
A Yeast GSK-3 Kinase Mck1 Promotes Cdc6 Degradation to Inhibit DNA Re-Replication
Cdc6p is an essential component of the pre-replicative complex (pre-RC), which binds to DNA replication origins to promote initiation of DNA replication. Only once per cell cycle does DNA replication take place. After initiation, the pre-RC components are disassembled in order to prevent re-replication. It has been shown that the N-terminal region of Cdc6p is targeted for degradation after phosphorylation by Cyclin Dependent Kinase (CDK). Here we show that Mck1p, a yeast homologue of GSK-3 kinase, is also required for Cdc6 degradation through a distinct mechanism. Cdc6 is an unstable protein and is accumulated in the nucleus only during G1 and early S-phase in wild-type cells. In mck1 deletion cells, CDC6p is stabilized and accumulates in the nucleus even in late S phase and mitosis. Overexpression of Mck1p induces rapid Cdc6p degradation in a manner dependent on Threonine-368, a GSK-3 phosphorylation consensus site, and SCFCDC4. We show evidence that Mck1p-dependent degradation of Cdc6 is required for prevention of DNA re-replication. Loss of Mck1 activity results in synthetic lethality with other pre-RC mutants previously implicated in re-replication control, and these double mutant strains over-replicate DNA within a single cell cycle. These results suggest that a GSK3 family protein plays an unexpected role in preventing DNA over-replication through Cdc6 degradation in Saccharomyces cerevisiae. We propose that both CDK and Mck1 kinases are required for Cdc6 degradation to ensure a tight control of DNA replication.
Published in the journal:
. PLoS Genet 8(12): e32767. doi:10.1371/journal.pgen.1003099
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1003099
Summary
Cdc6p is an essential component of the pre-replicative complex (pre-RC), which binds to DNA replication origins to promote initiation of DNA replication. Only once per cell cycle does DNA replication take place. After initiation, the pre-RC components are disassembled in order to prevent re-replication. It has been shown that the N-terminal region of Cdc6p is targeted for degradation after phosphorylation by Cyclin Dependent Kinase (CDK). Here we show that Mck1p, a yeast homologue of GSK-3 kinase, is also required for Cdc6 degradation through a distinct mechanism. Cdc6 is an unstable protein and is accumulated in the nucleus only during G1 and early S-phase in wild-type cells. In mck1 deletion cells, CDC6p is stabilized and accumulates in the nucleus even in late S phase and mitosis. Overexpression of Mck1p induces rapid Cdc6p degradation in a manner dependent on Threonine-368, a GSK-3 phosphorylation consensus site, and SCFCDC4. We show evidence that Mck1p-dependent degradation of Cdc6 is required for prevention of DNA re-replication. Loss of Mck1 activity results in synthetic lethality with other pre-RC mutants previously implicated in re-replication control, and these double mutant strains over-replicate DNA within a single cell cycle. These results suggest that a GSK3 family protein plays an unexpected role in preventing DNA over-replication through Cdc6 degradation in Saccharomyces cerevisiae. We propose that both CDK and Mck1 kinases are required for Cdc6 degradation to ensure a tight control of DNA replication.
Introduction
To constitute the pre-RC and initiate DNA replication, all six-components of the Origin Recognition Complex (Orc1-6p) bind to replication origins followed by Cdc6p, Cdt1p and the Mcm2-7p complex [1]. Then the pre-RC has to be activated by the Dbf kinase-Cdc7p complex, resulting in the formation of a bidirectional replication fork in which the Mcm complex acts as a replicative helicase [1]. Finally, DNA polymerase synthesizes new strands of DNA. The cell cycle progression is driven by the Cyclin/CDK complex. Of the nine cyclins in S. cerevisiae six are B-type cyclins (Clb1-6) [2] and there is a single CDK (Cdc28). Cdc28-Clb activity is required to initiate DNA replication [3]–[5].
Eukaryotes ensure that DNA is replicated once and only once per cell cycle. There are multiple overlapping mechanisms to prevent re-initiation of DNA replication. Pre-RC components such as Cdc6, Mcm2–7, and the ORC complex are phosphorylated by Cyclin/CDK to prevent a second round of DNA replication from occurring before mitosis. Cdc6 is phosphorylated by Cyclin/CDK complex at the N-terminal region and is targeted for ubiquitin-mediated proteolysis in S. cerevisiae [6]–[8]. The MCM complex is translocated to the cytoplasm after phosphorylation by Cdk activity [9], [10]. Orc2 and Orc6 are also phosphorylated in a CDK-dependent manner [11], [12]. In addition to these mechanisms, a direct recruitment of the cyclin-CDK complex Clb5p-Cdc28p to the origin of replication is an important component of re-replication control [13]. The Clb5p recruitment to the origin is accomplished by binding of the Clb5p hydrophobic patch substrate-targeting domain [14]–[16] to an Arg-X-Leu (RXL) target sequence in the Orc6p subunit of the ORC origin recognition complex [13]. This Clb5 binding to Orc6 after origin licensing serves as a local switch to inhibit DNA re-replication by preventing Cdt1/Mcm2–7 loading onto the origin [17]. The ORC6-rxl mutation strongly synergized with other mutations previously implicated in re-replication control including: N-terminal deletions in Cdc6 which stabilize the protein (CDC6ΔNT) [13], mutations which force nuclear localization of the Mcm complex (MCM7-NLS) [11], and mutations blocking Orc2 (ORC2-ps) and Orc6 phosphorylation (ORC6-ps) [18]. Such multiple mutant strains strongly over-replicate DNA within a single cell cycle [13].
ORC6-rxl GAL-CDC6ΔNT cells are viable, but show moderate DNA re-replication when incubated in galactose [19]. The cell cycle in the ORC6-rxl GAL-CDC6ΔNT cells arrest at G2/M phase due to DNA damage checkpoint activation [19]. Moderate cell viability in the ORC6-rxl GAL-CDC6ΔNT cells was heavily dependent on DNA damage checkpoint components such as MRE11 gene. Cell viability was reduced and DNA re-replication was enhanced in mre11 ORC6-rxl GAL-CDC6ΔNT cells [19]. It is known that Rad53 is phosphorylated upon DNA damage checkpoint activation. Rad53 was hyperphosphorylated in ORC6-rxl GAL-CDC6ΔNT cells [19], suggesting that DNA damage was induced. We concluded that DNA re-replication most likely causes double strand breaks which in turn activates the DNA damage checkpoint response [19].
To identify a new component that inhibits DNA re-replication in S. cerevisiae, synthetic genetic array (SGA analysis) [20] was performed using an ORC6-rxl strain to eliminate Clb5-Orc6 binding. We found that mck1 deletion cells combined with the ORC6-rxl mutation showed synthetic lethality. The MCK1 gene in S. cerevisiae encodes a serine/threonine protein kinase homologous to mammalian glycogen synthase kinase-3 (GSK-3) [21]. Mammalian GSK-3 was initially identified as an enzyme involved in the control of glycogen metabolism [22]. GSK-3 kinase is highly conserved through evolution and plays an important role in the Wnt signaling pathway in the mammalian system (for a review, see [23]). One of the interesting features of GSK-3 kinase is its role in protein degradation. GSK-3 phosphorylates cyclin D1 to promote its nuclear export and subsequent degradation in the mammalian system [24]. Yeast Mck1p has diverse biological functions. Mck1p stimulates calcineurin signaling [25]–[27] and binds stress-response elements to activate transcription [27] therefore cells lacking Mck1p are hot and cold sensitive [28]. Mck1 is also implicated in mitosis and meiosis. Yeast MCK1 has been isolated as a dosage suppressor of centromere (CEN) DNA mutation in CDEIII, suggesting that Mck1 has a role in centromere/kinetochore function [28]. The mck1 mutant exhibits poor sporulation [29], and sensitivity to benomyl, a microtubule destabilizing drug [28].
Cdc6 levels are regulated by three distinct mechanisms: transcription [30], ubiquitin-mediated proteolysis [7], [8], [31], [32] and nuclear localization [33]. Here we show that Mck1p has a novel function in inhibition of DNA re-replication by Cdc6p degradation through the GSK-3 consensus site at T368.
Results
Deletion of MCK1 causes synthetic lethality in the orc mutants
Synthetic genetic array (SGA analysis) [20] was performed using ORC6-rxl, to eliminate Clb5-Orc6 binding, in order to identify a new component in the regulation of DNA re-replication in S. cerevisiae. We found that mck1 deletion cells showed synthetic lethality in cells containing the ORC6-rxl mutation. It is interesting that mck1 was the only deletion strain that caused synthetic lethality in the ORC6-rxl cells among 4700 deletion strains tested, and that we did not obtain other GSK-3 orthologs in this screening. Tetrad analysis confirmed the genetic interaction between ORC6-rxl and mck1 deletion strains (Figure 1A). Haploid progenies, which contain both ORC6-rxl and Δmck1 mutations, were not able to grow on YEPD plates whereas single mutants grew fine. We also tested if the mck1 deletion genetically interacts with the other orc mutants such as the Orc6 phosphorylation site mutant (ORC6-ps) and the Orc2 phosphorylation site mutant (ORC2-ps). Deletion of MCK1 reduced cell growth in the ORC6-ps cells (Figure 1A). Furthermore, the mck1 deletion caused severe growth defects in the ORC2-ps cells (Figure 1A). Thus, mck1 deletion caused synthetic lethality or semi-lethality with DNA re-replication-prone orc mutants in general. This strongly suggests that Mck1p has a function in DNA replication control. The mck1 deletion strain did not have genetic interactions with other pre-RC mutants such as MCM7-NLS or CDC6ΔNT (data not shown).

Combination of mck1 deletion and ORC6-rxl mutation induced DNA damage checkpoint activation
To investigate the molecular basis of the synthetic lethality between Δmck1 and ORC6-rxl, we generated partial loss of function mutants of mck1 by PCR mutagenesis. Among them, mck1-16 allele exhibited semi-synthetic lethality at high temperature (36 degrees) when combined with ORC6-rxl mutation (Figure 1B). Consistent with this effect being due to the disruption of Clb5-Orc6 protein interaction by the ORC6-rxl mutation, the clb5 mck1-16 cells were also semi-lethal when incubated at 36 degrees (Figure 1B). To analyze the terminal phenotype of the mck1-16 ORC6-rxl strain, cells were incubated either at permissive or non-permissive temperatures and cell cycle profiles were analyzed by flow cytometry analysis. The mck1-16 ORC6-rxl cells showed G2/M arrest after 4 hours incubation at 36 degrees (Figure 1C, top right), with some cells showing a DNA content over 2C (Figure 1C, arrow), suggesting re-replicated DNA. Cell morphologies of the mck1-16 ORC6-rxl mutants were further analyzed. The mck1-16 ORC6-rxl cells incubated at 36 degrees for 4 hours showed large budded cells with a single nuclei visualized by propidium iodide staining of DNA (Figure 1D). This phenotype is reminiscent of cells with DNA re-replication found in our previous report [19]. Nuclear division did not occur in the mck1-16 ORC6-rxl cells. Their cell cycle is arrested during G2 or early mitosis, most likely due to DNA damage checkpoint activated by DNA re-replication. This is similar to our previous observation that mitotic arrest in the ORC6-rxl CDC6ΔNT cells was due to DNA damage [19].
Previously we have shown that the ORC6-rxl mutant causes semi-synthetic lethality with a CDC6ΔNT mutant. The ORC6-rxl CDC6ΔNT cells are arrested during mitosis with moderate DNA re-replication followed by DNA damage. Viability of the ORC6-rxl CDC6ΔNT cells was heavily dependent on an intact DNA damage checkpoint gene such as MRE11, a component of the MRX complex [19]. Rad53, a transducer kinase required for DNA damage checkpoint activation, was hyperphosphorylated in the ORC6-rxl CDC6ΔNT cells. To directly test if DNA damage checkpoint is activated in the mck-16 ORC6-rxl cells, Rad53 phosphorylation status was analyzed by Western blotting. Rad53 was only hyperphosphorylated in the mck-16 ORC6-rxl cells when incubated at 37 degrees (Figure 2A). We tested if the viability of the mck1-16 ORC6-rxl mutant also relies on DNA damage checkpoint. We found that cell viability of the mck-16 ORC6-rxl cells even at the permissive temperature (30 degrees) required MRE11 (Figure 2B). Next, the cell cycle profile of the mre11 mck-16 ORC6-rxl cells was examined. DNA re-replication was greatly enhanced in the mre11 mck-16 ORC6-rxl cells at the non-permissive temperature, indicating that DNA damage checkpoint activation limits DNA re-replication in the mck-16 ORC6-rxl cells (Figure 2C). Above all, we conclude that an induction of DNA re-replication in the mck-16 ORC6-rxl cells triggered DNA damage leading to cell cycle arrest by DNA damage checkpoint activation.

Mck1 prevents DNA re-replication in parallel to ORC and MCM complexes
Several parallel and partially overlapped molecular mechanisms ensure that cells do not re-initiate DNA replication at origins that have already fired. We have previously shown that ORC6-rxl CDC6ΔNT cells are mitotic arrested without extensive DNA re-replication [19]. However, multiple mutant strains such as ORC6-rxl,ps CDC6ΔNT MCM7-NLS ORC2-ps strongly over-replicate DNA within a single cell cycle [13]. We tested if mck1 deletion also synergizes with other pre-RC mutations. An addition of either MCM7-NLS or ORC2-ps mutation to the ORC6-rxl mck1-16 did not enhance lethality (Figure 3A). However, cells containing ORC6-rxl,ps mck1-16 MCM7-NLS and ORC2-ps mutations showed stronger lethality (Figure 3A). Flow cytometry analysis showed that DNA re-replication was enhanced in the ORC6-rxl,ps mck1-16 MCM7-NLS ORC2-ps mutant after 4 hours incubation at the non-permissive temperature (Figure 3B, bottom right). ORC6-rxl,ps MCM7-NLS ORC2-ps cells with wild type MCK1 grew normally and did not induce significant re-replication (Figure 3A and 3B bottom left). These results show that Mck1p contributes to the inhibition of DNA re-replication and suggest that the mechanism involved is likely to be distinct from the known mechanisms acting at the level of ORC and MCM proteins.

The mck1 deletion strain genetically interacted with S-phase cyclins, but not mitotic cyclins
The semi-lethal phenotype of ORC6-rxl Δmck1 cells (Figure 1D) was reminiscent of ORC6-rxl CDC6ΔNT cells [13]. Moreover, the deletion of MCK1 interacted genetically with ORC6-rxl (Figure 1A) but not CDC6ΔNT (data not shown). These observations led us to hypothesize that Mck1p could function in DNA replication control by regulating Cdc6. To further test this model, we examined if mck1 deletion behaved similarly to CDC6ΔNT in its interactions with mutations in the cyclin genes.
CDC6ΔNT genetically interacts with the clb5 deletion mutant, but not with other B-type cyclins [34]. We also tested if mck1 deletion cells genetically interact with other cyclin mutants in a similar way that CDC6ΔNT does. Table 1 summarizes the genetic interaction between mck1 and cyclin mutants. The mck1 deletion cells were semi-lethal in the ORC6-rxl mutant cells and also showed synthetic lethality with clb5 deletion cells because ORC6-rxl is a binding mutant for Clb5p. However, the mck1 deletion cells did not cause synthetic lethality with other B-type cyclin mutants such as clb1,2,3,4 or 6 (Table 1). Therefore, mck1 deletion genetically interacts specifically with clb5 deletion. It has been shown that Clb5p binds to Orc6p through the Clb5p hydrophobic patch substrate-targeting domain [14]. We tested if clb5-hpm (Clb5 hydrophobic patch mutant) causes synthetic lethality with Δmck1 cells and found that there was a genetic interaction between clb5-hpm and Δmck1 (Table 1). Moreover neither mck1 nor CDC6ΔNT caused lethality in clb5pCLB2, a mutant in which Clb2 is controlled under Clb5 promoter. Thus, we conclude that the Δmck1 cells require Clb5p-Orc6p protein binding for their survival. We also found that deletion of CLB6 rescues Δmck1 Δclb5 semi-lethality. We have previously shown that lethality in clb5 CDC6ΔNT cells can be rescued by the deletion of CLB6 [34] and proposed the idea that the S-phase cyclin Clb6 initiates DNA replication, but fails to inhibit DNA re-replication. Therefore, the DNA re-replication phenotype is suppressed if CLB6 is deleted by the reduction of initiation of DNA replication. Mitotic cyclins regulate DNA replication in the clb5 clb6 ORC6-rxl cells. We speculate that deletion of CLB6 rescues Δmck1 Δclb5 cells in the same manner.

From these results we conclude that the mck1 deletions genetically interacted with cyclin mutants in a way similar to that of stabilized CDC6ΔNT, reinforcing a model in which Mck1p acts in the same pathway as Cdc6p.
Mck1p kinase is required for Cdc6p degradation in mitosis
Because lack of Mck1p and stabilization of Cdc6p (Cdc6ΔNT) exhibited similar genetic interaction with DNA re-replication mutants, we speculated that Mck1p could control the stability of Cdc6p. To test this possibility, the Cdc6 protein (Cdc6-HA) expressed under inducible GAL1 promoter in mitotically arrested cells was examined in wild type or Δmck1 backgrounds. We found that the Cdc6 protein level was sustained at a higher level during mitosis in the mck1 deletion cells than in wild type cells even after Cdc6 expression was shut off by glucose (Figure 4A). It is important to mention that CDC6 was expressed under the GAL1 promoter, excluding possible involvement of CDC6 transcription by Mck1 in this experiment. To test if Mck1 regulates Cdc6p post-translational levels, endogenous Cdc6 synthesis was blocked by cycloheximide. In the mitotically arrested wild type cells, Cdc6 protein was rapidly depleted by addition of cycloheximide (Figure S1). In the mitotic mck1 deletion cells, the cdc6 protein level was high and remained stable after cycloheximide, excluding the possibility that Mck1p regulates Cdc6p by translation. These results strongly suggest that Mck1p controls Cdc6 protein levels by affecting degradation rates.

To further explore the possible involvement of Mck1p in Cdc6p degradation, Protein A-tagged Cdc6 protein integrated at the genome locus was examined in the wild type or mck1 deletion cells by Western blotting throughout a single cell cycle progression. We noticed a dramatic accumulation of Cdc6 protein in the mck1 deletion cells (Figure 4B). In wild type cells, Cdc6p was expressed transiently during G1 phase, 10 minutes after alpha-factor release, and suppressed throughout S-phase. Then Cdc6p was expressed again for a short time during mitosis, 70 minutes after alpha-factor release (Figure 4B, upper panel). This is consistent with a previous report by Drury et al [32]. While in the mck1 deletion cells, Cdc6p was not expressed during alpha-factor arrest but was expressed 10 min after alpha-factor release and continued to accumulate during S-phase and mitosis (Figure 4B, lower panel). The increase in Cdc6 protein level is unlikely to be due to an alteration in the cell cycle progression of Δmck1 cells because the kinetics of the cell cycle progression was similar in these two strains as judged by budding index (Figure 4B). To confirm that Cdc6p is stabilized during mitosis in the mck1 deletion strain, CDC6-ProteinA or mck1 CDC6-ProteinA strains were arrested in mitosis by nocodazole and were synchronously released into the cell cycle by washing. A small amount of Cdc6p was detectable at time zero in nocodazole arrested wild type cells (Figure 4D, left). This amount was transiently increased 10–20 minutes after release. This is consistent with a previous report that Cdc6 protein is expressed in late mitosis and degraded after the G1/S transition [7]. In contrast, Cdc6p was stabilized throughout mitotic progression in the mck1 deletion cells (Figure 4D, right).
To further confirm if Cdc6 is stabilized in the mck1 deletion cells, we visualized Cdc6p localization in vivo. We introduced a GFP-tag into the C-terminus of the chromosomal copy of the CDC6 gene to allow endogenous expression. The CDC6-GFP fusion appears to be fully functional as a CDC6-GFP strain and did not show any growth defect in any of the conditions tested (data not shown). Consistent with previously published localization patterns of overexpression, Cdc6-GFP [33], [35] protein localized and accumulated in the nucleus in late mitotic cells (large budded cells with divided nuclei) or in unbudded G1 cells (Figure 4C). The Cdc6-GFP signal was undetectable in the cells with small to large buds, confirming tight regulation of Cdc6 abundance by rapid degradation after S-phase onset. In sharp contrast, Cdc6-GFP was constitutively found in the nucleus throughout the cell cycle in mck1 deletion cells (Figure 4C). This localization analysis was consistent with Western blot results that Cdc6p is stabilized in mck1 deletion cells during S-phase and mitosis, as shown in Figure 4B and 4D.
We also tested if overexpression of Mck1 promotes rapid Cdc6p degradation. Exogenously expressed Mck1p under the GALL promoter significantly reduced Cdc6p protein levels 10 minutes after the addition of galactose (Figure 5A, top right). This result supports the idea that Mck1p promotes Cdc6p degradation.
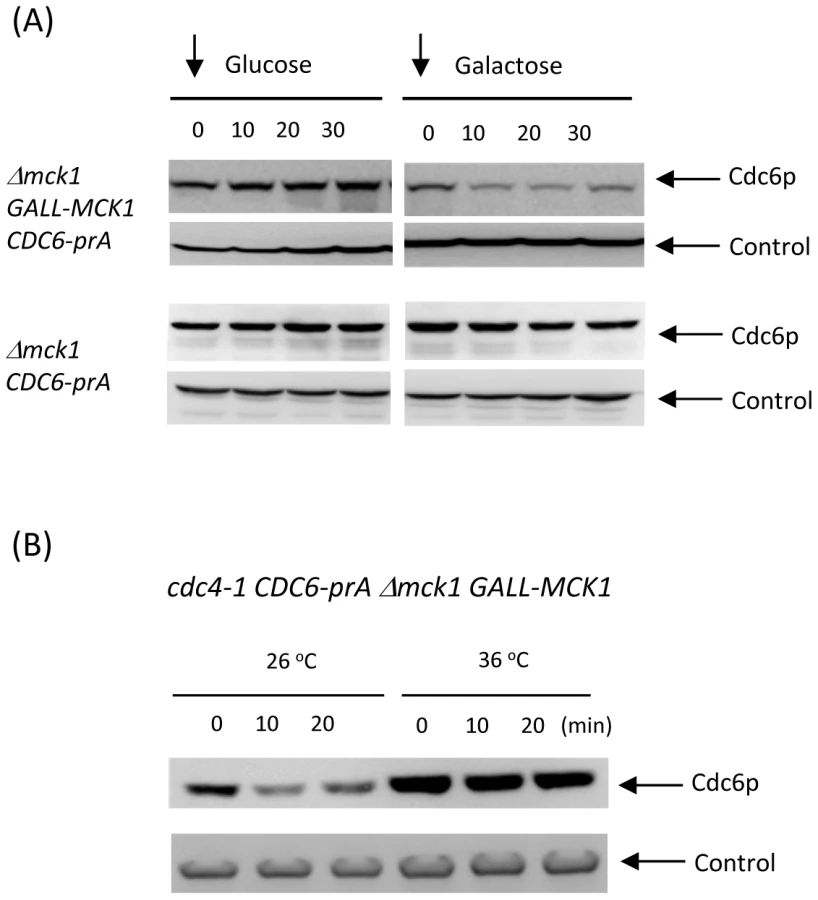
Mck1-mediated Cdc6 degradation was inhibited in cdc4-1 mutant
We next examined if Mck1-mediated Cdc6 degradation is due to SCFCDC4 ubiquitin ligase. When cdc4-1 CDC6-prA mck1 GALL-MCK1 strain was incubated at 26 degrees, Cdc6p was rapidly degraded followed by galactose addition (Figure 5B). This is consistent with results in Figure 5A. When Cdc4 was inactivated at 36 degrees, Cdc6 became stable and was not degraded even after Mck1 overexperssion (Figure 5B). This result suggests that Mck1p phosphorylates Cdc6p to be subsequently recognized by SCFCDC4 complex for degradation.
Mck1p binds to Cdc6p through a GSK-3 consensus site in the C-terminal region
GSK-3 kinases phosphorylate the first serine or threonine residues in the consensus site followed by a phospho-serine or phospho-threonine at the position +4 [S/T-XXX-pS/T] [36]. There are two potential GSK-3 consensus phosphorylation sites in Cdc6p, TPESS (39–43) and TPTTS (368–372) (Figure 6A). To test if Mck1p binds Cdc6p at the GSK-3 consensus sites, we performed a yeast two-hybrid assay. We examined whether Mck1p, fused with Gal4 activation domain (GAD), interacts with various truncated CDC6 mutants fused to the LexA DNA binding domain. Mck1p interacted with the C-terminal region of Cdc6p (aa341–390) and not with the N-terminus (aa 1–47) (Figure 6B). The mutation at T368M or S372A abolished two-hybrid interaction between Mck1p-Cdc6p indicating that Mck1p targets Cdc6p through the GSK consensus site at 368–372 (Figure 6B). The physical interaction between Mck1p and Cdc6p was also confirmed by co-immunoprecipitation (Co-IP) assay using the MCK1-MYC GAL-CDC6ΔNT-HA strain. Mck1p interacted with Cdc6ΔNTp, indicating that Mck1p interacts with Cdc6p, and the protein interaction was mediated through the C-terminal region in Cdc6p (Figure 6C). The protein binding between Mck1p and Cdc6p was observed only in mitotic arrested cells blocked by nocodazole and not in asynchronous culture or G1-arrested cells (data not shown). Therefore the physical interaction between Mck1p and Cdc6p is likely primed by mitotic CDK phosphorylation of the S372 site (see next section). We also noticed that Cdc6ΔNT migrates slower in the co-IP samples than the input, consistent with the idea that only the phosphorylated form of Cdc6, probably targeted by CDK, binds to Mck1 (Figure 6C).

CDC6 mutations that abrogate GSK-3 binding are lethal with the orc mutants
A GSK-3 kinase usually requires priming [36]. In Cdc6, the predicted priming site is located at S372 based on the amino acid sequence. After priming, the GSK-3 kinase phosphorylates the target site at the first serine or threonine that corresponds to T368 (see discussion). Next, we tested to see if mutations at the GSK-3 consensus phosphorylation site in CDC6 cause lethality in orc mutants like the mck1 deletion does. To prove that the C-terminus GSK-3 consensus site 368–372 in CDC6 was involved in the inhibition of DNA re-replication, the potential phosphorylation site (T368) and the priming phosphorylation site (S372) were altered to alanine. The CDC6-T368A S372A in a 2 micron plasmid was transformed into wild type, ORC6-rxl, ORC6-ps or ORC6-rxl,ps mutants. Colonies formed when either CDC6 wild type or CDC6 T368A S372A plasmids were transformed into the ORC6-wild type strain (Figure 7B, top left). In contrast, the CDC6 T368A S372A plasmid (but not CDC6-wt) was toxic in the ORC6-rxl cells, as transformants gave very few visible colonies (Figure 7B, top right). This effect was even more pronounced in ORC6-rxl,ps cells and, in this case, even the CDC6-wt plasmid appeared somewhat toxic (Figure 7B, bottom right). The CDC6-T368A S372A plasmid did not induce toxicity in the ORC6-ps cells (Figure 7B, bottom left) which confirmed the result that mck1 did not genetically interact with ORC6-ps mutation (Figure 1). The plasmid harboring CDC6-T368A or CDC6-S372A single mutation was also toxic in the ORC6-rxl strain (Figure S2). These results suggest that the interaction of Cdc6p with Mck1p and/or its phosphorylation by Mck1p contributes to the down-regulation of Cdc6p levels.

Mck1p phosphorylates Cdc6 for its degradation
To confirm that Cdc6p is phosphorylated by Mck1 in vivo, we analyzed the Mck1-dependent mobility shift of Cdc6p in the cdc4-1 mutant background by western blot. We used cdc4-1 mutant to prevent degradation of phosphorylated Cdc6 and examined the effect of Mck1 on the phosphorylation status of Cdc6p. Cdc6p in the wild type cells migrated slower that that in the Δmck1 deletion cells indicating that Cdc6p is hyper-phosphorylated in wild type cells. (Figure 7C). In the mck1 deletion cells, the signal of the higher molecular weight band was abrogated and the lower band was abundant suggesting that Cdc6p is less phosphorylated and more stable (Figure 7C right). To confirm that the slow migrating band of Cdc6p in the wild type cells is due to phosphorylation, protein extracts from wild type cells were treated with CIP (calf intestine phosphatase). After the CIP treatment, the slower migrating band of Cdc6p disappeared and the faster-migrating band was observed at the same level as that in Δmck1 cells. It suggests that the band shift between wild type and Δmck1 is due to phosphorylation (Figure 7C and 7D).
Finally we tested if Mck1p dependent destabilization of the Cdc6p is mediated by the T368 residue. The mck1 GALL-MCK1 CDC6-proteinA CDC6T368A strain contains both wild type Cdc6 (tagged with protein A) and Cdc6T368A (no tag). First the cells were arrested in mitosis with nocodazole and then released into galactose to overexpress Mck1p. Wild type Cdc6p was degraded rapidly after Mck1p overexpression, which is consistent with previous results in Figure 5A (Figure 7E, upper panel). In contrast, Cdc6T368A protein was resistant to degradation and was stable even after Mck1p overexpression (Figure 7E, lower panel). We also observed faster migration of Cdc6T368A protein than the wild type Cdc6p by western blot (Figure S3). We conclude that Cdc6p is phosphorylated at T368 by Mck1p to induce its degradation.
Discussion
In this study, we show that a GSK-3-like kinase, Mck1p, is involved in the inhibition of DNA re-replication through its role in Cdc6p turnover in S. cerevisiae. There are 8 CDK consensus sites in CDC6. The first 47 amino acids at the N-terminus of Cdc6 are targeted by Cyclin/CDK and are critical for SCFcdc4 dependent proteolysis [7]. Stabilization of Cdc6p in mck1 deletion cells suggests that CDK-dependent phosphorylation at the N-terminus of Cdc6 is not sufficient enough for CDC6p degradation in vivo, that Mck1-dependent phosphorylation through T368 site is also required. The Cdc6 T368A mutant was resistant to Mck1p-dependent degradation (Figure 7E). Nocodazole was added to the media throughout this experiment, therefore Cdc6 stabilization by the T368A mutation, even after Mck1p overexpression, is not due to a change in cell cycle progression. This is of particular interest because activation of CDK promotes both DNA replication and Cdc6p degradation at the same time. The requirement of Mck1 for Cdc6p degradation most likely ensures that degradation of Cdc6p occurs only after origin firing has been initiated.
Three distinct Cdc6p degradation modes have been proposed by Diffley's group [32]. Mode1 degradation during G1 phase is independent of Cdc6 CDK consensus sites and is mediated neither by SCF nor APC. The Cdc6p degradation by Mode 2 and Mode 3 are triggered later during the cell cycle. Mode3 is required for Cdc6 degradation during mitosis. The Cdc6p degradation by Mck1p accounts for the mode3 mechanism based on the Cdc6p stabilization pattern during mitosis in mck1 deletion (Figure 4A). Diffley's group has reported that the Cdc6 T368M mutation leads to Cdc6p stabilization during mitosis and the mutation is resistant to mode 3 proteolysis by SCFcdc4 complex [6]. In this study, we showed that Mck1-dependent Cdc6 phosphorylation is targeted by SCFCDC4 complex for degradation (Figure 5B). Therefore, Mck1, most likely, phosphorylates Cdc6 and the phosphorylation at T368 is recognized by Cdc4. It is not clear if mode 3 requires CDK activity. Therefore Mck1p may promote complete Cdc6 degradation during mitosis in addition to its degradation mechanism through CDK phosphorylation. Further studies are required to test if Mck1 could also promote Cdc6 degradation via Mode 1 or Mode 2.
There are two potential GSK-3 sites S/TXXXpS/T in Cdc6, at 39-43 and 368-372 amino acid residues. It has been reported that these sites share sequence similarities and are targeted for SCFCDC4 dependent proteolysis [6]. Our yeast-two hybrid assay showed a specific interaction between Mck1p and Cdc6p through the GSK-3 consensus site located at residues 368–372 (TPTTS). This GSK-3 site in Cdc6p, amino acid 368–372, is also shared by two potential CDK phosphorylation sites 368–371 (TPTT) and 372–275 (SPVK). The former partially matches with a minimal consensus CDK phosphorylation site (S/T-P) whereas the latter perfectly matches an optimal CDK site, with a basic residue at the +3 position. It is important to note that Cdc6 is a very good substrate of the B-type Cyclin/CDK complex [37]. The GSK-3 kinase and CDK could share substrate specificity [38]. GSK-3 kinases require “priming” phosphorylation by another kinase on their substrates [36]. The priming site is usually located C-terminally of the GSK-3 phosphorylation site, at the +4 position, which corresponds to S372 in Cdc6. After priming, GSK-3 recognizes its target and can phosphorylate the first serine or threonine residue, which corresponds to T368 in Cdc6. Thus, C-terminal Cdc6p (aa 341–390), including the GSK-3 consensus phosphorylation sequence, is sufficient for Mck1 binding and their interaction likely depends on phosphorylation of S372 by CDK (Figure 6B). We propose a model in which S372 is phosphorylated by cyclin/CDK first in order to induce phosphorylation at T368 by Mck1p kinase. This priming model allows Cdc6 to create Cdc4 diphospho-degrons which is an efficient Cdc4 recognition site. David Morgan's group shows that Eco1 is primed by CDK and DDK in order to be targeted by Mck1, which creates Cdc4 recognition site (personal communication). Mck1 is involved in the degradation of SCFCDC4 substrates such as Rcn1and Hsl1 [25], [26], [39]. Therefore, the priming model to create Cdc4 diphospho-degrons seems to be a universal mechanism to regulate protein degradation.
Mck1p protein levels are not cell cycle-regulated (data not shown) therefore Mck1 activation is not regulated by its own expression level. This result supports the idea of the priming hypothesis in which Mck1 can target its substrate, Cdc6p, only after Cdc6 is phosphorylated by cyclin/CDK in a cell cycle-dependent manner. Given the requirement of T368 for Mck1 dependent degradation of Cdc6, Mck1 most likely phosphorylates this residue directly in vivo. However, it is formally possible that Mck1 affects Cdc4 function other than Cdc6. We favor the model that Mck1 directly phosphorylates Cdc6 to promote Cdc4-dependent degradation based on our results in Figure 5B, Figure 7C and 7D. Whether or not SCFCDC4 or other targets such as Sic1 are also phosphorylated by Mck1 is an interesting future study.
The glycogen synthase kinase-3 (GSK-3) was originally identified as a kinase that inactivates glycogen synthase [40]. In higher eukaryotes, there are two isoforms, GSK-3α and GSK-3β, that regulate various cellular processes including Wnt signaling [41] and insulin signaling [42], [43]. The yeast homologue of GSK3, Mck1p, also has diverse biological functions (see introduction). This is the first evidence to show that Mck1p or any GSK-3 kinase controls DNA replication. Whether GSK-3 kinases contribute to the regulation of DNA replication at other targets should be investigated further.
Materials and Methods
SGA analysis
SGA analysis was performed as previously described [19], [20]. A query strain, MATalpha ORC6-rxl::LEU2 mfa::MFA1pr-HIS3 trp1 ade2 can1 leu2 his3 lys2 ura3, was placed on YEPD in rectangle plates. Then deletion mutant arrays (MATa geneX::KanMX TRP1 ADE2 met15 leu2 ura3 his3) were put on top of the query strains. The resulting diploid cells were sporulated on the plates containing 2% agar, 1% potassium acetate, 0.1% yeast extracts, 0.05% glucose, supplemented with uracil and histidine. After incubation at 22 degrees for 5 days, the spores were pinned onto haploid selection plates (SD-His/Leu/Arg plus canavanine) to select for MATa mfa::MFA1pr-HIS3 ORC6-rxl::LEU2 progeny, followed by pinning onto YEPD plates containing G418 to select out the deletion array mutants. Finally, double mutants were placed on SD-His/Leu/Arg plus canavanine plus G418 for 2 days. The proliferation of those that contained haploid cells was scored visually. The deletion sets used in this study were obtained from EuroScarf and are derivatives of BY4741 [44].
Cell cycle blocks
First, GAL-CDC6-HA or mck1 GAL-CDC6-HA strains were grown in raffinose-containing media and then galactose was added to express Cdc6-HA for 2 hours. The cell cycle was blocked during mitosis by nocodazole at the concentration of 15 µg/ml for 2 hours. Next, glucose was added to the media to shut off the GAL expression (Figure 4A). CDC6-PRA or mck1 CDC6-PRA strains were grown in liquid YEPD to log-phase at 30 degrees and then treated with alpha-factor at the concentration of 100 nM for 2 hours. The cells were washed with YEPD three times to release the cell cycle from G1. Samples were collected every 10 minutes for 80 minutes for Figure 4B. To block the cell cycle during mitosis, CDC6-PRA or mck1 CDC6-PRA strains were treated with nocodazole at the concentration of 15 µg/ml for 2.5 hours at 30 degrees. The mitotic block was released by washing cells with YEPD twice. Samples were collected every 10 minutes for 60 minutes for Figure 4D. For Figure 5 and 7D, cells were treated with nocodazole for 2 hours and then switched to YEPD or YEPG containing nocodazole at 15 µg/ml.
Plasmids and strains
All strains used, except for SGA analysis, are derivatives of W303 (strain list in Table S1). Standard methods were used for mating and tetrad analysis. DNA transformation was performed by the lithium acetate method [45]. To generate mck1 or mre11 deletion in the W303 background, genes disrupted by a KanMX cassette in BY4741 haploid deletion libraries (EuroScarf) were amplified by PCR. The PCR product containg the KanMX cassette with MCK1 flanking region was transformed into the wild type W303 strain. The resulting mck1 deletion cells in W303 were confirmed by PCR. The MCK1-MYC strain was generated by PCR genomic integration of a PCR product containing a MYC tag and a TRP gene [46]. GAL-CDC6ΔNT-HA strain and plasmid were kindly provided by Dr. Stephen Bell. The ORC6-rxl, ORC6-ps, ORC2-ps and MCM7-NLS mutations were described previously [11], [13], [34]. CDC6-proteinA strain was generated as previously described [47]. Rad53-FLAG strain was obtained from Dr. Petrini [48]. To generate GALL-MCK1, MCK1 gene was cloned into GALL-pRS405 plasmid at BamHI and SpeI sites using MCK1 plasmid provided by Dr. P. Hieter [28]. The resulting GALL-MCK1/pRS405 plasmid was cut with BstEII, and the linearized plasmid was transformed into bar1 mck1::KanMX CDC6-prA::HIS3 strain to integrate GALL-MCK1 at LEU locus. Cdc6-GFP strain was made by direct transformation of a GFP cassette [49] in BY4741 and subsequently back crossed to W303-1B three times for Figure 4C. CDC6 plasmid was generated by PCR method using W303 wild type genomic DNA. The resulting PCR product was cloned into pYES2.1 Topo TA plasmid (Invitrogen) for Figure 7B. The CDC6/pYES2.1 plasmid was subjected to site-directed mutagenesis using QuickChange Site-directed mutagenesis kit (Agilent Technologies, CA) to introduce T368A S372A mutation for Figure 7B. CDC6/pRS406 plasmid was generated by PCR cloning. CDC6 gene including the endogeneous promoter (300 bp upstream from the start codon) was amplified by PCR using primers that contain BamHI and XhoI, and cloned into pRS406 at BamH1and XhoI sites. The CDC6/pRS406 plasmid was used as a temperate to generate CDC6-T368A/pRS406. Site-directed mutagenesis was performed as described above. The resulting plasmids were cut with NcoI to integrate the mutated CDC6 at URA3 locus in mck1 GALL-MCK1 CDC6-proteinA strain for Figure 7E.
Making a temperature-sensitive mutant of mck1
A temperature sensitive mutant of MCK1 was generated using a previously described method [34]. MCK1 gene was cloned into pRS414 at BamHI and SpeI sites from MCK1 plasmid provided by Dr. P. Hieter [28]. The MCK1/pRS414 plasmid was mutagenized by PCR mutagenesis to introduce random mutations in the MCK1 gene as previously described [50]. The mutagenized mck1/pRS414 plasmid was transformed into mck1 orc6-rxl strain containing MCK1/pRS416 plasmid. The mck1 orc6-rxl cells containing mutagenized mck1 plasmid were tested for its viability at 37 degrees. The mutagenized mck1/pRS414 plasmid (mck1-16 mutation) was isolated from the strain and was inserted into the pRS406 plasmid at BamHI SpeI sites. The resulting mck1-16/pRS406 plasmid was cut with BstEII restriction enzyme and was integrated at the URA3 genome locus. Sequence analysis identified two mutations in the temperature sensitive mck1-16 allele, resulting in P275L and E357G.
Co-IP and Western blotting
A 50-ml culture of each strain was grown to log-phase an OD595 of 0.5 was reached. The cell pellets were washed in cold TE buffer, and resuspended with 400 µl of protein extraction buffer [20 mM HEPES, pH 7.4, 110 mM potassium acetate, 2 mM MgCl2, 0.1% Tween 20, 1 mM DTT, 2 µg/ml DNaseI, protease inhibitor cocktail (Sigma-Aldrich, MO) and phosphatase inhibitor (Sigma-Aldrich, MO)]. Acid-washed glass beads (0.15 g) were added, and cells were disrupted by FastPrep (MP Biomedicals, OH) for 20 seconds, twice, at speed 6. Samples were centrifuged and 10 µl of supernatants were kept for Western blotting as “INPUT”. The remaining protein extracts were subjected to co-immunoprecipitation (Co-IP). Agarose beads conjugated with anti-MYC antibody (A7470) (Sigma-Aldrich, MO) were pre-incubated with 5% BSA in protein extraction buffer for 1 hour at 4 degrees to reduce non-specific binding first. Then the beads were mixed with the protein extract supernatants and rotated for 2 hours at 4 degrees. Beads were washed with protein extraction buffer five times. After the final wash, 30 µl of 2× sample buffer was added to the beads, and the protein was denatured at 95 degrees for 5 minutes. Proteins were separated by SDS-PAGE with Novex 4–20% Tris-Glycine polyacrylamide gel (Invitrogen, CA) except Figure 7C with 7% acrylamide large gel. The proteins on the gels were transferred to PVDF membrane (Millipore, MA). Western blot analysis was performed using anti-MYC antibody 9E10 (M4439) (Sigma-Aldrich, MO) at 1∶4000 dilution, anti-HA antibody 3F10 (Roche, IN) at 1∶4000 dilution and anti-FLAG antibody (A8592) (Sigma-Aldrich, MO) at 1∶4000 dilution. Cdc6-proteinA was visualized using anti-peroxidase soluble complex antibody produced in rabbit (P1291) (Sigma-Aldrich, MO) at 1∶4000 dilution. Cdc6 was detected using anti-Cdc6 antibody (9H8/5) (Abcam, MA) at 1∶500 dilution.
Fluorescence microscopy
Log phase cultures of Cdc6-GFP expressing cells in SC medium supplemented with 20 mg/L adenine were imaged live with an Eclipse E600 fluorescence microscope (Nikon) equipped with a DC350F CCD camera (Andor) and 100×, NA 1.45, or 60×, NA 1.4, oil objectives. The images were captured with NIS-Elements software (Nikon) and prepared using Photoshop software.
Flow cytometry
DNA content analysis by FACScanto (BD Biosciences, NJ) was performed as described previously [51].
Yeast two-hybrid assay
The pBTM116 constructs containing various Cdc6 mutants were obtained from Dr. J. Diffley's lab [6]. Full length MCK1 was cloned into pACT at BamHI and XhoI sites by PCR method. The MCK1/pACT and each of the various CDC6/pBMT116 plasmids were co-transformed into L40 strain and plated on SD-Leu/Trp plates [52]. The colonies were transferred to nitrocellulose membrane and kept at −80 degrees overnight. The membrane was placed on whatman paper soaked with 3 ml of Z buffer, [60 mM Na2HPO4, 40 mM NaH2PO4, 10 mM KCl, 1 mM MgSO4] with 300 µg/ml X-gal and 0.044 M 2-mercaptoethanol. The membrane was incubated at 30 degree overnight to visualize the blue colonies.
Supporting Information
{kind=link}
{kind=link}
Zdroje
1. BellSP, DuttaA (2002) DNA replication in eukaryotic cells. Annu Rev Biochem 71 : 333–374.
2. NasmythK (1993) Control of the yeast cell cycle by the Cdc28 protein kinase. Curr Opin Cell Biol 5 : 166–179.
3. SchwobE, NasmythK (1993) CLB5 and CLB6, a new pair of B cyclins involved in DNA replication in Saccharomyces cerevisiae. Genes Dev 7 : 1160–1175.
4. TanakaS, UmemoriT, HiraiK, MuramatsuS, KamimuraY, et al. (2007) CDK-dependent phosphorylation of Sld2 and Sld3 initiates DNA replication in budding yeast. Nature 445 : 328–332.
5. ZegermanP, DiffleyJF (2007) Phosphorylation of Sld2 and Sld3 by cyclin-dependent kinases promotes DNA replication in budding yeast. Nature 445 : 281–285.
6. PerkinsG, DruryLS, DiffleyJF (2001) Separate SCF(CDC4) recognition elements target Cdc6 for proteolysis in S phase and mitosis. Embo J 20 : 4836–4845.
7. DruryLS, PerkinsG, DiffleyJF (1997) The Cdc4/34/53 pathway targets Cdc6p for proteolysis in budding yeast. Embo J 16 : 5966–5976.
8. ElsasserS, ChiY, YangP, CampbellJL (1999) Phosphorylation controls timing of Cdc6p destruction: A biochemical analysis. Mol Biol Cell 10 : 3263–3277.
9. LabibK, DiffleyJF, KearseySE (1999) G1-phase and B-type cyclins exclude the DNA-replication factor Mcm4 from the nucleus. Nat Cell Biol 1 : 415–422.
10. NguyenVQ, CoC, IrieK, LiJJ (2000) Clb/Cdc28 kinases promote nuclear export of the replication initiator proteins Mcm2–7. Curr Biol 10 : 195–205.
11. NguyenVQ, CoC, LiJJ (2001) Cyclin-dependent kinases prevent DNA re-replication through multiple mechanisms. Nature 411 : 1068–1073.
12. VasA, MokW, LeatherwoodJ (2001) Control of DNA rereplication via Cdc2 phosphorylation sites in the origin recognition complex. Mol Cell Biol 21 : 5767–5777.
13. WilmesGM, ArchambaultV, AustinRJ, JacobsonMD, BellSP, et al. (2004) Interaction of the S-phase cyclin Clb5 with an “RXL” docking sequence in the initiator protein Orc6 provides an origin-localized replication control switch. Genes Dev 18 : 981–991.
14. CrossFR, JacobsonMD (2000) Conservation and function of a potential substrate-binding domain in the yeast Clb5 B-type cyclin. Mol Cell Biol 20 : 4782–4790.
15. SchulmanBA, LindstromDL, HarlowE (1998) Substrate recruitment to cyclin-dependent kinase 2 by a multipurpose docking site on cyclin A. Proc Natl Acad Sci U S A 95 : 10453–10458.
16. WohlschlegelJA, DwyerBT, TakedaDY, DuttaA (2001) Mutational analysis of the Cy motif from p21 reveals sequence degeneracy and specificity for different cyclin-dependent kinases. Mol Cell Biol 21 : 4868–4874.
17. ChenS, BellSP CDK prevents Mcm2-7 helicase loading by inhibiting Cdt1 interaction with Orc6. Genes Dev 25 : 363–372.
18. TannyRE, MacalpineDM, BlitzblauHG, BellSP (2006) Genomewide Analysis of Rereplication Reveals Inhibitory Controls That Target Multiple Stages of Replication Initiation. Mol Biol Cell
19. ArchambaultV, IkuiAE, DrapkinBJ, CrossFR (2005) Disruption of mechanisms that prevent rereplication triggers a DNA damage response. Mol Cell Biol 25 : 6707–6721.
20. TongAH, EvangelistaM, ParsonsAB, XuH, BaderGD, et al. (2001) Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science 294 : 2364–2368.
21. PuzissJW, HardyTA, JohnsonRB, RoachPJ, HieterP (1994) MDS1, a dosage suppressor of an mck1 mutant, encodes a putative yeast homolog of glycogen synthase kinase 3. Mol Cell Biol 14 : 831–839.
22. PlyteSE, HughesK, NikolakakiE, PulvererBJ, WoodgettJR (1992) Glycogen synthase kinase-3: functions in oncogenesis and development. Biochim Biophys Acta 1114 : 147–162.
23. AliA, HoeflichKP, WoodgettJR (2001) Glycogen synthase kinase-3: properties, functions, and regulation. Chem Rev 101 : 2527–2540.
24. DiehlJA, ChengM, RousselMF, SherrCJ (1998) Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev 12 : 3499–3511.
25. HiliotiZ, GallagherDA, Low-NamST, RamaswamyP, GajerP, et al. (2004) GSK-3 kinases enhance calcineurin signaling by phosphorylation of RCNs. Genes Dev 18 : 35–47.
26. MizunumaM, HirataD, MiyaokaR, MiyakawaT (2001) GSK-3 kinase Mck1 and calcineurin coordinately mediate Hsl1 down-regulation by Ca2+ in budding yeast. Embo J 20 : 1074–1085.
27. HirataY, AndohT, AsaharaT, KikuchiA (2003) Yeast glycogen synthase kinase-3 activates Msn2p-dependent transcription of stress responsive genes. Mol Biol Cell 14 : 302–312.
28. SheroJH, HieterP (1991) A suppressor of a centromere DNA mutation encodes a putative protein kinase (MCK1). Genes Dev 5 : 549–560.
29. NeigebornL, MitchellAP (1991) The yeast MCK1 gene encodes a protein kinase homolog that activates early meiotic gene expression. Genes Dev 5 : 533–548.
30. ZwerschkeW, RottjakobHW, KuntzelH (1994) The Saccharomyces cerevisiae CDC6 gene is transcribed at late mitosis and encodes a ATP/GTPase controlling S phase initiation. J Biol Chem 269 : 23351–23356.
31. SanchezM, CalzadaA, BuenoA (1999) The Cdc6 protein is ubiquitinated in vivo for proteolysis in Saccharomyces cerevisiae. J Biol Chem 274 : 9092–9097.
32. DruryLS, PerkinsG, DiffleyJF (2000) The cyclin-dependent kinase Cdc28p regulates distinct modes of Cdc6p proteolysis during the budding yeast cell cycle. Curr Biol 10 : 231–240.
33. HoneyS, FutcherB (2007) Roles of the CDK phosphorylation sites of yeast Cdc6 in chromatin binding and rereplication. Mol Biol Cell 18 : 1324–1336.
34. IkuiAE, ArchambaultV, DrapkinBJ, CampbellV, CrossFR (2007) Cyclin and cyclin-dependent kinase substrate requirements for preventing rereplication reveal the need for concomitant activation and inhibition. Genetics 175 : 1011–1022.
35. LuoKQ, ElsasserS, ChangDC, CampbellJL (2003) Regulation of the localization and stability of Cdc6 in living yeast cells. Biochem Biophys Res Commun 306 : 851–859.
36. FiolCJ, MahrenholzAM, WangY, RoeskeRW, RoachPJ (1987) Formation of protein kinase recognition sites by covalent modification of the substrate. Molecular mechanism for the synergistic action of casein kinase II and glycogen synthase kinase 3. J Biol Chem 262 : 14042–14048.
37. KoivomagiM, ValkE, VentaR, IofikA, LepikuM, et al. Dynamics of Cdk1 substrate specificity during the cell cycle. Mol Cell 42 : 610–623.
38. FrameS, CohenP, BiondiRM (2001) A common phosphate binding site explains the unique substrate specificity of GSK3 and its inactivation by phosphorylation. Mol Cell 7 : 1321–1327.
39. KishiT, IkedaA, NagaoR, KoyamaN (2007) The SCFCdc4 ubiquitin ligase regulates calcineurin signaling through degradation of phosphorylated Rcn1, an inhibitor of calcineurin. Proc Natl Acad Sci U S A 104 : 17418–17423.
40. EmbiN, RylattDB, CohenP (1980) Glycogen synthase kinase-3 from rabbit skeletal muscle. Separation from cyclic-AMP-dependent protein kinase and phosphorylase kinase. Eur J Biochem 107 : 519–527.
41. WuD, PanW GSK3: a multifaceted kinase in Wnt signaling. Trends Biochem Sci 35 : 161–168.
42. WelshGI, MillerCM, LoughlinAJ, PriceNT, ProudCG (1998) Regulation of eukaryotic initiation factor eIF2B: glycogen synthase kinase-3 phosphorylates a conserved serine which undergoes dephosphorylation in response to insulin. FEBS Lett 421 : 125–130.
43. WelshGI, ProudCG (1993) Glycogen synthase kinase-3 is rapidly inactivated in response to insulin and phosphorylates eukaryotic initiation factor eIF-2B. Biochem J 294 (Pt 3) 625–629.
44. WinzelerEA, ShoemakerDD, AstromoffA, LiangH, AndersonK, et al. (1999) Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science 285 : 901–906.
45. GietzD, St JeanA, WoodsRA, SchiestlRH (1992) Improved method for high efficiency transformation of intact yeast cells. Nucleic Acids Res 20 : 1425.
46. KnopM, SiegersK, PereiraG, ZachariaeW, WinsorB, et al. (1999) Epitope tagging of yeast genes using a PCR-based strategy: more tags and improved practical routines. Yeast 15 : 963–972.
47. ArchambaultV, LiCX, TackettAJ, WaschR, ChaitBT, et al. (2003) Genetic and biochemical evaluation of the importance of Cdc6 in regulating mitotic exit. Mol Biol Cell 14 : 4592–4604.
48. UsuiT, PetriniJH (2007) The Saccharomyces cerevisiae 14-3-3 proteins Bmh1 and Bmh2 directly influence the DNA damage-dependent functions of Rad53. Proc Natl Acad Sci U S A 104 : 2797–2802.
49. LongtineMS, McKenzieA3rd, DemariniDJ, ShahNG, WachA, et al. (1998) Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14 : 953–961.
50. IkuiAE, CrossFR (2009) Specific genetic interactions between spindle assembly checkpoint proteins and B-Type cyclins in Saccharomyces cerevisiae. Genetics 183 : 51–61.
51. EpsteinCB, CrossFR (1992) CLB5: a novel B cyclin from budding yeast with a role in S phase. Genes Dev 6 : 1695–1706.
52. HollenbergSM, SternglanzR, ChengPF, WeintraubH (1995) Identification of a new family of tissue-specific basic helix-loop-helix proteins with a two-hybrid system. Mol Cell Biol 15 : 3813–3822.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2012 Číslo 12
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Population Genomics of Sub-Saharan : African Diversity and Non-African Admixture
- Dnmt3a Protects Active Chromosome Domains against Cancer-Associated Hypomethylation
- Excessive Astrocyte-Derived Neurotrophin-3 Contributes to the Abnormal Neuronal Dendritic Development in a Mouse Model of Fragile X Syndrome
- Pre-Disposition and Epigenetics Govern Variation in Bacterial Survival upon Stress
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy