinkristin, etoposid a prednison).
V roce 2010 byla prokázána mutace BRAFV600E u pacientů s LCH a bylo prokázáno, že proliferace těchto buněk je závislá na MAPK aktivační cestě. U 25–65 % případů LCH je nalézána patogenní varianta BRAFV600E. Protein BRAF hraje důležitou roli v MAPK signální dráze. Proto americká agentura Food and Drug Administration (FDA) schválila dva inhibitory BRAFV600E kinázy, vemurafenib a dabrafenib, pro léčbu ECD, jejíž buňky obsahují uvedenou mutaci. Tyto léky se používají i pro léčbu LCH, pokud mají tuto mutaci [29]. U nás použití této léčby popsali Gabanec et al. z Hradce Králové [31].
Sledování po léčbě
LCH může přejít do chronického stadia, ale může také recidivovat i po dosažení remise nemoci. Proto je třeba pacienty po ukončené léčbě sledovat. A protože LCH je také spojena s vyšším rizikem dalších malignit ve srovnání s průměrnou populací, je cílem kontrol jak včasné podchycení recidivy této nemoci, tak i včasná diagnostika jiných maligních onemocnění [29].
Histiocytóza z indeterminovaných dendritických buněk
Morfologická podstata
Histiocytóza z indeterminovaných buněk (indeterminate dendritic cell tumor nebo též indeterminate histiocytosis) je odvozena od indeterminovaných dendritických buněk, které jsou považovány za prekurzory Langerhansových buněk. Morfologicky se tedy velmi podobá LCH. Transformované dendritické buňky vykazují pozitivitu S100 a CD1a, na rozdíl od LCH nemají Birbeckovy granule a neexprimují langerin (CD207). V některých případech histiocytózy z indeterminovaných buněk byla prokázána BRAFV600E mutace [2].
Zkušenosti našeho pracoviště
Na našem pracovišti jsme léčili jednoho pacienta kombinovanou léčbou kladribinem a elektronovým ozářením. Pacient se dostal do remise. Délku remise není možné vyhodnotit, pacient asi rok od zahájení léčby zemřel na koronární příhodu [32].
Klinické projevy a léčba
Choroba je podstatně vzácnější než LCH. V 80 % je limitována pouze na kožní povrch, na němž vytváří infiltráty, které lze charakterizovat jako makuly, papuly, noduly a plaky. Výjimečně nemoc postihuje lymfatické uzliny a slezinu. U pacientů s histiocytózou z indeterminovaných dendritických buněk bývají často přítomny dalšími krevní nemoci, nejčastěji typu myeloproliferací. Pro léčbu lze použít kladribin, ale také ozáření elektrony s nízkou energií (electron beam irradiation) nebo UV světlem. Lokálně i systémově bývají podávány steroidy. V případě prokázané BRAF mutace lze použít cílenou léčbu podobně jako u LCH [33–39]. Kožní formu této nemoci ilustruje obr. 11.

Juvenilní xantogranulom
Morfologická podstata
Xantogranulom je označení pro žlutou či žlutooranžovou tkáň, která tvoří uzlíky (granulomy) a může výjimečně i ulcerovat.
Xantom je označení pro žlutou či oranžovožlutavou plochu, mírně vyvýšenou nad povrch, s ostrou hranicí oproti nepoškozené pokožce. Xantomy postihují nejčastěji oblasti očních víček, ale také flexorové části končetin a trupu. Mohou být průvodním znakem hyperlipoproteinemie, ale existují také normolipemické formy.
Histologický obraze xantogranulomů je charakteristický histiocyty, které obsahují fagocytované lipidy. Ty při klasickém barvení tvoří struktury nazývané pěnité buňky (foamy cells). Přítomny jsou také zánětlivé buňky (lymfocyty, plazmocyty) a obrovské mnohojaderné Toutonovy buňky, někdy i krystaly cholesterolu. Pro nekrobiotický xantogranulom jsou charakteristické mapovité oblasti nekrobiózy kolagenu. V okolí ložisek xantogranulomu vznikají zánětlivé, fibrotické a občas i nekrotické struktury. Diagnostická pro tuto chorobu je opět imunohistochemie [2].
Juvenilní xantogranulom je po LCH druhou nejčastější histiocytózou. Jak již napovídá název, juvenilní xantogranulom je typická choroba dětského věku, která často spontánně vymizí. Kožní morfy mají barvu od červené po žlutou a většinou tvoří noduly či papuly, které se objevují na obličeji, krku, trupu.
Pro mimokožní ložiska se používá termín systémový juvenilní xantogranulom, pokud se manifestuje v dětském věku, nebo označení systémový xantogranulom při pozdější manifestaci. Ten může postihovat CNS a také játra a slezinu.
Xantogranulomová ložiska se výjimečně mohou objevit i v dospělém věku a jejich generalizovaná forma, tj. ECD, tvoří samostatnou jednotku WHO klasifikace [2].
Choroby, které jsou patology označeny jako xantogranulom, mohou mít tedy velmi variabilní průběh, od spontánně regredujícího izolovaného ložiska po více ložisek, která progredují.
V rámci skupiny xantomatózních a xantogranulomatózních nemocí dospělých se v literatuře nejčastěji uvádějí následující klinické jednotky, z nichž poslední dvě jsou zřejmě reaktivní formy onemocnění indukované monoklonálním imunoglobulinem [2,3,39].
Klinické formy xantogranulomatózních chorob
Klinickými formami xantogranulomatózních chorob jsou:
- periokulární xantogranulom dospělých (adult onset periocular xantoranuloma) bez vazby na monoklonální imunoglobulin;
- periokulární xantogranulom u dospělých spojený s astmatem (adult onset asthma and periocular xanto-granulomula);
- nekrobiotický xantogranulom (nekrobiotic xanthogranuloma), často (v 80 %) asociovaný s monoklonální gamapatií;
- normolipemická plošná xantomatóza (xanthoma planum), často asociovaná s monoklonální gamapatií;
- ECD – dnes představuje separátní jednotku V. verze WHO klasifikace krevních chorob.
Rozdíly v uvedených jednotkách jsou spíše ve formě klinické prezentace než v morfologii. Možná proto se tyto názvy neodrazily ve WHO klasifikaci krevních (tedy i histiocytárních) nemocí, která uvádí pouze jednotku diseminovaný juvenilní xantogranulom a pod tento termín shrnuje jako synonyma další jednotky, které mají podobný morfologický vzhled, obsahující pěnité histiocyty s tukovými inkluzemi [2,3,39].
Zkušenosti našeho pracoviště
Na našem pracovišti jsme se setkali s kombinací xantogranulomu s nemocí asociovanou s imunoglobulinem IgG4, jejíž projevy vymizely po léčbě rituximabem a cyklofosfamidem. Na kontrolním 18FDG-PET/CT zobrazení však zůstaly morfy v podkoží paží. Ty byly biop-továny a vyšel z nich xantogranulom. Jako léčbu jsme v tomto případě zvolili operační odstranění xantogranulomu. V literatuře je více podobných případů souběhu xantogranulomu s onemocněním asociovaným s imunoglobulinem IgG4 [40,41]. Příčina asociace těchto dvou chorob čeká na objasnění.
Na našem pracovišti jsme se setkali s vícero případy nekrobiotického xantogranulomu asociovaného s monoklonálním imunoglobulinem, v případě dosažení kompletní remise mnohočetného myelomu vymizel i nekrobiotický xantogranulom [42,43].
Léčba a sledování po léčbě
Juvenilní xantogranulom u malých dětí obvykle spontánně regreduje. Pro systémové xantogranulomatózní onemocnění zatím nebyla publikována oficiální mezinárodní doporučení. V jednotlivých v literatuře popsaných případech byl přínosem kladribin nebo klofarabin, ale i jiné léky. Při hledání optimální léčby pro konkrétního pacienta bude nutné tedy vždy konzultovat nové léčebné možnosti v databázi PubMed. Efekt léčby je možné sledovat pomocí 18FDG-PET/CT zobrazení [44–48].
Erdheimova-Chesterova choroba (ECD)
Morfologická podstata
ECD je systémové xantogranulomatózní onemocnění, které tvoří v poslední WHO klasifikaci krevních chorob již samostatnou jednotku. V přechozí ještě bylo řazeno do skupiny juvenilního xantogranulomu. ECD je považována za blízkou formu juvenilního xantogranulomu, histologicky mohou být obě jednotky neodlišitelné. Histologickým podkladem jsou opět xantogranulomová ložiska infiltrující skelet a/nebo další orgány.
Patologické infiltráty tvoří pěnité histiocyty (histiocyty s bohatými tukovými inkluzemi – foamy cells), dále histiocyty s eozinofilní cytoplazmou a příměs malých reaktivních lymfocytů, plazmocytů a neutrofilů. Jsou i případy, kdy dominuje fibróza. Nález připomíná zánět nebo reparativní změny. Diagnosticky cenné jsou vícejaderné buňky Toutonova typu s věnečkem jader kolem eozinofilního středu a s lemem pěnité cytoplazmy na periferii. Histiocyty vykazují pozitivitu CD68, CD163 a faktoru XIIIa a jsou negativní při barvení na S100, CD1a a na langerin [2,49].
Zkušenosti našeho pracoviště
V průběhu posledních 35 let jsme léčili tři pacienty s touto nemocí, kteří neměli prokázanou mutaci BRAFV600E. Pro iniciální léčbu jsme zvolili kladribin. Ve dvou případech jsme dosáhli dlouhodobé kompletní remise této nemoci, ale v jednom z těchto případů jsme nyní prokázali myelodysplastický syndrom. V jednom případě choroba na zvolenou léčbu nereagovala, pořád přetrvávaly známky zánětu indukované touto nemocí a progredovaly perirenální fibrotické změny. Tento pacient je na dlouhodobé udržovací léčbě preparátem anakinra [50–52]. V současnosti jsme přijali do léčby nového pacienta s xantogranulomatózním onemocněním BRAFV600E negativním, u něhož byla předchozí léčba interferonem alfa na jiném pracovišti neúspěšná. Léčbu 2. linie jsme zahájili kladribinem. Ten však neovlivnil intenzivní zánětlivou reakci, s níž mimo jiné souvisí exsudativní perikarditida vyžadující drenáž. Vysoké dávky dexametazonu (20 mg denně) utlumily tvorbu výpotku v perikardu. Takovou dávku dexametazonu je možno podávat jen krátkodobě, proto jsme přešli na tlumení zánětlivé reakce anakinrou – denně si pacient aplikuje jednu ampulku podkožně.
Klinické projevy
Choroba se projevuje symetrickou osteosklerózou postihující diafýzu i metafýzu dlouhých kostí a šetřící epifýzu. Radiologický nález je pro tuto nemoc patognomický. Nicméně 5–8 % pacientů může mít také postiženy ploché kosti, vč. kostí obličejových. Často je chronicky zvýšena hodnota C-reaktivního proteinu (CRP) při normálním prokalcitoninu.
Mimokostní postižení je u této nemoci popisováno v 50 % případů. Byly popsány následující komplikace:
- postižení hypotalamu s následným diabetem insipidem a hypopituarizmem;
- retroperitonální infiltrace s postižením ledvin s perirenální fibrózou;
- ložiska na očních víčkách vzhledu xantalezmat a xantomů, exoftalmus;
- postižení plic, plicní fibróza;
- zesílení cévní stěny velkých cév (periaortitida);
- infiltrace CNS způsobující ataxii či parézy;
- infiltrace viscerálních orgánů, kostí;
- infiltráty na kůži, v orbitě a v paranazálních dutinách.
Klinicky se nemoc projevuje bolestmi končetin a může způsobit klasické zánětlivé projevy zvané v tomto případě B-symptomy, úbytek hmotnosti, subfebrilie či febrilie, noční pocení, patologickou únavu. Laboratorně tomu odpovídají vysoké hodnoty CRP, obvykle s normální hodnotou prokalcitoninu [49].
Průběh nemoci je velmi individuální a odpovídá stupni poškození organizmu, nezřídka byl popsán fatální konec [53–61]. Četnost projevů ilustruje graf 1.
![Grafické znázornění frekvence jednotlivých projevů dle [49].](https://pl-master.mdcdn.cz/media/image/03f505639de09af85a15fcbd0070ac0e.png?version=1781542429)
Typické projevy ECD ilustruje obr. 12.

Stanovení diagnózy
Z klinických projevů a zobrazovacích nálezů lze na tuto chorobu vznést podezření. Osteosklerotizující ložiska lze velmi citlivě znázornit Na18F-PET/CT zobrazením, ale ložiska jsme viděli i na 18FDG-PET/CT zobrazení, protože xantogranulomová ložiska zvýšeně akumulují fluorodeoxyglukózu. Stanovení diagnózy však vyžaduje odběr vzorku pro morfologické vyšetření.
Diagnózu lze provést odběrem z ložiska osteosklerotické tkáně, to obvykle provádí ortoped. Pokud zobrazovací vyšetření prokáže infiltráty v jiných částech těla, velmi často v oblasti retroperitonea, tak o způsobu diagnostického výkonu rozhoduje urolog, který musí volit mezi diagnostickou punkcí pod CT nebo odebráním histologického vzorku z retroperitonea metodou laparoskopie. Retroperitonální fibróza může mít více příčin (primární retroperitoneální fibróza, s imunoglobulinem IgG4 asociované onemocnění, ECD, Castlemanova choroba, inflamatorní myofibroblastový tumor, takže bez odběru vzorku pro histomorfologické vyšetření není možné přesné stanovení diagnózy.
Současná kritéria ECD uvádí tab. 2.

Léčba ECD
Pohled na léčbu se neustále vyvíjí, v tomto textu se přidržíme posledního mezinárodního doporučení:
Pro pacienta s multisystémovým onemocněním s prokázanou BRAFV600 mutací a život ohrožujícím průběhem, zejména s postižením kardiovaskulárním či neurologickým, se dnes pro léčbu 1. linie doporučují BRAF-inhibitory, vemurafenib nebo dabrafenib. Výběr BRAF-inhibitoru by se měl řídit dle nežádoucích účinků preparátů a dle zkušeností ošetřujícího lékaře s preparátem.
U pacientů, jejichž stav umožňuje podání systémové chemoterapie, nebo u pacientů netolerujících cílenou léčbu se doporučuje léčba kladribinem. Výhodou této léčby je její časově omezený interval a naděje na dlouhodobější remisi.
Pro pacienty s menším rozsahem choroby, která postihuje kosti a retroperitoneum, se doporučují biologické léky blokující interleukin-1, ponejvíce anakinra [53–60].
Problémem výše uvedené biologické léčby inhibitory BRAF a MEK je ne zcela dobrá tolerance dlouhodobé biologické léčby. Goyal et al. popsali v souboru 64 pacientů sice 85 % léčebných odpovědí, ale u 61 % z nich bylo nutné léčbu přerušit pro nežádoucí účinky [61]. Takže pokud je účinná klasická léčba kladribinem, je to pro pacienty výhodou.
Sledování po léčbě
18FDG-PET/CT je metodou volby, doporučuje se ve 3. a 6. měsíci po zahájení léčby. Dle nejvíce postiženého orgánu (srdce, mozek, orbity) se pak doporučuje cílené vyšetřování (CT nebo magnetická rezonance). Vzhledem k tomu, že fibróza je součástí patologického procesu, tak ani při maximálním potlačení aktivity nemoci nevymizí fibrotické změny postižených orgánů.
CRP je zvýšen u 80 % případů a jeho pokles signalizuje léčebnou odpověď.
Endokrinopatie jsou ale obvykle neměnné. Naopak, pokud na počátku nebyl přítomný diabetes insipidus, může se v průběhu sledování objevit. V průběhu sledování se mohou projevit také myeloidní neoplazie, a proto je třeba kontinuálně sledovat krevní obraz [49].
Rosaiova-Dorfmanova nemoc (RDD)
Morfologická podstata
RDD (Rosai-Dorfman disease, někdy též zvaná Rosai-Dorfman-Destombes disease), synonymum sinusová histiocytóza s masivní lymfadenopatií, typicky postihuje krční uzliny a často tvoří masivní lymfadenopatii. Choroba byla v době publikace IV. WHO klasifikace krevních chorob považována za reaktivní, proto do ní nebyla zahrnuta. Metody molekulární biologie prokázaly, že jde o klonální proliferaci. To vedlo k jejímu zařazení do V. verze WHO klasifikace krevních chorob.
Morfologickou podstatou ložisek RDD je infiltrace uzliny a jejich sinusů velkými bledými makrofágy. Sinusoidy obsahují množství velkých histiocytárních buněk s hladkými obrysy hypochromatických jader. Výjimečná je dvojjadernost, atypie a vzácné mitotické figury. Většina buněk však obvykle zachovává hladké jaderné obrysy a bohatou bledou cytoplazmu. Diagnostickým, ale nikoliv specifickým znakem je přítomnost neporušených hematolymfoidních buněk uvnitř vakuol nebo volně plovoucích v cytoplazmě histiocytů. Tento jev – průnik jiné krvinky do histiocytu, aniž by došlo k její destrukci – se nazývá emperipolesis. Je typickým znakem této nemoci. Emperipolesis je tedy jiný jev než fagocytóza. Při fagocytóze dochází k destrukci pohlcené buňky.
Velké histiocytární buňky exprimujíznak S100 a histiocytární markery (CD68,CD163) a dle definice jsou negativní na průkaz znaků LCH, CD1a a langerinu (CD207). Plazmatické buňky kortexu paknesou znaky CD38, CD138 a MUM1 a mohou zde být velmi četné IgG4-pozitivní plazmatické buňky. Uvádí se, že v případě RDD obsahují ložiska často vyšší počet IgG4+ plazmocytů, a proto je odlišení od IgG4-related disease (s imunoglobulinem IgG4 asociovaným onemocněním) velmi obtížné, ne-li nemožné [2,3,62].
Zkušenosti našeho pracoviště
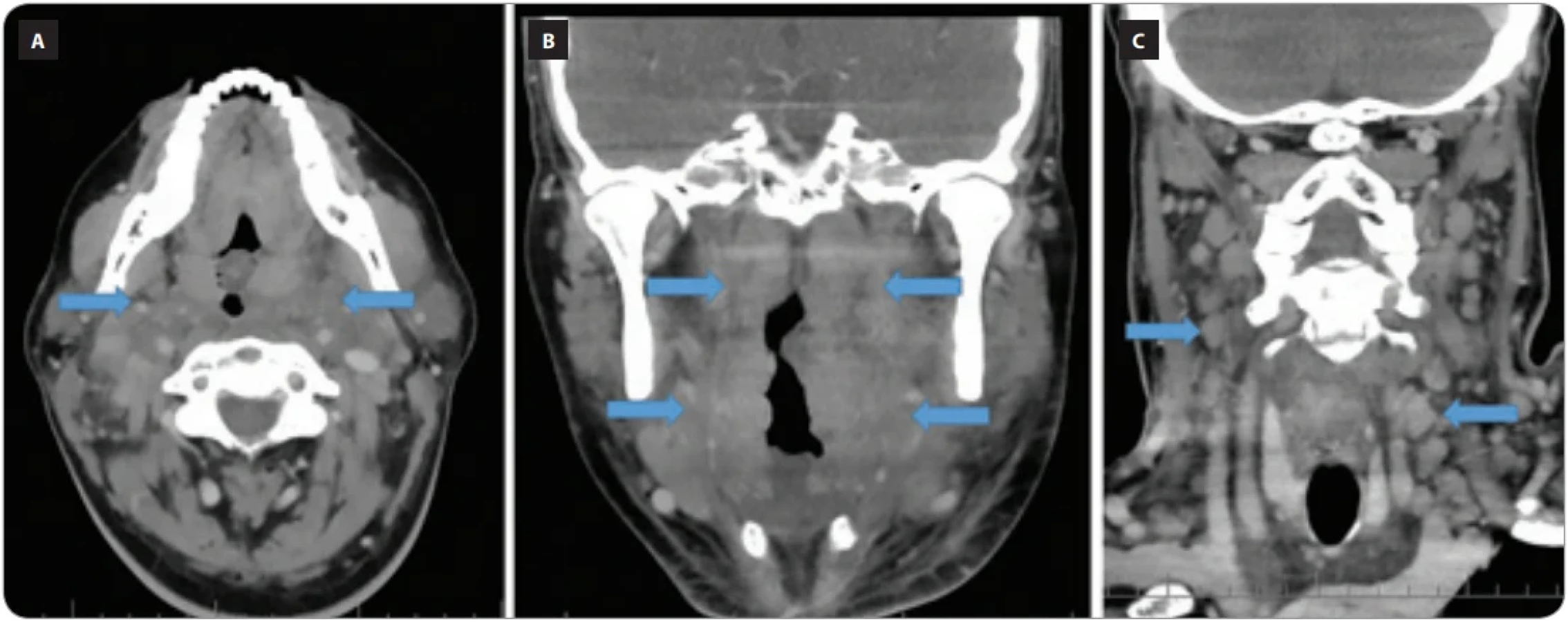
Na našem pracovišti jsme léčili dva případy. V prvním případě léčba kladribinem dosáhla dlouhodobé parciální remise [63,64] (obr. 13). V druhém případě, který byl diagnostikován v říjnu 2025, nás překvapila choroba prudkostí nástupu. Pacient si zvětšování uzliny všiml v září 2025, uzliny na krku se mu rychle zvětšovaly, začaly komprimovat dýchací cesty, což si vyžádalo dočasnou tracheostomii a plicní ventilaci (obr. 14). Překvapivé ale bylo také to, že po léčbě prednisonem 1 mg/kg hmotnosti v průběhu týdne uzliny vymizely a ultrazvukové vyšetření krku, provedené 10 dní po zahájení léčby, již neprokázalo žádnou lymfadenopatii, ačkoliv, jak ukazuje obr. 14, postižení bylo masivní.

Klinické projevy
Lymfadenopatie
Ze synonyma RDD sinusová histiocytóza s masivní lymfadenopatií vyplývá, že nemoc nejčastěji způsobuje nebolestivou cervikální lymfadenopatii. Ta může být asociována se systémovou zánětlivou reakcí a typickými B-symptomy. Postižení inguinálních a abdominálních uzlin je méně časté [62]. Lymfadenopatickou formu v domácí literatuře popsali Králík et al. [65].
Z laboratorních znaků provází systémové onemocnění zvýšená sedimentace erytrocytů, leukocytóza, polyklonální hypergamaglobulinemie, zvýšený feritin a někdy i autoimunitní hemolytická anemie [62].
Extranodální RDD
Extranodální lokalizace RDD je nalézána asi u 40 % případů a většinou je přítomna i s lymfadenopatií. Výjimečné jsou extranodální lokalizace RDD bez lymfadenopatie. K častým mimouzlinovým lokalizacím patří kůže (10 %), paranazální dutiny (11 %), kosti (5–10 %), orbity a retrobulbární prostor (11 %) a CNS s predominantním postižení tvrdé pleny mozkové (5 %) [62].
Kožní RDD
Kožní forma RDD postihuje obvykle osoby starší 40 let. Kožní forma bývá izolovaná bez extrakutánního postižení a nemívá tendence k rozšiřování, takže u této formy nemoci je chirurgická excize kurativní [62].
Pachymeningitida a jiné postižení CNS
Postižení tvrdé pleny mozkové chorobou RDD vytváří obraz pachymeningitidy nebo meningeomu, jak v domácí literatuře popsali Medek et al. [66]. Při lokalizaci RDD v mozkových obalech nebývá současná lymfadenopatie [62,67].
Zahraniční analýza histiocytárních projevů v CNS uvádí, že mnohočetná tumorózní ložiska s predominantním postižením mening jsou obrazem RDD anebo ECD, vaskulární postižení je typické pouze pro ECD. LCH postihuje ve většině případů hypothalamo-pituitární osu [68].
V literatuře lze nalézt četné popisy případů, u nichž příčinou závažných neurologických problémů byla postižení mozkových obalů RDD [69–71].
Pachymeningitida představuje vždy obtížný diferenciální diagnostický problém, protože ji může způsobit jak RDD, tak se velmi často popisuje jako projev choroby asociované s imunoglobulinem IgG4 [72,73].
A někdy od sebe tyto dvě příčiny pachymeningitidy nejdou odlišit. Proto také klasifikace Histiocyte Society [3] zdůrazňuje vazbu RDD s onemocněním asociovaným s imunoglobulinem IgG4 a uvádí několik forem RDD (tab. 3).
Vaskulitida
Postižení kardiovaskulárního systému RDD je vzácné, ale vyskytuje se. Odborné literatura eviduje více než 30 popisů případů s kardiovaskulární manifestací. V některých popsaných případech byla také postižena aorta. U některých pacientů nebyla přítomna ani lymfadenopatie a diagnóza RDD byla stanovena až po cévní operaci histologicky, která prokáže histiocytární proliferaci s jevem zvaným emperipolesis, zánětlivou infiltrací a imunohistochemickým průkazem CD.
U těchto forem se velmi osvědčilo sledování pomocí FDG-PET/CT zobrazení [74–77].
Překryv RDD s nemocí asociovanou s imunoglobulinem IgG4
V odborné literatuře je hodně publikací jak o morfologickém překryvu, tak o překryvu klinických projevů RDD s IgG4-related disease. V popsaných případech nemoc vykazovala znaky obou těchto chorob. A proto i klasifikace Histiocyte Society zahrnuje tyto případy [78–82] (tab. 1).
Léčba RDD
V nezávažných případech je vhodnou alternativou sledování, protože 20–50 % pacientů s nodálními či kožními manifestacemi má spontánní remise. V případě izolované kožní formy nemoci je nejúčinnější léčbou operační odstranění ložiska. Chirurgická intervence může přinést profit při závažné multifokální formě nemoci. Léčebné doporučení pro tuto nemoc je z roku 2018 a zatím nebylo upgradováno [62].
Glukokortikoidy obvykle zmenší jak velikost ložiska, tak i symptomy. V případech orbitálního poškození měl v ně-kterých případech léčebný účinek prednison v dávce 40–70 mg denně. Při srovnání se sarkoidózou se zde používá vyšší dávky prednisonu (> 0,5 mg/kg/den) nebo dexametazonu (8–20 mg/den). Léčba pomocí klasické chemoterapie má smíšené výsledky, testovány byly velmi pestré režimy se střídavými úspěchy a neúspěchy [62].
Nukleosidová analoga, kladribin a klofarabin mají vyšší potenciál léčebné odpovědi. Kladribin se podává v dávce 5 mg/m2/den 5 dní po sobě v 28denních intervalech, 4–6 cyklů. Tato léčba přináší remise i u pacientů, jejichž nemoc je refrakterní na glukokortikoidy [83–86].
Klofarabin se podává v dávce 25 mg/m2/den také 5 dní po sobě v podobném počtu cyklů. Oba dva léky se doporučují v závažnějších případech, kdy potenciální přínos převáží nežádoucí účinky těchto léků [62]. Výjimečně byl testován sirolimus s pozitivním výsledkem [84,85]. Lenalidomid má taktéž prokázán léčebný přínos [62,86]. Na rozdíl od ECD a LCH u této nemoci mutace BRAFV600E nejsou pozorovány, a tak není relevantní použití inhibitorů BRAF. MEK inhibitory se v této indikaci teprve testují. V refrakterním případech se doporučuje vyšetření mutace signální cesty MAPK a v případě prokázání této mutace cílená léčba [87,88].
Literatura popisuje ve více případech také léčebný účinek rituximabu, v popsaných případech, kdy pomohl rutiximab, však byla obvykle ještě přítomna zánětlivá reakce, automunita či znaky IgG-related disease [89–91].
Radioterapie je u této nemoci středně účinná, obvykle se používají dávky 30–50 Gy [92].
ALK-pozitivní histiocytózy
ALK-pozitivní histiocytózy jsou vzácným podtypem histiocytárních neoplazií, poprvé popsaným v roce 2008 u tří dětí s multisystémovým onemocněním postihujícím játra a hematopoetický systém.
Tato jednotka byla následně předmětem sérií dalších popisů případů, což ukázalo, že spektrum ALK-pozitivních histiocytóz je relativně pestré.
Největší skupina pacientů s ALK-pozitivní histiocytózou byla popsána nedávno. Histologie odpovídala ve třetině případů klasickému obrazu xantogranulomu, zatímco u ostatních pacientů měla ložiska charakter denzních monomorfních infiltrátů bez přítomnosti histiocytů obsahujících lipidy. Ložiska ALK-pozitivní histiocytózy měla někdy vřetenovitou či epiteloidní morfologii.
Histiocyty u ALK-pozitivních histiocytóz mohou nabývat různých tvarů, vč. velkých oválných buněk, pěnitých buněk, vřetenovitých buněk, a někdy jsou mnohojaderné, vč. Toutonových gigantických buněk. Neoplastické histiocyty vykazují pozitivitu makrofágových markerů a často vykazují znaky aktivace MAPK signální cesty. I v případech ALK-pozitivních histiocytóz může být pozorována emperipolesis.
Morfologická diagnostika ne vždy dochází k jednoznačnému závěru, a zásadní je tedy molekulárně biologický průkaz ALK translokace. Fúzní gen KIF5B-ALK byl detektován u 27 pacientů, zatímco CLTC-ALK, TPM3-ALK, TFG-ALK, EML4-ALK a DCTN1-ALK fúzní geny byly identifikovány jen v jednotlivých případech. Průkaz uvedených mutací pak opravňuje k podání cílené léčby [93,94].
Histiocytární sarkom
Morfologická podstata
Morfologická diagnostika histiocytárního sarkomu je vzhledem k široké diferenciální diagnostice velmi obtížná. Nádor se totiž v základním barvení podobá jak velkobuněčným lymfomům, tak nediferencovanému karcinomu, melanomu, vřetenobuněčnému nebo pleomorfnímu sarkomu. Jedině aplikace širokého panelu protilátek pro imunohistochemické vyšetření fenotypu neoplazie dokáže tyto jednotky vyloučit. Musí být pozitivní alespoň některé histiocytární znaky (CD68, CD163, lysozym). Znak CD4 bývá v histiocytárních sarkomech atypicky cytoplazmaticky pozitivní, ale není původním histiocytárním markerem. Komplikované je také odlišení myelosarkomu, v čemž může pomoci klinická anamnéza.
Před zavedením imunofenotypizace byla diagnóza maligní histiocytózy stanovena mnohem častěji, protože četné B - i T-buněčné lymfoproliferace byly považovány za histiocytární malignity. V současné době jsou patology diagnostikovány velice zřídka.
Přibližně třetina histiocytárních sarkomů se manifestuje lokalizovanou lymfadenopatií, třetina se manifestuje kožními ložisky (solitární či mnohočetná) a poslední třetina vzniká extranodálně, často v oblasti zažívacího traktu.
Někteří nemocní mají systémové postižení (s mnohočetnými ložisky), jehož popis se může shodovat s dřívějšími popisy maligní histiocytózy. WHO klasifikace by nyní pro tento stav použila termínu generalizovaná či diseminovaná forma histiocytárního sarkomu [2,3].
Lokalizovaný histiocytární sarkom
Tato jednotka je odvozena od fagocytujících mononukleárních buněk ve stadiu tkáňové fixace a diferenciace, tj. zralých makrofágů. Může vzniknout jak v kůži, v podkoží, tak i v nadklíčku, mediastinu či v zažívacím traktu nebo v kostech. Pokud se nepodaří totální operační odstranění s lemem zdravé tkáně, tak je tento tumor poměrně rezistentní k následné chemoterapii i radioterapii [2,3].
Diseminovaný histiocytární sarkom, synonymem maligní histiocytóza
Někteří pacienti s histiocytárním sarkomem mají mnohočetné postižení vč. hepatomegalie a splenomegalie, což odpovídá staršímu popisu maligní histiocytózy. Tento termín se dnes již nepoužívá a místo něj se používá termín diseminovaný histiocytární sarkom.
Tato diseminovaná forma histiocytárního sarkomu je velmi agresivně probíhající nemoc. Klinické příznaky se podobají projevům lymfoblastické leukemie s generalizovaným postižením orgánů. Maligní histiocytózu velmi často provází vysoké horečky nad 39 °C, splenomegalie (100 %), lymfadenopatie (92 %), hepatomegalie (67 %). Mohou však být infiltrovány i jiné orgány, např. plíce, mozek, kůže, což k výše uvedeným příznakům může přidat dušnost či bolesti hlavy. Někdy způsobuje osteolýzu a s ní spojené bolesti kostí. Kožní manifestace může nabývat různých podob, od benigně vyhlížejícího exantému až po četné kožní tumory trupu a končetin. Postižení střeva se často projeví obstrukčními příznaky.
Nemoc charakterizují následující laboratorní změny: trombocytopenie (92 %), anemie (92 %), leukocytopenie (67 %). V biochemickém vyšetření jsou u těchto pacientů velmi často detekovány vysoké hodnoty laktátdehydrogenázy a bilirubinu, přičemž jaterní enzymy a renální funkce bývají jen nepatrně zhoršené. Při postižení CNS lze často nalézt v mozkomíšním moku patologické fagocytující neoplastické histiocyty. Klinicky se tyto diseminované histiocytární sarkomy chovají velmi agresivně [2,3].
Analýza National Cancer Database v USA z let 2004–2015 hodnotila 409 případů. Medián věku stanovení diagnózy byl 61 let. Nejčastější lokalizací byla kůže, podkoží a pojivová tkáň (41 %) následovaná lymfatickými uzlinami (14 %) gastrointestinálním traktem (12 %) a hematopoetickým systémem (8 %). Medián přežití byl jen 6 (rozmezí 1–127) měsíců [95].
Optimální léčba histiocytárního sarkomu není definována. Uplatňují se agresivní polychemoterapeutické režimy, používané jinak pro léčbu agresivních lymfomů, ale jejich účinnost je malá, jak vyplývá z uvedené analýzy. Proto jsou v posledních letech u pacientů s histiocytárním sarkomem prováděny analýzy genetických mutací. Pokud se prokáže některá z blokovatelných mutací, je použita cílená léčba. Při průkazu senzitivity na PD-L1 blokádu je možno použít nivolumab či jiné check-point inhibitory [96–98].
Zkušenosti našeho pracoviště
Léčbou vysoce agresivních histiocytárních chorob se na našem pracovišti od roku 2015 zabývá oddělení solidních tumorů pod vedením MUDr. Michala Eida, který rozvíjí léčbu histiocytárních sarkomů, sarkomů z folikulárně dendritických buněk a sarkomů z interdigitujících dendritických buněk a uplatňuje u nich možnosti precizní medicíny. Všechny tyto tři klinické jednotky spadaly dle starší klasifikace do kategorie vysoce agresivních histiocytárních chorob. Poslední, V. verze WHO klasifikace vytvořila z posledních dvou entit samostatnou skupinu.
Folikulární dendritické buňky (follicular dendritic cells – FDC) jsou mezenchymálního původu a jsou umístěné v B-folikulech, kde zachycují a prezentují antigeny B-lymfocytům.
Interdigitující dendritické buňky (interdigitating dentritic cells – IDC) jsou taktéž mezenchymální původu a také se podílejí na prezentaci antigenů. Sarkomy od nich odvozené tvoří nyní samostatnou skupinu, zatímco v přechozí, IV. verzi WHO klasifikace krevních chorob patřily do skupiny histiocytárních malignit.
Sarkom z FDC se může vyskytovat kdekoliv po těle a jeho průběh je těžko predikovatelný. Velká část sarkomů z FDC exprimuje PD-L1. Sarkom z IDC postihuje zejména lymfatické uzliny, méně pak slezinu, tenké střevo či tonzily. Sarkomy z FDC a IDC většinou postihují pacienty ve středním věku a prognóza je špatná.
V roce 2024 referoval MUDr. Eid o výsledcích léčby této skupiny pacientů na Brněnských onkologických dnech. Léčba histiocytárního sarkomu, sarkomu z FDC a IDC není standardizována. Mimo režimy pro agresivní lymfomy lze pro sarkom z FDC otestovat účinnost tyrozinkinázových či mTOR inhibitorů nebo imunoterapii.
Od roku 2015 do září 2025 bylo na ambulancích pro solidní nádory Interní hematologické a onkologické kliniky FN Brno diagnostikováno a léčeno celkem 11 pacientů s histiocytárním sarkomem, čtyři pacienti se sarkomem z FDC a jeden pacient se sarkomem z IDC. Z toho počtu bylo osm pacientů (50 %) diagnostikováno s lokalizovaným či lokálně pokročilým onemocněním. Druhá polovina pacientů byla vstupně diagnostikována s metastatickým postižením.
Ve skupině pacientů s nemetastatickou chorobou byli dva léčeni režimem osahujícím gemcitabin, cisplatinu a dexametazon (GDP) s následnou radioterapií (25 × 2,0 Gy) v rámci neoadjuvance a u obou bylo dosaženo dlouhotrvající kompletní patologické remise. Další jedna pacientka měla po chemoterapii a radioterapii jen částečnou odpověď a byla indikována k dalším liniím systémové terapie vč. pazopanibu a imunoterapie nivolumabem pro PD-L1 expresi 100 %. Oba dva preparáty vedly k dlouhodobé regresi choroby navzdory výrazné předléčenosti. Finálně bylo u této pacientky dosaženo sekundární resekability solitárního postižení v oblasti pravé mandibuly s nutností rekonstrukčního výkonu. Pacientka je v dlouhodobé remisi choroby. Zbylí pacienti v této skupině byli primárně resekováni s nebo bez adjuvantní radioterapie.
Ve skupině pacientů s iniciálně metastatickou chorobou byly použity chemoterapeutické léčebné režimy jinak používané pro léčbu lymfomů. Nejlepší efekt byl pozorován u pacientů léčených již zmíněným režimem GDP. U jedné pacientky byla pozorována regrese oligometastatického postižení po indukční chemoterapii a následně byla provedena radioterapie. Dle recentního přešetření ze září 2025 došlo k výrazné regresi mediastinální masy a infiltrace supraklavikulárně vlevo. Bude zvažována sekundární resekabilita. Pro systémovou terapii je doporučen i režim s cyklofosfamidem, doxorubicinem, vinkristinem a prednisonem (CHOP), event. obohacený o etoposid (CHOEP). S těmito režimy jsme u našich pacientů dosáhli nejlepší odpovědi ve formě stabilizace choroby. Dva pacienti se sarkomem z FDC byli léčeni pazopanibem a bylo dosaženo dlouhotrvající regrese. U pěti pacientů bylo provedeno prediktivní panelové sekvenování s následujícími nálezy:
- fúze NTRK u histiocytárního sarkomu – léčba entrektinibem, bez efektu (progrese po 3 měsících terapie);
- vysoká mutační nálož TMB-high (18 mutací/megabázi) u histiocytárního sarkomu – léčba pembrolizumabem, časné úmrtí na influenzu-A;
- mutace PALB2 u histiocytárního sarkomu – léčba cisplatinou, dlouhodobá stabilizace;
- mutace JAK2 u sarkomu z FDC – léčba ruxolitinibem, bez efektu (progrese po 3 měsících terapie).
Všichni pacienti léčení cílenou terapií byli výrazně předléčení [99,100].
Přehled důležitých mutací pro léčbu histiocytárních chorob
Signální dráha MAPK (mitogen-aktivovaná proteinkináza) spojuje extracelulární a intracelulární procesy, které kontrolují proliferaci, migraci a apoptózu buněk. Jakékoliv abnormality v této signální dráze vedou k progresi nebo vývoji nádorů. Somatické mutace v této dráze hrají roli v diferenciální diagnostice ECD a jiných histiocytóz. Mutace v RAS a BRAF genech jsou kritické u ECD, protože ovlivňují terapii MAPK inhibitory.
ALK (anaplastic lymphoma kinase) je receptorová tyrozin kináza. Mezi jejími aktivujícími ligandy byly zatím identifikovány dva proteiny, pleiotrofin a midkine, které ale nejsou specifické pro ALK. ALK kináza aktivuje několik signálních drah (mTOR, JAK/STAT, MAPK a jiné), které ovlivňují proliferaci, transformaci a antiapoptotické dráhy. Zatím bylo identifikováno 21 genů, které jsou translokačními partnery ALK. Všechny tyto translokace vedou k aktivaci proteinkinázové domény ALK, a tedy k tumorigenicitě. U buněk ovlivňují různé procesy, např. proliferaci, přežití, diferenciaci a migraci.
V některých případech se u stejného pacienta mohou nacházet lymfatické neoplazie se stejnou genetickou změnou. Pro tento jev byl zaveden termín transdiferenciace [2,3].
Závěr
Z uvedeného popisu je zřetelná nezměrná pestrost klinických projevů, ale i morfologických obrazů histiocytárních chorob. A poslední roky přinesly důraz na vyšetření mutací, které se u těchto nemocí vyskytují. Při průkazu mutace ovlivnitelné cílenou léčbou je možné tuto léčbu podat jen se schválením plátce zdravotní péče. Proto je při odběru materiálu na histologické vyšetření žádoucí odebrat nativní vzorek i k pozdějšímu cílenému vyšetření možných mutací, jejichž průkaz by mohl nasměrovat další terapeutické kroky. Pro molekulární diagnostiku histiocytárních neoplazií i pro účely prediktivní onkologie lze využít standardní formolparafinový materiál biopsie.
Vzhledem k tomu, že jde o velmi vzác-né choroby, je vhodné při stanovení diagnózy vždy přihlížet k publikovaným popisům nemoci, a pokud nekoreluje popis nemoci a histologie, tak znovu prověřovat, jak nyní právě provádíme u pacientky zobrazené na obr. 4. Na diagnóze multicentrická retikulohistiocytóza se shodlo více patologů. Literatura popisuje tuto nemoc jako zánětlivé onemocnění asociované s erozivní artritidou, postihující zejména akrální části těla, které jsou vystaveny slunci [101,102]. Zajímavé je, že tato diagnóza není uvedena ve WHO klasifikaci histiocytárních chorob, ale je uvedena ve WHO klasifikaci kožních chorob a v klasifikaci histiocytárních chorob dle Histiocyte Society. Zobrazená pacientka nemá vyjma kožních projevů žádné další patologie, což potvrdilo i PET/CT, proto je v plánu další biopsie s histologickým ověřením.
Dedikace
Podpořeno: MZ ČR – RVO (FNBr, 65269705 a MOÚ, 00209805).
Zdroje
1. Adam Z, Hermanová M, Horváth T et al. Přehled histiocytárních a dendritických neoplazií dle 5. verze WHO klasifikace krevních chorob z roku 2022. Klin Onkol 2024; 37 (3): 164–172. doi: 10.48095/ccko2024164.
2. Khoury JD, Solary E, Abla O et al. The 5th edition of the World Health Organization classification of haematolymphoid tumours: myeloid and histiocytic/dendritic neoplasms. Leukemia 2022; 36 (7): 1703–1719. doi: 10.1038/s41375-022-01613-1.
3. Emile JF, Abla O, Fraitag S et al. Histiocyte Society. Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood 2016; 127 (22): 2672–2681. doi: 10.1182/blood-2016-01-690636.
4. Liu H, Stiller CA, Crooks CJ et al. Incidence, prevalence and survival in patients with Langerhans cell histiocytosis: a national registry study from England, 2013–2019. Br J Haematol 2022; 199 (5): 728–738. doi: 10.1111/bjh.18459.
5. Makras P, Stathi D, Yavropoulou M et al. The annual incidence of Langerhans cell histiocytosis among adults living in Greece. Pediatr Blood Cancer 2020; 67 (9): e28422. doi: 10.1002/pbc.28422.
6. Adam Z, Rehák Z, Koukalová R et al. Plicní forma histiocytózy z Langerhansových buněk – hodnocení aktivity nemoci a léčebné odpovědi pomocí PET-CT (indexu SUV (max) Pulmo/SUV (max) Hepar). Popis vlastních zkušeností a přehled literatury. Vnitr Lek 2010; 56 (12): 1228–1250.
7. Řehák Z, Koukalová R, Szturz P et al. Measuring diffuse metabolic activity on FDG-PET/CT: new method for evaluating Langerhans cell histiocytosis activity in pulmonary parenchyma. Nucl Med Biol 2012; 39 (3): 429–436. doi: 10.1016/j.nucmedbio.2011.10.002.
8. Adam Z, Krejčí M, Pour L et al. Odlišné průběhy recidivující anebo multisystémové formy histiocytózy z Langerhansových buněk u dospělých osob – popis 22 případů z jednoho pracoviště. Vnitr Lek 2010; 56 (6): 542–556.
9. Adam Z, Nebeský T, Szturz P et al. Postižení plic u pacientů s multiorgánovou formou histiocytózy z Langerhansových buněk. Popis 8 pacientů a přehled literatury. Vnitr Lek 2010; 56 (Suppl 2): 2S105–2S122.
10. Doubková M, Adam Z, Doubek M et al. Diagnostika a léčba plicní formy histiocytózy z Langerhansových buněk. Stud Pneumol Phtiseol 2020; 80 (2): 70–75.
11. Adam Z, Balšíková K, Krejčí M. et al. Centrální diabetes insipidus u dospělých osob – první příznak histiocytózy z Langerhansových buněk a Erdheimovy-Chesterovy choroby. Popis tří případů a přehled literatury. Vnitr Lek 2010; 56 (2): 138–148.
12. Reisi N, Raeissi P, Harati et al. Unusual sites of bone involvement in Langerhans cell histiocytosis: a systematic review of the literature. Orphanet J Rare Dis 2021; 16 (1): 1. doi: 10.1186/s13023-020-01625-z.
13. Mlynček M, Uharček P. Vulvárna histiocytóza z Langerhansových buniek. Klin Onkol 2005; 18 (4): 134–137.
14. Mecherová J, Matějek T, Kamarádová K et al. Nehojící se atopický ekzém. Čes-slov Pediat 2017; 72 (6): 352–360.
15. Bui AN, Larocca C, Giobbie-Hurder A et al. Cutaneous Langerhans cell histiocytosis in adults: a retrospective cohort study of adult patients presenting to a single academic cancer center between 2003 and 2017. J Am Acad Dermatol 2022; 86 (6): 1413–1416. doi: 10.1016/j.jaad.2021.06.016.
16. Jouenne F, Benattia A, Tazi A. Pulmonary Langerhans cell histiocytosis – an update on pathogenesis and treatment. Curr Opin Pulm Med 2023; 29 (5): 451–458. doi: 10.1097/MCP.0000000000000988.
17. Singla A, Kopras EJ, Gupta N. Spontaneous pneumothorax and air travel in pulmonary Langerhans cell histiocytosis: a patient survey. Respir Investig 2019; 57 (6): 582–589. doi: 10.1016/j.resinv.2019.07.004.
18. Keane RJ, Subramaniam A, Varghese C et al. Initial presentation of pulmonary Langerhans cell histiocytosis as recurrent spontaneous pneumothoraces. Respir Med Case Rep 2020; 31 : 101280. doi: 10.1016/j.rmcr.2020.101280.
19. Montefusco L, Harari S, Elia D et al. Endocrine and metabolic assessment in adults with Langerhans cell histiocytosis. Eur J Intern Med 2018; 51 : 61–67. doi: 10.1016/j.ejim.2017.11.011.
20. García Gallo MS, Martínez MP, Abalovich MS et al. Endocrine manifestations of Langerhans cell histiocytosis diagnosed in adults. Pituitary 2010; 13 (4): 298–303. doi: 10.1007/s11102-010-0233-8.
21. Melzer JM, Winters J, Mitchell AO. Isolated adult lymphadenopathy: a rare presentation of Langerhans cell histiocytosis. Am J Otolaryngol 2015; 36 (1): 103–105. doi: 10.1016/j.amjoto.2014.10.016.
22. Guo Y, Ning F, Wang G et al. Retrospective study of Langerhans cell histiocytosis in ear, nose and neck. Am J Otolaryngol 2020; 41 (2): 102369. doi: 10.1016/j.amjoto.2019.102369.
23. Smilek P, Pažourková M. Projevy histocytózy z Langerhansových buněk v ORL oblasti. Vnitr Lek 2010; 56 (Suppl 2): 2S76–2S84.
24. Kiratli H, Tarlan B, Söylemezoglu F. Langerhans cell histiocytosis of the orbit. Eur J Ophthalmol 2013; 23 (4): 578–583. doi: 10.5301/ejo.5000244.
25. Li J, Wu H, Wang Z et al. Langerhans cell histiocytosis of the jaw: clinical analysis of 68 cases. Orphanet J Rare Dis 2025; 20 (1): 191. doi: 10.1186/s13023-025-03680-w.
26. Panis V, Nikitakis N, Daskalopoulos A et al. Langerhans cell histiocytosis mimicking aggressive periodontitis: challenges in diagnosis and management. Quintessence Int 2016; 47 (9): 731–738. doi: 10.3290/j.qi.a36568.
27. Fassmann A, Izakovičová Hollá L, Augustin P et al. Projevy histocytózy z Langerhansových buněk v orofaciální oblasti. Vnitr Lek 2010; 56 (Suppl 2): 85–90.
28. Goyal G, Young JR, Koster MJ et al. The Mayo Clinic Histiocytosis Working Group consensus statement for the diagnosis and evaluation of adult patients with histiocytic neoplasms: Erdheim-Chester disease, Langerhans cell, histiocytosis, and Rosai-Dorfman disease. Mayo Clin Proc 2019; 94 (10): 2054–2071. doi: 10.1016/j.mayocp.2019.02.023.
29. Goyal G, Tazi A, Go RS et al. International expert consensus recommendations for the diagnosis and treatment of Langerhans cell histiocytosis in adults. Blood 2022; 139 (17): 2601–2621. doi: 10.1182/blood.2021014343.
30. Adam Z, Szturz P, Vaníček J et al. Cladribine (2-chlorodeoxyadenosine) in frontline chemotherapy for adult Langerhans cell histiocytosis: a single-center study of seven cases. Acta Oncol 2013; 52 (5): 994–1001. doi: 10.3109/0284186X.2012.716164.
31. Gabanec F, Šimkovič, M Zavřelová A et al. Treatment of multifocal multisystem BRAF positive Langerhans cell histiocytosis with cladribine, surgery and allogenic stem cell transplantation. Acta Medica (Hradec Králové) 2017; 60 (4): 152–156. doi: 10.14712/18059694.2018.11.
32. Adam Z, Ježová M, Šlampa P et al. Histiocytóza z indeterminovaných buněk – vymizení kožní infiltrace po ozáření elektronovým svazkem a aplikace 2-chlorodeoxyadenozinu: kazuistika. Vnitr Lek 2017; 63 (4): 284–288.
33. Fischer AS, Zaladonis AG, Subrt P et al. Indeterminate cell histiocytosis mimicking rosacea. Cureus 2021; 13 (1): e12850. doi: 10.7759/cureus.12850.
34. Lie E, Jedrych J, Sweren R et al. Generalized indeterminate cell histiocytosis successfully treated with methotrexate. JAAD Case Rep 2022; 25 : 93–96. doi: 10.1016/j.jdcr.2022.05.027.
35. Liu T, Cai HC, Cai H et al. Intermediate-dose cytarabine is an effective therapy for adults with non-Langerhans cell histiocytosis. Orphanet J Rare Dis 2022; 17 (1): 39. doi: 10.1186/s13023-022-02193-0.
36. Zanella S, Berti E, Bonometti A. Indeterminate cell histiocytosis: a systematic review of the literature with a comprehensive revision of clinical, histopathological, and molecular features. J Eur Acad Dermatol Venereol 2023. Ahead of Print. doi: 10.1111/jdv.19095.
37. Sulejmani P, Brunsgaard EK, Decker ME et al. Indeterminate cell histiocytosis and a review of current treatment. Cutis 2025; 115 (1): 26–27. doi: 10.12788/cutis.1150.
38. Ozkaya N, Melloul Benizri S, Venkataraman G et al. Indeterminate DC histiocytosis is distinct from LCH and often associated with other hematopoietic neoplasms. Blood Adv 2024; 8 (22): 5796–5805. doi: 10.1182/bloodadvances.2024013545.
39. Höck M, Zelger B, Schweigmann G et al. The various clinical spectra of juvenile xanthogranuloma: imaging for two case reports and review of the literature. BMC Pediatr 2019; 19 (1): 128. doi: 10.1186/s12887-019-1490-y.
40. Andron AA, Nair AG, Della Rocca D et al. Concomitant adult onset xanthogranuloma and IgG4-related orbital disease: a rare occurrence. Orbit 2022; 41 (1): 108–111. doi: 10.1080/01676830.2020.1814353.
41. Sahu AK, Tripathy SR, Parida MK et al. Adult-onset asthma with periocular xanthogranuloma (AAPOX) associated with IgG4-related disorder: a case report and review of current literature. Cureus 2025; 17 (4): e82617. doi: 10.7759/cureus.82617.
42. Adam Z, Veselý K, Motyčková I et al. Oční víčka se žlutými granulomy a kašel – periokulární xantogranulom dospělých spojený s astmatem. Popis případu a přehled klinických forem juvenilního xantogranulomu a terapie. Vnitr Lek 2012; 58 (5): 365–377.
43. Adam Z, Pour L, Řehák Z et al. FDG-PET/CT dokumentované vymizení nekrobiotického xantogranulomu při potlačení tvorby monoklonálního imunoglobulinu bortezomibem, lenalidomidem a dexametazonem: popis případu a přehled literatury o léčbě nekrobiotického xantogranulomu. Vnitr Lek 2021; 67 (6): 352–356.
44. Eissa SS, Clay MR, Santiago T et al. Dasatinib induces a dramatic response in a child with refractory juvenile xanthogranuloma with a novel MRC1-PDGFRB fusion. Blood Adv 2020; 4 (13): 2991–2995. doi: 10.1182/bloodadvances.2020001890.
45. Mavi ME, Turan BS, Salanci BV et al. Role of FDG PET/CT in the evaluation of therapy response in systemic juvenile xanthogranuloma. Clin Nucl Med 2022; 47 (5): e395–e396. doi: 10.1097/RLU.0000000000004085.
46. Maintz L, Wenzel J, Irnich M et al. Successful treatment of systemic juvenile xanthogranulomatosis with cytarabine and 2-chlorodeoxyadenosine: case report and review of the literature. Br J Dermatol 2017; 176 (2): 481–487. doi: 10.1111/bjd.14813.
47. Picarsic J, Pysher T, Zhou H et al. BRAF V600E mutation in juvenile xanthogranuloma family neoplasms of the central nervous system (CNS-JXG): a revised diagnostic algorithm to include pediatric Erdheim-Chester disease. Acta Neuropathol Commun 2019; 7 (1): 168. doi: 10.1186/s40478-019-0811-6.
48. Zou T, Wei A, Ma H et al. Systemic juvenile xanthogranuloma: a systematic review. Pediatr Blood Cancer 2023; 70 (5): e30232. doi: 10.1002/pbc.30232.
49. Goyal G, Heaney ML, Collin M et al. Erdheim-Chester disease: consensus recommendations for evaluation, diagnosis, and treatment in the molecular era. Blood 2020; 135 (22): 1929–1945. doi: 10.1182/blood.2019003507.
50. Řehák Z, Koukalová R, Vašina J et al. 18F-FDG PET/CT obraz Erdheimovy-Chesterovy nemoci – přehled českých pacientů. NuklMed 2018; 7 (3): 50–52.
51. Král Z, Řehák Z, Krejčí M et al. Léčba tří pacientů s Erdheimovou-Chesterovou chorobou kladribinem, případně kombinací kladribinu a cyklofosfamidu a přehled léčby této nemoci. Vnitr Lek 2021; 67 (3): 157–164.
52. Adam Z, Petrášová H, Řehák Z. Hodnocení pětileté léčby Erdheimovy-Chesterovy nemoci anakinrou – kazuistika a přehled literatury. Vnitr Lek 2016; 62 (10): 820–832.
53. Al Bayati A, Plate T, Al Bayati M et al. Dabrafenib and trametinib treatment for Erdheim-Chester disease with brain stem involvement. Mayo Clin Proc Innov Qual Outcomes 2018; 2 (3): 303–308. doi: 10.1016/j.mayocpiqo.2018.05.001.
54. Cohen Aubart F, Emile JF, Carrat F et al. Targeted therapies in 54 patients with Erdheim-Chester disease, including follow-up after interruption (the LOVE study). Blood 2017; 130 (11): 1377–1380. doi: 10.1182/blood-2017-03-771873.
55. Cohen-Aubart F, Emile JF, Carrat F et al. Phenotypes and survival in Erdheim-Chester disease: results from a 165-patient cohort. Am J Hematol 2018; 93 (5): E114–E117. doi: 10.1002/ajh.25055.
56. Franconieri F, Deshayes S, de Boysson H et al. Superior efficacy and similar safety of double dose anakinra in Erdheim-Chester disease after single dose treatment. Oncoimmunology 2018; 7 (8): e1450712. doi: 10.1080/2162402X.2018.1450712.
57. Haroche J, Cohen-Aubart F, Amoura Z. Erdheim-Chester disease. Blood 2020; 135 (16): 1311–1318. doi: 10.1182/blood.2019002766.
58. Tesi M, Pegoraro F, Peyronel F et al. Cluster analysis reveals the clinical spectrum of Erdheim-Chester disease. Leukemia 2025; 39 (8): 1987–1996. doi: 10.1038/s41375-025-02656-w.
59. Oneal PA, Kwitkowski V, Luo L et al. FDA approval summary: vemurafenib for the treatment of patients with Erdheim-Chester disease with the BRAFV600 mutation. Oncologist 2018; 23 (12): 1520–1524. doi: 10.1634/theoncologist.2018-0295.
60. Tamura S, Kawamoto K, Miyoshi H et al. Cladribine treatment for Erdheim-Chester disease involving the central nervous system and concomitant polycythemia vera: a case report. J Clin Exp Hematop 2018; 58 (4): 161–165. doi: 10.3960/jslrt.18015.
61. Goyal G, Reiner AS, Bossert D et al. Long-term outcomes with single-agent BRAF inhibitor therapy in Erdheim-Chester disease. Blood 2025; 145 (18): 2100–2103. doi: 10.1182/blood.2024028032.
62. Abla O, Jacobsen E, Picarsic J, Krenova Z et al. Consensus recommendations for the diagnosis and clinical management of Rosai-Dorfman-Destombes disease. Blood 2018; 131 (26): 2877–2890. doi: 10.1182/blood-2018-03-839753.
63. Adam Z, Mašlaň J, Křen L et al. Sinusová histiocytóza s masivní lymfadenopatií: FDG-PET/CT dokumentovaná parciální remise po léčbě 2-chlorodeoxyadenozinem. Vnitr Lek 2016; 62 (6): 491–499.
64. Adam Z, Adamová Z, Pour L et al. Rosai-Dorfman--Destombesova choroba – histiocytární onemocnění se zánětlivými projevy. Klin Onkol 2022; 35 (4): 262–270. doi: 10.48095/ccko2022262.
65. Králík R, Jadrníčková P, Michálek J et al. Generalizovaná lymfadenopatie při Rosai-Dorfmanově nemoci. Kazuistiky v alergologii, pneumologii a ORL 2022; 19 (1): 9–12.
66. Medek O, Kašparová P, Bartoš M et al. Meningeal Form of Rosai-Dorfman Disease. Cesk Slov Neurol N 2021; 84 (5): 493–495. doi: 10.48095/cccsnn2021493.
67. Shimizu A, Noguchi-Shinohara M, Komatsu J et al. Multifocal intracranial Rosai-Dorfman disease mimicking immunoglobulin G4-related pachymeningitis. Neurology 2024; 103 (5): e209741. doi: 10.1212/WNL.0000000000209741.
68. Fan X, Liu T, Zhang Z et al. Comparison of neuroimaging features of histiocytic neoplasms with central nervous system involvement: a retrospective study of 121 adult patients. Eur Radiol 2023; 33 (11): 8031–8042. doi: 10.1007/s00330-023-09724-8.
69. Almási S, Pancsa T, Tiszlavicz L et al. Cerebral manifestation and diagnostic dilemma of Rosai-Dorfman disease. CNS Oncol 2023; 12 (4): CNS103. doi: 10.2217/cns-2023-0006.
70. da Silva MG, Cunha de Souza E, Jacobsen IF et al. Rosai-Dorfman-Destombes disease in adolescence with hearing and vision loss involvement: a multidisciplinary approach. Eur J Ophthalmol 2025; 35 (2): NP22–NP27. doi: 10.1177/11206721241300634.
71. Tezuka T, Nukariya T, Takahashi N et al. Repeated intravenous methylprednisolone may prevent deterioration of hypertrophic pachymeningitis in Rosai-Dorfman disease. Intern Med 2025; 64 (3): 459–462. doi: 10.2169/internalmedicine.3884-24.
72. Yim SH, Yoon JS, Lee CH et al. Hypertrophic pachymeningitis and interstitial lung disease in IgG4-related disease. J Clin Neurol 2022; 18 (4): 481–483. doi: 10.3988/jcn.2022.18.4.481.
73. Gavioli F, Capogna A, Terribili R et al. IgG4-related disease presenting as hypertrophic pachymeningitis. Clin Rheumatol 2025; 44 (9): 3773–3774. doi: 10.1007/s10067-025-07585-8.
74. Li L, Wang Z, Liu L et al. Rosai-Dorfman disease involving the descending aorta. Circ Cardiovasc Imaging 2023; 16 (3): e014582. doi: 10.1161/CIRCIMAGING.122.014582.
75. Baassiri W, Moussalem CK, Massaad E et al. Craniocervical Rosai-Dorfman disease involving the vertebral artery: case report and literature review. World Neurosurg 2020; 133 : 69–73. doi: 10.1016/j.wneu.2019.09.072.
76. Wang R, Zhang Z, Zhang J et al. Multimodality imaging of a rare extranodal Rosai-Dorfman disease involving great vessels. Circ Cardiovasc Imaging 2024; 17 (12): e016746. doi: 10.1161/CIRCIMAGING.124.016746.
77. Bouhani M, Bouaziz H, Boujelbene N et al. Spontaneous regression of Rosai-Dorfman disease presenting as a thigh mass with vascular involvement: a case report. J Investig Med High Impact Case Rep 2025; 13 : 23247096251374516. doi: 10.1177/23247096251374516.
78. Chen LY, Slack GW, Carruthers MN. IgG4-related disease and Rosai-Dorfman-Destombes disease. Lancet 2021; 398 (10307): 1213–1214. doi: 10.1016/S0140-6736 (21) 01812-2.
79. Cetin O, Yetisir S, Akpinar TS et al. Rosai-Dorfman disease mimicking IgG4-related disease. Wien Med Wochenschr 2026; 176 (1–2): 60–63. doi: 10.1007/s10354-025-01110-x.
80. Liu L, Perry AM, Cao W et al. Relationship between Rosai-Dorfman disease and IgG4-related disease: study of 32 cases. Am J Clin Pathol 2013; 140 (3): 395–402. doi: 10.1309/AJCPFH0SJ6YILXJU.
81. Shi J, Sun K, Kong F. Primary intraosseous Rosai--Dorfman disease: clinicopathological features and an assessment of a possible relationship with IgG4--related disease. Ann Diagn Pathol 2025; 75 : 152435. doi: 10.1016/j.anndiagpath.2024.152435.
82. Zhang X, Hyjek E, Vardiman J. A subset of Rosai-Dorfman disease exhibits features of IgG4-related disease. Am J Clin Pathol 2013; 139 (5): 622–632. doi: 10.1309/AJCPARC3YQ0KLIOA.
83. Averitt AW, Heym K, Akers L et al. Sinus histiocytosis with massive lymphadenopathy (Rosai Dorfman disease): diagnostic and treatment modalities for this rare entity revisited. J Pediatr Hematol Oncol 2018; 40 (4): e198–e202. doi: 10.1097/MPH.0000000000001044.
84. Golwala ZM, Taur P, Pandrowala A et al. Sirolimus-Atargeted therapy for Rosai-Dorfman disease. Pediatr Blood Cancer 2019; 66 (12): e27994. doi: 10.1002/pbc.27994.
85. Tirado-Sánchez A. Recalcitrant primary cutaneous Rosai-Dorfman disease. Efficacy of sirolimus and intralesional methylprednisolone. Skin Health Dis 2023; 3 (5): e273. doi: 10.1002/ski2.273.
86. Ghawas MS, Ng T, Chen LYC. Confirmed efficacy of lenalidomide and dexamethasone in unresectable cutaneous facial Rosai-Dorfman-destombes disease. Mayo Clin Proc Innov Qual Outcomes 2019; 3 (1): 94–96. doi: 10.1016/j.mayocpiqo.2018.11.002.
87. Go RS, Jacobsen E, Baiocchi R et al. Histiocytic Neoplasms, Version 2.2021, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw 2021; 19 (11): 1277–1303. doi: 10.6004/jnccn.2021.0053.
88. Jacobsen E, Shanmugam V, Jagannathan J. Rosai--Dorfman disease with activating KRAS mutation – response to cobimetinib. N Engl J Med 2017; 377 (24): 2398–2399. doi: 10.1056/NEJMc1713676.
89. Pagel JM, Lionberger J, Gopal AK et al. Therapeutic use of rituximab for sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease). Am J Hematol 2007; 82 (12): 1121–1122. doi: 10.1002/ajh.21024.
90. Alqanatish JT, Houghton K, Bond M et al. Rituximab treatment in a child with Rosai-Dorfman disease and systemic lupus erythematosus. J Rheumatol 2010; 37 (8): 1783–1784. doi: 10.3899/jrheum.091275.
91. Razanamahery J, Humbert S, Emile JF et al. Immune thrombocytopenia revealing enriched IgG-4 peri-renal Rosai-Dorfman disease successfully treated with rituximab: a case report and lterature review. Front Med (Lausanne) 2021; 8 : 678456. doi: 10.3389/fmed.2021.678456.
92. Thompson E, Indelicato DJ, Scarborough MT et al. Rosai-Dorfman disease: the role of radiation therapy. Pract Radiat Oncol 2025; 15 (5): 481–484: doi: 10.1016/j.prro.2025.06.001.
93. Kemps PG, Picarsic J, Durham BH et al. ALK-positive histiocytosis: a new clinicopathologic spectrum highlighting neurologic involvement and responses to ALK inhibition. Blood 2022; 139 (2): 256–280. doi: 10.1182/blood.2021013338.
94. Liu W, Liu HJ, Wang WY et al. Multisystem ALK-positive histiocytosis: a multi-case study and literature review. Orphanet J Rare Dis 2023; 18 (1): 53. doi: 10.1186/s13023-023-02649-x.
95. Kommalapati A, Tella SH, Go RS et al. Predictors of survival, treatment patterns, and outcomes in histiocytic sarcoma. Leuk Lymphoma 2019; 60 (2): 553–555. doi: 10.1080/10428194.2018.1492128.
96. Kemps PG, Woei-A-Jin FJ, Schöffski P et al. Real-world experience with targeted therapy in patients with histiocytic neoplasms in the Netherlands and in Belgium. Blood Neoplasia 2024; 1 (3): 100023. doi: 10.1016/j.bneo.2024.100023.
97. Venkataraman V, Massoth LR, Sullivan RJ et al. Secondary histiocytic sarcoma with BRAFV600E mutation responsive to MAPK-targeted therapy presenting with recurrence with mTOR mutation responsive to mTOR-targeted therapy. Pediatr Blood Cancer 2021; 68 (10): e29166. doi: 10.1002/pbc.29166.
98. Zhao Y, Deng Y, Jiang Y et al. Targeting the PD-1 receptor and genetic mutations validated in primary histiocytic sarcoma with hemophagocytic lymphohistiocytosis. Front Immunol 2023; 14 : 1127599. doi: 10.3389/fimmu.2023.1127599.
99. Eid M, Slabý O, Adámková D. Histiocytární sarkomy, sarkomy z folikulárně dendritických buněk a sarkomy z interdigitujících dendritických buněk: vzácné a agresivní malignity léčené ve Fakultní nemocnici Brno. Klin Onkol 2024; 37 (Suppl 1): S72.
100. Eid M, Slabý O. Uplatnění precizní medicíny u pacientů s pokročilými solidními nádory – zkušenosti FN Brno. Klin Onkol 2022; 35 (Suppl 1): S48.
101. Waked JA, Geng R, Sood S et al. Efficacy and safety of treatments for multicentric reticulohistiocytosis: an evidence-based review. J Cutan Med Surg 2026. In press. doi: 10.1177/12034754261436041.
102. Fend F, Dirnhofer S, Egan C et al. Histiocytoses and reactive proliferations of histiocytes: current state of the art and evolving concepts-a report from the joint CSHP EA4HP-SH workshop 2024, Hefei, China. Virchows Arch 2026; 488 (2): 245–262. doi: 10.1007/s00428-025-04096-4.
Štítky
Dětská onkologie Chirurgie všeobecná OnkologieČlánek vyšel v časopise
Klinická onkologie

2026 Číslo 3
- Profesionální expozice azbestu a riziko vzniku karcinomu plic
- Aktuální možnosti léčby mnohočetného myelomu
- Zeolit-jodový komplex pomáhá v péči o infikované rány
- Proces hojení ran krok za krokem a co ho může zkomplikovat
- Umělý svěrač anu po deseti letech
Nejčtenější v tomto čísle
- Rozdíly v zobrazení Erdheimovy-Chesterovy choroby FDG-PET/CT, NaF-PET/CT vyšetřením a scintigrafií skeletu. Ústup nemoci po léčbě kladribinem s cyklofosfamidem a anakinrou
- Druhá a vyšší linie systémové léčby karcinomu endometria – zkušenosti z reálné klinické praxe
- Histiocytární choroby dospělých – pestrost jejich klinických projevů vyžaduje spolupráci lékařů mnoha oborů
- Chronicky zvýšená teplota či horečka s nevelkou lymfadenopatií může být Castlemanova choroba, pokud se neprokáže maligní, autoimunitní anebo infekční příčina lymfadenopatie
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy