
NEUROFIBROMATÓZA 1. TYPU A GLIOM OPTIKU
Autoři:
A. Siwá 1; R. Autrata 1; K. Vejmělková 2; Z. Pavelka 2; K. Zitterbart 2
Působiště autorů:
Dětská oční klinika LF MU a FN Brno, přednosta prof. MUDr. Rudolf Autrata, CSc., MBA
1; Klinika dětské onkologie LF MU a FN Brno, přednosta prof. MUDr. Jaroslav Štěrba, Ph. D.
2
Vyšlo v časopise:
Čes. a slov. Oftal., 75, 2019, No. 4, p. 200-208
Kategorie:
Původní práce
doi:
https://doi.org/10.31348/2019/4/4
Souhrn
Cíl: Zhodnocení efektivity léčby gliomu optiku na zrakovou ostrost.
Metodika a soubor: Rešerše literatury zabývající se neurofibromatózou 1. typu a gliomem optiku, porovnaná se souborem dětských pacientů s gliomem optiku sledovaných na Dětské oční klinice LF MU a FN Brno od ledna 2013 do června 2018.
Diskuse: Hlavním problémem této i řady dalších retrospektivních studií je variabilní časový interval očních vyšetření. U některých dětských pacientů se navíc zrakové funkce nedají spolehlivě zhodnotit. Rizikovými faktory jsou věk pacienta při zahájení léčby a lokalizace nádoru. Samotná progrese nádoru nemusí mít vliv na zrakovou ostrost a je otázkou, zda je v některých případech žádoucí zahajovat léčbu. Prozatím neexistují klinické biomarkery, které by dokázaly nadcházející ztrátu zraku předpovídat.
Závěr: Výsledky souhlasí s literaturou. Standardní léčbou je chemoterapie, jejímž nejčastějším výsledkem je stabilita zrakové ostrosti. Pro důkladné zhodnocení léčby je nutná užší mezioborová spolupráce a pravidelné standardizované vyhodnocování zrakových funkcí.
Klíčová slova:
neurofibromatóza – deti – gliom optiku – chemoterapie – léčba
NEUROFIBROMATÓZA 1. TYPU
Při incidenci 1/3000 živě narozených dětí je neurofibromatóza 1. typu (NF1) jedním z nejčastěji se vyskytujících autosomálně-dominantně dědičných onemocnění u člověka [30]. Zhruba polovina případů je dědičná a zbylých 50 % se vyskytuje na základě nových mutací [26]. Nové mutace se obvykle nacházejí na paternálních chromozomech. Jedním z vysvětlení je, že vyšší věk otců v době početí vede k dlouhodobé metylaci DNA některých spermií se vznikem bodových mutací, které mají za následek zvýšený výskyt sporadické NF1. “Příčinou rozvoje onemocnění je mutace tumor supresorového genu NF1 (17q11.2) a následná porucha tvorby cytoplazmatického proteinu neurofibrominu. Tato porucha je u NF1 dávána do souvislosti s výskytem mnohočetných nádorových procesů, které histologicky odpovídají hamartomům nebo benigním nádorům. Nádory centrálního nervového systému (CNS) jsou především gliomy nízkého gradu (pilocytární astrocytom, grade I), zvláště u zrakové dráhy [33].”
KLINICKÝ OBRAZ
Kávové skvrny (café-au-lait) – tento kožní příznak téměř nikdy nechybí. Jedná se o vícečetné, světle hnědé, hladké, ostře ohraničené pigmentace v úrovni kůže. Nejčastěji jsou lokalizované na trupu, ale také na končetinách a obličeji.
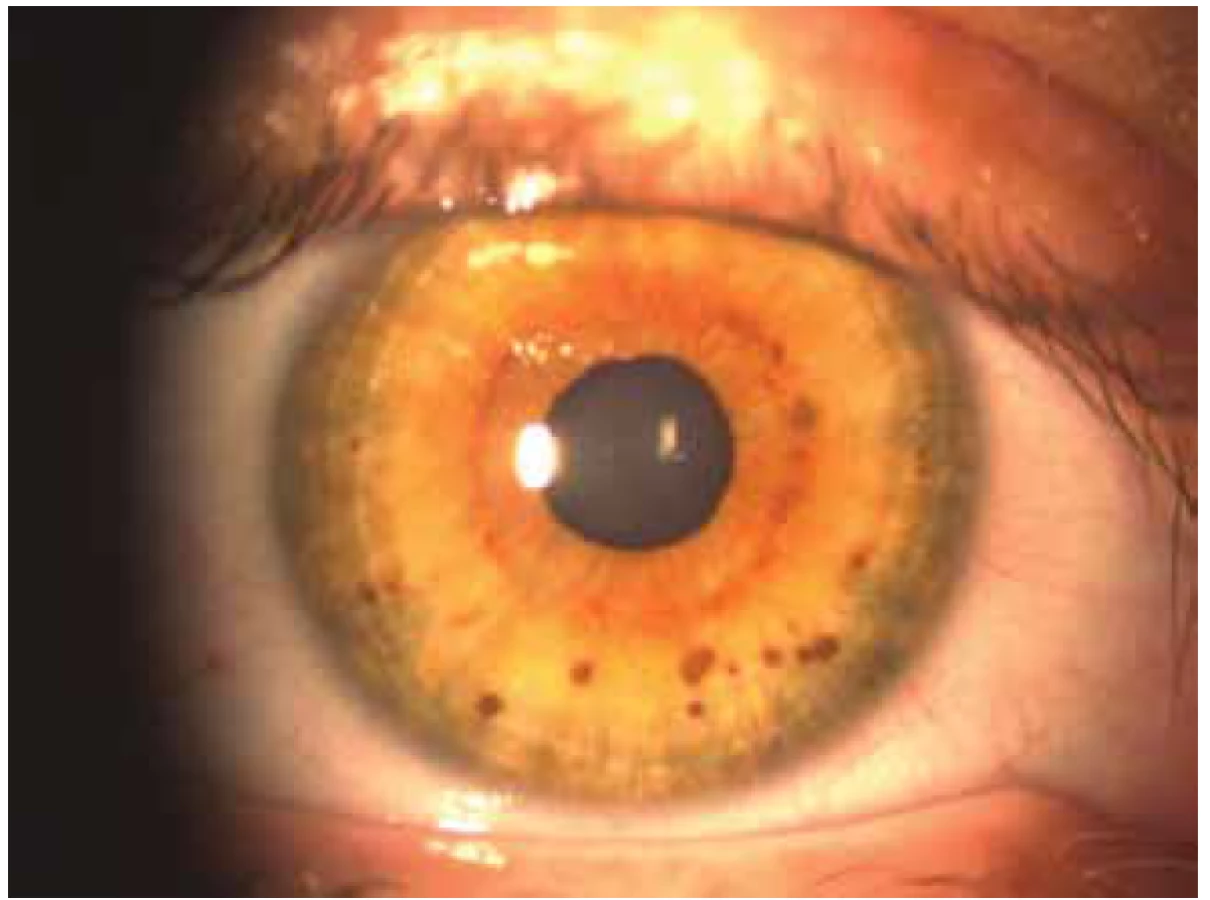
Vznikají nahromaděním pigmentových melanoblastů na bazální vrstvě epidermis a následnou poruchou vývoje melanocytů [21]. Objevují se už u novorozenců, ale jejich velikost a počet se s věkem může zvětšit [19,21,30]. Pro definitivní diagnózu NF1 stačí 6 skvrn s průměrem větším než 5mm u dětí a 15mm u dospělých [30,40]. Neexistuje vztah mezi počtem skvrn a závažností NF1 [27]. Lischovy noduly jsou užitečným diagnostickým kritériem NF1. Jde o mnohočetné hamartomy na duhovce a většinou se jedná o bilaterální nález.
Lze je nalézt u malého procenta dětí, ale vyskytují se u více než 90 % jedinců starších 6 let [23,27]. Zpočátku jsou melanocytární hamartomy zbarvené světle, ale s věkem tmavnou [19,21,30]. Freckling – různý počet drobných okrouhlých makul se zvýrazněnou pigmentací. Nachází se v podpaží a v tříslech a objevují se mezi 3. až 5. rokem života.
Neurofibrom se může objevit kdekoliv v periferním nervu. Pokud se vyskytnou více než 2, představují jedno z diagnostických kritérií NF1. Neurofibromy se manifestují před začátkem puberty nebo během ní a mohou být nalezeny i u osob bez diagnózy NF1. Kožní neurofibromy je v případě malých lézí možné odstranit pomocí laseru, u větších se upřednostňuje chirurgické odstranění, ale je zde riziko hojení hypertrofickou jizvou a rekurence neurofibromů [33].
Plexiformní neurofibrom (PN) se může objevit na všech částech těla, kde jsou periferní nervy. Je bohatě vaskularizovaný, neopouzdřený a nemetastazuje. Často se chová infiltrativně [2]. Transformace benigního PN na maligní nádor pochvy periferního nervu (MPNST, z anglického Malignant Peripheral Nerve Sheath Tumor) představuje 8-13 % celoživotního rizika [14]. Příznakem maligní povahy PN může být bolest, změna konzistence a agresivní růst [2]. Pacienti se základní diagnózou NF1 mají od stanovení diagnózy MPNST 5letou prognózu přežití 21 %, bez diagnózy NF1 42 %. MPNST je nejčastější příčinou úmrtí dospělých s diagnózou NF1 [14]. PN se vyskytuje převážně v orbitopalpebrální formě. Klinické příznaky mohou být vyjádřeny u novorozenců, častá je ale manifestace v prvních letech života a progrese bývá patrná během puberty. Postižení je vždy jednostranné a častěji se vyskytuje u chlapců. Predilekční oblastí je zevní polovina horního víčka a strop očnice. Typické jsou deformace a hypertrofie horního víčka s hmatnými uzlíky, protruze a dislokace bulbu směrem dolů, zvětšení očnice a vyklenutí spánkové jámy z dlouhotrvající expanze objemu měkkých tkání. Neurofibrom je hlavní příčinou zvětšování očnice, jejíž strop může jevit známky těžké kostní dysplazie. V případě defektu orbitálního stropu se do očnice vyklenuje meningoencefalokéla, která přenáší mozkovou pulzaci na orbitální obsah a dává vznik pulzujícímu exoftalmu. Kůže víčka je pigmentovaná, tažná (cutis lata), s prosvítajícími angiektáziemi. Může dojít k postižení okohybných svalů a intrakonálních senzorických a motorických nervů. Na snímcích magnetické rezonance (MRI, z anglického Magnetic Resonance Imaging) je T1 hypointenzní a T2 hyperintenzní [2]. U neurofibromů, které vadí svou lokalizací, bolí i v klidu, rychle rostou a ovlivňují okolí, se nejčastěji přistupuje k chirurgickému zákroku. Léčba PN je ale problematická a u dětí dochází až v 75 % k recidivě [32]. Gliom optické dráhy (OPG, z anglického Optic Pathway Glioma) je z histologického hlediska spongioblastom, astrocytom či oligodendrocytom. Nádor vzniká hyperplazií neuroglie v kmeni optiku, proniká piální pochvou a šíří se v intervaginálních prostorách, kde způsobuje mohutnou proliferaci mezoteliálních buněk. Jedná se o charakteristickou vlastnost tohoto nádoru. OPG se makroskopicky jeví jako vřetenovité zduření optiku v očnici nebo v lebeční dutině, nebo má činkovitý vzhled, pokud je přítomen v obou dutinách. Počátečním příznakem je porucha zraku, která ale nebývá včas zaznamenána. Prvním důvodem vyšetření u dětí je strabismus ex anopsia, častěji konvergentní než divergentní, a progredující axiální protruze oka bez poruchy hybnosti, edému či zarudnutí. Při vyšetření lze na oku s exoftalmem pozorovat relativní aferentní pupilární defekt (RAPD). Na snímcích z počítačové tomografie (CT, z anglického Computed Tomography) je typické vřetenovité ztluštění nervu v celém orbitálním průběhu, hladce a ostře konturované. Na RTG snímcích je vyjádřeno koncentrické rozšíření kanálu nad 5,5 mm průměru bez uzurace a destrukce okolní tkáně. Za patologickou hodnotu rozšíření se považuje hodnota od 6 mm výše, stejně jako stranový rozdíl 20 %, tedy větší než 1mm. Diagnostickým obrazem gliomu chiazmatu je v boční projekci patrné hruškovité sedlo, způsobené rozšířením ventrální části tureckého sedla důsledkem tlakového působení gliomu.
U orbitálních gliomů může být na očním pozadí prostá atrofie nebo edém terče zrakového nervu. Nález oboustranně městnavé papily nebo kombinace městnání a atrofie poukazuje na postižení hypothalamu a spodiny III. komory mozkové poukazuje s blokádou likvorové cirkulace a s vnitřním hydrocefalem.
Gliomy chiazmatu se dělí na dva klinické typy. Přední optiko-chiasmatický tumor má stejný klinický obraz, diagnostiku a prognózu jako gliom optiku. Zadní hypothalamo-chiasmatický představuje primární gliom hypothalamu a má tudíž jinou biologickou povahu. Jedná se o velmi maligní nádor, chiasma infiltruje sekundárně, jeho rychlý a agresivní růst vede k letálnímu konci. Mezi funkční příznaky gliomu chiazmatu patří progresivní porucha zraku, výpadky zorného pole, a porucha barvocitu. Na očním pozadí je vyjádřen obraz bilaterálních městnavých papil, přecházející v atrofii papil. Často je přítomný vnitřní hydrocefalus s obrnou n. abducentis, s diencefalickou symptomatologií a poruchou růstu [2,21,29,30,31]. Nejlepší metodou pro odhadování rozsahu OPG je MRI s gadoliniem [38]. Dalšími diagnostickými metodami jsou CT, difuzní tenzorové zobrazování a difúzní tenzorová traktografie. Gliomy optiku jsou při MRI vyšetření T1 hypointenzní nebo T1 izointenzní a T2 hyperintenzní [35]. V současnosti se gliom nejčastěji léčí chemoterapií podle protokolu SIOP pro nádory nízkého gradu, používající karboplatinu a vinkristin. Více o terapii a její problematice v retrospektivní studii. Další klinické příznaky – až 85 % dětí s NF1 má FASI nebo UBO (z anglických Foci of Abnormal Signal Intensity, Unidentified Bright Object); hypersignální ložiska v T2 vážených obrazech na MRI zobrazení mozku a míchy [18,32]. Nejspíše se jedná o aberantní myelinizace s vakuolární změnou myelinu nacházející se v oblasti bazálních ganglií, globus pallidus, v thalamu, mozečku, v mozkovém kmeni a subkortikálně, převážně v temporální oblasti. Nesytí se po podání kontrastní látky a nezpůsobují tlak na okolní struktury, čímž se odlišují od gliomů nízkého gradu. FASI jsou z ložiskového neurologického hlediska asymptomatické, ale mohou souviset s kognitivními poruchami u NF1. V dospělosti mají tendenci se zmenšovat nebo mizet, u jedinců starších 30 let jsou vzácné. Náhodným nálezem při MRI zobrazení jsou intraspinální nádory, manifestující se pomaleji než nádory nitrolební. K nenádorovým projevům NF1 se řadí malý vzrůst s abnormálními hodnotami růstového hormonu, makrocefalie a anomálie skeletu na podkladě kongenitální dysplazie dlouhých kostí, nejčastěji tibie, projevující se v novorozeneckém nebo kojeneckém věku. Vyskytuje se i skolióza, která se zhoršuje s věkem. Systémová hypertenze je nejčastěji renovaskulární, kdy na podkladě fibromuskulární dysplazie dochází ke stenóze renální arterie. Tyto cévní změny se mohou vyskytovat i jinde. Vzácný je moya-moya syndrom s rizikem ischemické cévní mozkové poruchy. Jeho vznik je dáván do souvislosti s předchozí radioterapií nádorů CNS [32]. Lehká mentální retardace postihuje 29-35 % jedinců s NF1 [13,39]. Objevují se specifické vývojové poruchy učení a chování. Epilepsie se vyskytuje až u 7 % dětí a je převážně sekundární při expanzivních procesech CNS [40,18]. Přehled diagnostických kritérií pro NF1 je uveden v tabulce 1.
Zdroj: Pediatrická klinika LF MU a FN Brno

Zdroj: Dětská oční klinika LF MU a FN Brno
Zdroj: Pediatrická klinika LF MU a FN Brno

Zdroj: Pediatrická klinika LF MU a FN Brno

National Institute of Neurological Disorders and Stroke: Neurofibromatosis Fact Sheet (USA), 2011 [41]
![Neurofibromatóza 1. typu - diagnostická kritéria<br>
National Institute of Neurological Disorders and Stroke: Neurofibromatosis Fact Sheet (USA), 2011 [41]](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image_pdf/723e8c48ea4be78d453d72157e53538a.png)
GLIOM OPTICKÉ DRÁHY
Gliom optické dráhy (OPG) je nejčastější nádor zrakové dráhy. Tvoří 3-5 % dětských mozkových nádorů [25,35], 75 % nádorů optiku a 10 % nádorů očnice [2]. Řadí se do skupiny gliomů s nízkou malignitou (LGG, z anglického Low Grade Gliomas), jež jsou nejběžnější dětské mozkové nádory. Prognóza přežití pro pacienty s LGG je 94 % po 10 letech od diagnózy [20]. Zhruba polovina všech LGG je asymptomatických. První volbou léčby bývá chemoterapie, po níž se nádor stabilizuje ve 44 % případů a radioterapie, která má za následek stabilizaci v 62 % případů [20]. Tato čísla se však nevztahují k výsledné zrakové ostrosti (ZO) a kvalitě života. Dívky jsou k nádorům náchylnější než chlapci [12], kteří také vykazují delší stabilitu nádoru po léčbě chemoterapií [20]. Biologické faktory určující progresi nádorů však zůstávají neobjasněny [4].
OPG se může vyskytnout kdekoliv v průběhu zrakové dráhy. V roce 1958 byla navržena Dodge klasifikace, která kategorizovala nádory do tří typů podle lokalizace a sloužila k volbě léčby a výběru pacientů k resekci. Nástup MRI umožnil přesnější lokalizaci OPG podle modifikované Dodge klasifikace, viz obrázek 7 [38]. Nejčastějšími funkčními příznaky OPG jsou zhoršená ZO a nystagmus [36]. Mohou se vyskytnout i poruchy zorného pole, barvocitu a strabismus. Pokud není ohrožen život pacienta, léčba se indikuje s cílem zastavit ztrátu ZO a předejít oslepnutí, protože tyto faktory jsou spojeny s výrazným poklesem kvality života. Riziko ztráty ZO se zvyšuje, pokud je nádor bilaterální, nebo se vyskytuje v chiasmatické oblasti [12,18,16,35]. Pokud je nádor v chiazmatu, může navíc dojít k propagaci do hypothalamu a tím k zasažení hypothalamo-hypofyzární-gonadální osy, způsobující předčasnou pubertu a diencefalický syndrom. OPG se vyskytuje ve spojení s NF1 (NF1+) nebo sporadicky (NF1-). V literatuře se podíl sporadického OPG pohybuje od 30-60 % [11,16,17,25,29,37]. U NF1+ pacientů se v 91 % případů objeví symptomy, zatímco u sporadického typu je to pouze v 29 % [36]. Jejich projevy se různí a vyžadují individuální péči.
Zdroj: Klinika dětské radiologie
LF MU a FN Brno

Zdroj: Dětská oční klinika LF MU a FN Brno

Zdroj: vlastní ilustrace

VÝSKYT
NF1 je nejdůležitějším rizikovým faktorem pro OPG. U dětí s NF1 se OPG obvykle vyskytuje v 15-25 % případů [6, 17], ale v jedné studii to bylo dokonce v 76 % [12]. Objevuje se zpravidla v prvním desetiletí života, ale okolo 50 % pacientů je v tomto období asymptomatických [4, 18]. NF1 mutace postihuje častěji dívky [17,36]. a při diagnóze NF1 se u nich OPG vyskytuje častěji [5]. U dívek je pravděpodobnější výskyt nádoru v postchiasmatické oblasti, ale mají lepší výsledky léčby než chlapci [36]. Z literatury vyplývá, že ke zhoršení ZO dojde u 20-70 % pacientů navzdory léčbě [5]. Rizikovým faktorem je věk pod 2 roky a nad 5 let [17]. U pacientů s NF1 jsou ve 46 % případů zasaženy oba optické nervy, zatímco u sporadického OPG je to 17 % a podle některých studií se sporadický OPG nachází častěji v chizmatu [36,37,38] a podle některých studií se OPG nachází častěji v chiazmatu [36,37]. Pokud je nádor v postchiasmatické oblasti je riziko ztráty zraku 2x vyšší [5]. U pacientů s NF1 a OPG bylo navíc zdokumentováno riziko sekundárních nádorů po léčbě radioterapií [37].
Sporadický OPG má agresivnější charakter a horší prognózu na vývoj ZO. I když má zpočátku sporadický OPG horší prognózu přežití [37], výsledky dlouhodobého sledování naznačují, že se po 15 letech rozdíl mezi NF1+ a NF1 - pacienty stírá [34]. Nystagmus je charakteristickým příznakem sporadické formy OPG lokalizované v chiasmatické oblasti a v hypothalamu [36,38].
LÉČBA
Pacienti s OPG mají relativně dobrou prognózu přežití, která se ale časem snižuje z 95 % po 5 letech od diagnózy, na 92 % po 10, na 81 % po 15, a na 76 % po 18 letech. Délka sledování pacientů se v literatuře pohybuje do 10 let a je proto možné, že plně nevystihuje opravdovou povahu tohoto onemocnění. Riziko smrti je dále 3x vyšší u pacientů diagnostikovaných před prvním rokem života a až 5x vyšší při výskytu intrakraniální hypertenze, jež se nabízí jako prognostický faktor přežití [34]. Jelikož OPG u pacientů s NF1 nemají agresivní povahu, je hlavním cílem léčby zachování ZO [16]. Léčba se odvíjí od věku pacienta, od lokalizace nádorů a od vyjádřených klinických příznaků. V určitých případech se přistupuje k resekci za účelem zmenšení objemu nádoru. Resekce je spojována s lepší prognózou přežití a stabilizací nádoru, ale může mít za následek slepotu, poškození hypothalamu, vaskulární komplikace a zhoršení endokrinních funkcí [1,3]. Studie 109 NF1+ pacientů s OPG poukazuje na lokalizaci nádoru a chirurgický zákrok jako rizikové faktory pro progresi nádoru [12]. V roce 2011 bylo na mezinárodní konferenci pediatrické neurochirurgie v Paříži vydáno usnesení, že chirurgický zákrok není standardní primární léčbou, i když u některých případů s menším rizikem je resekce přijatelná [41]. Biopsie se doporučuje, protože pomáhá s jistotou určit diagnózu a je užitečná, pokud se na MRI objeví atypický nález [1]. Chemoterapie je v dnešní době standardním postupem. OPG je u 60-80 % pacientů diagnostikován do 5 let věku a chemoterapie se indikuje, pokud je zaznamenán pokles ZO a nebo nepřijatelná progrese nádoru [5,9,11,25,29,34,36,37]. Samotná asymptomatická progrese nádoru k zahájení léčby nestačí, neboť OPG může být benigní a korelace mezi progresí nádoru a ZO není spolehlivá [5,9,11,25,28]. Rizikovými faktory jsou nízký věk a nádory v chiazmatu nebo hypotalamu, u kterých bylo zaznamenáno zhoršení ZO ve 32 % případů [11]. Nejlepší výsledky ZO po chemoterapii mají pacienti mezi 2. a 5. rokem života [17]. Efektivita chemoterapie na zachování zraku je však mnoha studiemi zpochybňována. Nejčastějším výsledkem léčby, ve zhruba 50 % případů, je stabilizace ZO na úrovni před chemoterapií [9,11,28]. U pacientů, kteří začali chemoterapii se vstupní ZO horší než 6/24, bylo zaznamenáno další zhoršení ZO [25]. V jedné studii se dokonce uvádí, že žádný z pacientů nezaznamenal po chemoterapii zlepšení ZO [17]. Jako nejúčinnější léčba pro zachování ZO se jeví radioterapie [28]. Tato léčba má ale řadu nežádoucích účinků, jako jsou vaskulární, endokrinní dysfunkce a zhoršení kognitivních funkcí u dětí mladších 5 let [3,11]. U pacientů mladších 5 let se obvykle nasazuje chemoterapie s cílem oddálit radioterapii. NF1 syndrom ale způsobuje poruchy učení, chování a lehkou mentální retardaci [13,39]. Je proto otázkou, zda je u NF1 pacientů riziko zhoršení kognitivních funkcí dostatečným důvodem oddalování radioterapie. Pro potvrzení této hypotézy je potřeba dalších klinický zkoušek [3,28].
RETROSPEKTIVNÍ STUDIE
ÚVOD
Cílem této retrospektivní studie je analýza souboru pacientů s OPG a porovnání získaných dat s mezinárodními publikacemi. Ve zdravotnické dokumentaci Dětské oční kliniky a Kliniky dětské onkologie LF MU a FN Brno bylo identifikováno 47 dětí s OPG. Všichni pacienti byli sledováni na Dětské oční klinice LF MU a FN Brno v období od ledna 2013 do června 2018.
METODIKA
Kritéria pro zařazení do souboru zahrnovaly: věk v době stanovení diagnózy, NF1 status, pohlaví, lokalizaci nádoru podle MDC provedenou radiologem a ZO před a po léčbě. Navíc musel být v dokumentaci popsán vývoj radiologických nálezů a klinických příznaků. ZO byla hodnocena na Snellenových optotypech, u dětí v předškolním věku byly použity obrázkové optotypy nebo Pflügerovy háky. U děti mladších 3 let se vyšetřovalo preferenční vidění pomocí Cardiff a Single Book testu. Tato kritéria splnilo 37 pacientů.
VÝSLEDKY
Z 37 pacientů s OPG bylo 15 dívek a 22 chlapců. Sporadický OPG se vyskytl ve 27 % případů (n = 10) a obě pohlaví byla zastoupena stejně. Většina OPG (73 %) byla spojena s NF1, mezi dívkami byl výskyt NF1 v 66 % případů (n = 10), u chlapců v 77 % (n = 17). Ve skupině pacientů NF1+ byl medián věku 2 roky a 9 měsíců a u skupiny NF1+ byl medián 4 roky a 3 měsíce. U obou skupin byl nejčastěji postižen optický nerv. Sporadický OPG měl vyšší pravděpodobnost manifestace v optickém nervu, chiasmatické a postchiasmatické oblasti. U obou skupin se bilaterální nádor vyskytoval ve srovnatelném zastoupení, viz graf 1 a tabulka 2.

![Modifikovaná Dodge klasifikace – vysvětlení. Zdroj: Taylor T. et al., 2008 [37]](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image_pdf/06c796a5e97d9f9a47c146c31906c1dd.png)
Nystagmus byl charakteristickým nálezem u skupiny NF1 - (n = 3 vs. n = 0 u NF1+). Hydrocefalus se vyskytl u 3 pacientů, z toho jeden byl ze skupiny NF1+.
Prognóza ZO u pacientů se sporadickým nádorem je špatná. Při posledním vyšetření bylo z 20 očí (n = 10 pacientů) pouze 5 očí se stabilní ZO 6/6 (25 %). Jedno oko zaznamenalo zlepšení ZO z 6/24 na 6/6. 13 očí (65 %) bylo amaurotických a podle World Health Organization spadaly do jedné ze tří kategorií slepoty. Jedno oko muselo být enukleováno. Průběh nemoci a výsledná ZO byly u NF1+ pacientů podstatně lepší. Z 27 NF1+ pacientů bylo 59 % (n = 17) pouze sledováno. Z 34 očí v této skupině bylo 20 očí se ZO 6/6 po celou dobu sledování a 7 očí se spontánně zlepšilo o 1 až 2 řádky. U 7 očí byla zaznamenána stabilita ZO. Zbývajících 10 pacientů (20 očí) podstoupilo léčbu chemoterapií. U 6 pacientů byl zvolen protokol SIOP (karboplatina a vinkristin), u 3 protokol ACNS (karboplatina, vinkristin, temolozomid) a jeden pacient byl léčen monoterapií karboplatinou. Výsledky léčby byly u 3 očí zhoršení ZO a u jednoho oka zlepšení ZO o jeden řádek, 80 % očí zaznamenalo stabilitu. Oko se vstupní ZO lepší než 6/24 zaznamenalo po chemoterapie stabilizaci a u oka se vstupní ZO 6/24 a méně, došlo po léčbě ke zhoršení ZO.
DISKUSE
Ve skupině pacientů s NF1 se OPG na rozdíl od literatury vyskytoval častěji u chlapců (63 % vs. 37 %). U této skupiny se neprokázal ani zvýšený výskyt OPG v chiazmatu v porovnání s ostatními lokalizacemi, ani častější výskyt bilaterálního OPG v porovnání se skupinou NF1 - [12,17,36,37]. Hlavními problémemy, se kterými se potýkala i tato studie, byly nízká frekvence očních kontrolních vyšetření a variabilní časový interval mezi nimi [17,28]. Tato vyšetření jsou důležitá ze tří důvodů.
Zaprvé slouží ke zjištění, zda pacient podstupuje léčbu s dlouhodobě stabilní ZO nebo v průběhu jejího zhoršování. U všech pacientů v této studii byly jen zřídka před zahájením léčebného postupu k dispozici výsledky ZO ze dvou a více vyšetření. Nebylo tudíž možné určit, zda probíhalo v době zahájení léčby zároveň zhoršování ZO. V literatuře se uvádí, že zlepšení zraku po léčbě je spíše výjimkou (0-32 %), což se shoduje s touto studií (16 %) [17,25]. Je nutné zvážit, zda není u pacientů se stabilní ZO léčba chemoterapií zbytečnou zátěží. Z literatury totiž vyplývá, že se možná zhoršení ZO nedá chemoterapií v určitých případech předejít [9]. Je obecným zájmem tyto případy identifikovat a nezahajovat léčbu, pokud by měl přirozený vývoj choroby stejný výsledek na ZO jako chemoterapie. Protože genetická analýza v dnešní době nedokáže předpovědět závažnost OPG, je potřeba pravidelného, standardizovaného oftalmologického sledování NF1 pacientů a pacientů s diagnostikovaným OPG [5]. Tato studie navíc dokládá, že u skupiny s NF1 došlo nejčastěji ke zlepšení ZO spontánně (32 %). U pacientů bez NF1 však tato statistika není k dispozici, protože všech 10 pacientů podstoupilo léčbu a ke zlepšení ZO došlo u jednoho z nich (10 %). Potvrzeným charakteristickým nálezem u skupiny NF1 - byl nystagmus [36]. Výsledky dále potvrzují, že sporadická forma OPG má daleko rychlejší a závažnější průběh, který zpravidla vyžaduje okamžitou léčbu [36,37].
Druhým cílem pravidelných očních vyšetření je zhodnocení vlivu nádoru na ZO. Špatná korelace progrese nádoru a ZO, která je v literatuře dobře zdokumentována, vylučuje radiologický nález jako jediné kritérium pro zahájení léčby, protože nemůže vyvážit rizika s ní spojené [6,9,11,17]. Za klinicky významné se považuje 1) zhoršení ZO o 0.2 logMAR nebo více (z anglického Logarithm of the Minimum Angle of Resolution), 2) potvrzení zhoršující se ZO nebo 3) nový nález [10]. Tato korelace nebyla z důvodu nedostatku dat v této studii zkoumána. Pravidelné oční kontroly dále slouží k identifikaci pacientů s rizikovými faktory. Prvním rizikovým faktorem je ZO horší než 6/24 před zahájením léčby, protože výsledkem bývá další zhoršení [25]. Druhým faktorem je věk pod 2 roky a nad 5 let. Ve studii 115 pacientů došlo ke zhoršení po chemoterapii u 50 % pacientů mladších 2 let a také u 36 % starších 5 let [17]. Většina dětí s OPG je léčena před 1. rokem života [10,35]. Počáteční ZO horší než 6/24 a nízký věk byly rizikovými faktory i u tohoto souboru pacientů. U NF1 - pacientů byl medián věku 2 roky a 9 měsíců a medián ZO 6/24, u NF1+ 4 roky a 3 měsíce a ZO 6/6. Výsledky potvrdily nejen predikční schopnost těchto faktorů, ale dokládají i agresivnější průběh sporadické formy OPG. Třetím rizikovým faktorem je bilaterální postižení nebo lokalizace nádoru v chiazmatu/postchiazmatu nebo v hypotalamu, kdy dochází k dlouhodobému zhoršení ZO u 32 % pacientů [11,36]. Vliv lokalizace nádoru na výslednou ZO, který je v dnešní době předmětem zkoumání nebylo možné zhodnotit [38]. S výjimkou nádorů intrakonálních, chiasmatických a v hypotalamu je ZO po první chemoterapii prediktivní dlouhodobě, a je tudíž nutné zvážit sekundární léčbu, pokud je první výsledek neuspokojivý [11]. Třetím argumentem pro častější oftalmologické vyšetření je zhodnocení léčby a dlouhodobé prognózy, která je sice krátkodobě horší pro skupinu NF1-, ale může být dlouhodobě stejná jako u skupiny NF1+ [34]. Častými problémy, na které poukazovali i jiní autoři retrospektivních studií, jsou nestandardní a nedostačující hodnocení ZO. U několika pacientů léčených chemoterapií převzatých do naší péče z jiných pracovišť nebyla zaznamenána vstupní hodnota ZO, a tudíž nemohla být potvrzena účinnost léčby. Důležitá je v tomto případě spolupráce pacienta s lékařem a respektování termínu kontrolních vyšetření. Pokud nelze objektivně zhodnotit ZO kvůli nespolupráci pacienta, musí se vyšetření ZO zopakovat do 2 týdnů a pokud je i napodruhé vyšetření nevýtěžné, je nutné to dokumentovat [16]. Žádný ze souboru pacientů v této studii nebyl sledován dostatečně dlouho na to, aby bylo možné objektivně zhodnotit účinnost léčby a porovnat dlouhodobou prognózu přežití mezi skupinou NF1 - and NF1+. Protože chiasmatická a postchiazmatická oblast je spojena s horší prognózou, je důležité jednotlivé lokalizace rozlišit podle modifikované Dodge klasifikace. Některé studie však tuto klasifikaci nepoužívají a existují i rozdíly v interpretaci mezi jednotlivými radiology. Je možné, že špatná korelace objemu nádorů a jeho vlivu na ZO je způsobená nepřesností v odhadu velikosti nádorů z 2D MRI snímků. Volumetric Magnetic Imaging (VMI) je alternativní metodou k objektivnímu hodnocení, ale je limitována problémy s měřením nádorů v postchiasmatické oblasti [10]. Jelikož genetická analýza ani biomarkery nedokáží spolehlivě předpovědět vývoj OPG, je nutné pravidelné standardizované sledování. U dětí s NF1 se doporučuje každoroční oftalmologické vyšetření [4]. To by se mělo vykonávat každé 3 měsíce, pokud se na MRI .potvrdí OPG [5,15], tabulka 3.
![Doporučené intervaly sledování pacientů s NF1. Zdroj: de Blank P.M.K. et al., 2017 [10]](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image_pdf/a0dfd3c3a46bfbd88ce5cc0744f01b8c.png)
U každého vyšetření je nutné kvantitativně zhodnotit ZO a na očním pozadí papilu zrakového nervu [16,35]. U malých dětí se ZO hodnotí použitím Teller Cards (Cardiff test, Single Book), u starších HOTV optotypy. Vyšetření ZO a zorného pole by se měly provádět při každé návštěvě oftalmologa. U perimetrického vyšetření je však nutná pacientova spolupráce, která je limitována věkem a délkou vyšetření (5-7 minut na jedno oko) a komplikována zvýšeným výskytem poruch pozornosti u pacientů s NF1 [13,35,39]. Pokud je zaznamenán nystagmus, strabismus, ztráta barvocitu, edém zrakového nervu, nebo nablednutí terče, měl by být pacient monitorován se zvýšenou pozorností, stejně jako v případě asymptomatického růstu nádoru [10]. Více vyšetření se pro statistické účely nedoporučuje, i když existují další metody, které mají potenciál biomarkeru ZO. Optická koherentní tomografie (OCT), měřící tloušťku vrstvy nervových vláken sítnice RNFL (z anglického Retinal Nerve Fiber Layer) má slibné výsledky. Použití OCT jako biomarkeru ZO se ale nedoporučuje, dokud nebude prokázána korelace úbytku RNFL s budoucí ztrátou ZO. Ačkoli ztráta ZO často koreluje s úbytkem RNFL, zdá se, že k úbytku RNFL dochází dlouhodobě a v některých případech pokračuje i po stabilizaci zrakové ostrosti [10]. Vizuální evokované potenciály (VEP) mají vysokou senzitivitu (90-100 %) a proto mohou být použity k časné identifikaci nádoru. Zároveň však mají nízkou specificitu (60-69 %) a korelují se zrakovou ostrostí pouze v 50 % případů [10,16]. Někteří pacienti s NF1 vykazují abnormální výsledky, i když se u nich nevyskytuje OPG [24]. VEP navíc nejsou schopny určit, který OPG vyžaduje léčbu, ani monitorovat změnu ZO v závislosti na léčbě. Proto nemohou sloužit jako klinický biomarker, protože ten musí být schopen identifikovat a předpovídat ztrátu ZO před zahájením léčebného postupu [10,16].
ZÁVĚR
V současné době je standardní léčbou chemoterapie, která se indikuje na základě klinické a radiologické progrese nádoru. Vliv chemoterapie na ZO je však diskutabilní, jelikož jejím nejčastějším výsledkem je stabilizace ZO a v určitých případech se ztrátě ZO pomocí chemoterapie nejspíš nedá předejít. Pro tyto pacinety je chemoterapie možná zbytečnou zátěží. Bohužel neexistuje klinický biomarker, který by dokázal spolehlivě předpovědět nadcházející ztrátu ZO. Budoucí studie budou muset porovnat výsledky radiologických nálezů a oftalmologických vyšetření s potenciálními klinickými biomarkery ZO, sledovat jejich změny ve stejných časových intervalech a v závislosti na léčbě. Nutné je také dlouhodobé sledování pacientů, aby se diferencovalo krátkodobé zlepšení od dlouhodobého trendu vývoje nemoci. To však bude možné pouze tehdy, dojde-li ke koordinaci vyšetření mezi jednotlivými specialisty.
Autoři práce prohlašují, že vznik i téma odborného sdělení a jeho zveřejnění není ve střetu zájmů a není podpořeno žádnou farmaceutickou firmou.
Přijato do redakce: 18. 2. 2019
Přijato do tisku: 8. 7. 2019

MUDr. Aneta Siwá
Dětská oční klinika LF MU a FN Brno
Černopolní 9613 00 Brno
Zdroje
1. Aihara, Y., Chiba, K., Eguchi, S. et al.: Pediatric Optic Pathway/Hypothalamic Glioma. Neurol Med Chir, 58(1); 2018 : 1–9.
2. Autrata, R.: Choroby víček a orbity. In Autrata, R. (Ed), Dětská oftalmologie I. část. Sborník prací lékařské fakulty Masarykovy univerzity č. 122. Masarykova univerzita, Brno, 2008, s. 16–17.
3. Awdeh, RM., Keihna, EN., Drewry, RD. et al.: Visual outcomes in pediatric optic pathway glioma after conformal radiation therapy. Int J Radiat Oncol Biol Phys, 84(1); 2012 : 46–51.
4. Bakker, AC., La Rosa, S., Sherman, LS. et al.: Neurofibromatosis as a gateway to better treatment for a variety of malignancies. Prog Neurobiol, 2017(152): 149–165.
5. Balcer, LJ., Liu, GT., Heller, G. et al.: Visual loss in children with neurofibromatosis type 1 and optic pathway gliomas: relation to tumor location by magnetic resonance imaging. Am J Ophthalmol, 131(4); 2001 : 442–445.
6. Cameron, F., Parsa, R.: Neurogenic tumors. In Pediatric Ophthalmology and Strabismus Fourth Edition. Eds Hoyt, C.S., Taylor, D. Edinburgh: Elsevier Ltd., 2012, s. 206–215.
7. Campen, CJ., Gutmann, DH.: Optic Pathway Gliomas in Neurofibromatosis Type 1. J Child Neurol, 33(1); 2017 : 73–81.
8. Ceballos-Quintal, JM., Pinto-Escalante, D., Castillo-Zapata, I. et al.: A new case of Klippel-Trenaunay-Weber (KTW) syndrome: Evidence of autosomal dominant inheritance. Am J Med Genet, 63(3); 1996 : 426–427.
9. Dalla Via, P., Opocher E., Pinello, ML. et al.: Visual outcome of a cohort of children with neurofibromatosis type 1 and optic pathway glioma followed by a pediatric neuro-oncology program. Neuro Oncol, 9(4); 2007 : 430–437.
10. de Blank, PMK., Fisher, MJ., Liu, GT. et al.: Optic Pathway Gliomas in Neurofibromatosis Type 1: An Update: Surveillance, Treatment Indications, and Biomarkers of Vision. J Neuroophthalmol, 37(Suppl); 2017 : 23–32.
11. Dodgshun, AJ., Elder, JE., Hansford, JR. et al.: Long‐term visual outcome after chemotherapy for optic pathway glioma in children: Site and age are strongly predictive. Cancer, 121(23); 2015 : 4190–4196.
12. Driever, PH., von Hornstein, S., Pietsch, T. et al.: Natural history and management of low-grade glioma in NF-1 children. J Neurooncol, 100(2); 2010 : 199–207.
13. Erdoğan-Bakar, E., Cinbis, M., Ozyürek, H. et al.: Cognitive functions in neurofibromatosis type 1 patients and unaffected siblings. Turk J Pediatr, 51(6); 2009 : 565–71.
14. Evans, DGR., Baser, ME., McGaughran, J. et al: Malignant peripheral nerve sheath tumors in neurofibromatosis. J Med Genet, 39(5); 2002 : 311–314.
15. Ferner, RE., Huson, SM., Thomas, N. et al.: Guidelines for the diagnosis and management of individuals with neurofibromatosis 1. J Med Genet, 44(2); 2007 : 81–88.
16. Fisher, MJ., Avery, RA., Allen, JC. et al.: Functional outcome measures for NF1-associated optic pathway glioma clinical trials. Neurology, 81(21 Suppl 1); 2013 : 15–24.
17. Fisher, MJ., Loguidice, M., Gutmann, DH. et al.: Visual outcomes in children with neurofibromatosis type 1–associated optic pathway glioma following chemotherapy: a multicenter retrospective analysis. Neuro Oncol, 14(6); 2012 : 790–797.
18. Friedrich, RE., Nuding, MA.: Optic Pathway Glioma and Cerebral Focal Abnormal Signal Intensity in Patients with Neurofibromatosis Type 1: Characteristics, Treatment Choices and Follow-up in 134 Affected Individuals and a Brief Review of the Literature. Anticancer Res, 36(8); 2016 : 4095–4121.
19. Gerinec, A.: Fakomatózy. In Gerinec, A. (Ed), Detská oftalmológia. Martin, Osveta, 2005, s. 517–520.
20. Gnekow, AK., Falkenstein, F., von Horstein, S. et al.: Long-term follow-up of the multicenter, multidisciplinary treatment study HIT-LGG-1996 for low-grade glioma in children and adolescents of the German Speaking Society of Pediatric Oncology and Hematology. J Neurooncol, 14(10); 2012 : 1265–1284.
21. Grigg, J., Jamieson, R.: Phakomatoses. In Pediatric Ophthalmology and Strabismus Fourth Edition. Eds Hoyt, C.S., Taylor, D. Edinburgh: Elsevier Ltd., 2012, s. 675–689.
22. Happle, R.: Klippel-Trenaunay syndrome: is it a paradominant trait? Br J Dermatol, 128; 1993 : 465.
23. Hudson, S., Beck, L.: Ophthalmic manifestations of neurofibromatosis. Br J Dermatol, 71(3); 1987 : 235–238.
24. Iannaccone, A., McCluney, RA., Brewer, VR. et al.: Visual evoked potentials in children with neurofibromatosis type 1. Doc Ophthalmol 105; 2002 : 63–81.
25. Kalin-Hajdu, E., Décarie, JC., Marzouki, M. et al.: Visual acuity of children treated with chemotherapy for optic pathway gliomas. Pediatr Blood Cancer, 61(2); 2013 : 223–227.
26. Kissil, J., Blakeley, J., Ferner, R. et al.: What’s New in Neurofibromatosis? Proceedings from The 2009 NF Conference: New Frontiers. Am J Med Genet A, 152 A(2); 2010 : 269–283.
27. Korf, BR.: Clinical features and pathobiology of neurofibromatosis. J Child Neurol, 17(8); 2002 : 573–577.
28. Moreno, L., Bautista F., Ashley S. et al.: Does chemotherapy affect the visual outcome in children with optic pathway glioma? A systematic review of the evidence. Eur J Cancer, 46(12); 2010 : 2253–2259.
29. Otradovec, J.: Ložiskové příznaky nitrolebních nádorů. In Otradovec, J. (Ed), Klinická neurooftalmologie. Praha, Grada, 2003, s. 364–367.
30. Otradovec, J.: Fakomatózy. In Otradovec, J. (Ed), Klinická neurooftalmologie. Praha, Grada, 2003, s. 396–403.
31. Otradovec, J.: Choroby zrakového nervu. In Otradovec, J. (Ed), Klinická neurooftalmologie. Praha, Grada, 2003, s. 193–196.
32. Petrák, B., Plevová, P., Novotný, J. et al.: Neurofibromatosis von Recklinghausen. Klin Onkol, 22(Suppl); 2009 : 38–44.
33. Petrák, B., Bendová, Š., Lisý, J. et al.: Neurofibromatosis von Recklinghausen typ 1 (NF1) – klinický obraz a molekulárně-genetická diagnostika. Cesk Patol, 51(1); 2015 : 34–40.
34. Rakotonjanahary, J., De Carli, E., Delion, M. et al.: Mortality in Children with Optic Pathway Glioma Treated with Up-Front BB-SFOP Chemotherapy. PLoS ONE [online], 10(6); 22. června 2015. [cit. 15. prosince 2018]. Dostupné na WWW: < https://doi.org/10.1371/journal.pone.0127676 >.
35. Rasool, N., Odel, JG., Kazim, M.: Optic pathway glioma of childhood. Curr Opin Ophthalmol, 28(3); 2017 : 289–295.
36. Robert-Boire, V., Rosca, L., Samson, Y. et al.: Clinical Presentation and Outcome of Patients With Optic Pathway Glioma. Pediatr Neurol, 75; 2017 : 55–60.
37. Singhal, S., Birch, JM., Kerr, B. et al.: Neurofibromatosis type 1 and sporadic optic gliomas. Arch Dis Child, 87(1); 2002 : 65–70.
38. Taylor, T., Jaspan, T., Milano, G. et al.: Radiological classification of optic pathway gliomas: experience of a modified functional classification system. Br J Radiol, 81(970); 2008 : 761–766.
39. Torres Nupan, MM., Van Meerbeke, AV., López Cabra, CA. et al.: Cognitive and Behavioral Disorders in Children with Neurofibromatosis Type 1. Front Pediatr [online]. Frontiers Media S.A, 5(227); 2017. [cit. 15. prosince 2018]. Dostupné na WWW:<https://www.frontiersin.org/articles/10.3389/fped.2017.00227/full>
40. Vivarelli, R., Grosso, S., Calabrese, F. et al.: Epilepsy in neurofibromatosis 1. J Child Neurol, 18(5); 2003 : 338–42.
41. Walker, DA., Liu, J., Kieran, M. et al.: A multi-disciplinary consensus statement concerning surgical approaches to low-grade, high-grade astrocytomas and diffuse intrinsic pontine gliomas in childhood (CPN Paris 2011) using the Delphi method. Neuro Oncol, 15(4); 2013 : 462–468.
42. National Institute of Neurological Disorders and Stroke: Neurofibromatosis Fact Sheet. National Institute of Neurological Disorders and Stroke [online]. Květen 2011. National Institute of Health, 11-2126.; 6. července 2018. [cit. 15. prosince 2018]. Dostupné na WWW: https://www.ninds.nih.gov/Disorders/Patient-Caregiver-Education/Fact-Sheets/Neurofibromatosis-Fact-Sheet
Štítky
OftalmologieČlánek vyšel v časopise
Česká a slovenská oftalmologie

2019 Číslo 4
- Selektivní laserová trabekuloplastika nesnižuje nitroční tlak více než argonová laserová trabekuloplastika
- Progresi glaukomu je třeba hodnotit strukturálními i funkčními parametry
- Ztráta centrálního vidění po filtrujících operacích glaukomu
- Od PGF-2 alfa-isopropyl esteru k latanoprostu: přehled vývoje Xalatanu
- Compliance u pacientů s glaukomem
Nejčtenější v tomto čísle
- NEUROFIBROMATÓZA 1. TYPU A GLIOM OPTIKU
- Léčba vitreomakulární trakce intravitreální aplikací perfluoropropanu
- Lichen planus jako možná vzácná příčina očního onemocnění
- Doc. MUDr. Vladimír Krásnik, PhD oslavuje 60-tku
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy