
Multicentrická Castlemanova choroba. Příznaky, diagnostika a léčba
Multicentric Castleman’s disease. Symptoms, diagnostics and therapy
Castleman disease (CD) describes a group of heterogeneous hematologic disorders with characteristic histopathological features. CD can present with unicentric (UCD) or multicentric (MCD) regions of lymph node enlargement. Some cases of MCD are caused by human herpesvirus-8 (HHV-8), whereas others are HHV-8–negative/idiopathic (iMCD). Treatment of iMCD is challenging, and outcomes can be poor. In this paper, we briefly report about symptoms of iMCD and about the International, evidence‑based consensus diagnostic criteria for HHV-8-negative/idiopathic multicentric Castleman disease and International evidence based consensus treatment guidelines for idiopathic multicentric Castleman disease.
Keywords:
Castleman disease – siltuximab
Autoři:
Zdeněk Adam 1; Zdeněk Řehák 2; Zuzana Adamová 3; Renata Koukalová 2; Luděk Pour 1; Marta Krejčí 1; Ivanna Boichuk 1; Viera Sandecká 1; Martin Krejčí 1; Martin Štork 1; Sabina Ševčíková 4; Zdeněk Král 1
Působiště autorů:
Interní hematologická a onkologická klinika LF MU a FN Brno
1; Oddělení nukleární medicíny Masarykův onkologický ústav Brno
2; Chirurgické oddělení nemocnice Frýdek-Místek
3; Ústav patologické fyziologie LF MU, Brno
4
Vyšlo v časopise:
Vnitř Lék 2022; 68(1): 41-53
Kategorie:
Přehledové články
Souhrn
Castlemanova choroba (CD) představuje heterogenní skupinu poruch krvetvorby s charakteristickými histopatologickými znaky. CD může tvořit unicentrickou (UCD) anebo multicentrickou formu (MCD). Některé případy MCD jsou způsobeny infekcí herpesvirem-8 (HHV-8), zatímco jiné jsou HHV-8 negativní (idiopatická multicentrická Castlemanova choroba – iMCD). Léčba iMCD není jednoduchá a výsledky mohou být neuspokojivé. V tomto článku stručně informujeme o symptomech iMCD a mezinárodních diagnostických kritériích pro HHV-8 negativní iMCD a o mezinárodním doporučení pro léčbu iMCD.
Klíčová slova:
Castlemanova choroba – siltuximab
Úvod
Multicentrická forma Castlemanovy nemoci (multicentric Castleman disease) patří mezi velmi vzácné choroby. Epidemiologické údaje z ČR nemáme k dispozici. Incidence unicentrické formy Castlemanovy nemoci (UCD) v USA je 16–19 případů na 1 milion obyvatel. Incidence multicentrické formy Castlemanovy choroby je v USA 5 případů na 1 milion obyvatel (1). Pokud by tomu bylo podobně i v ČR, tak by u nás ročně mělo být diagnostikováno 160–190 případů unicentrické formy a 50 případů multicentrické formy této nemoci. Vývoj poznání a postupné definování jednotlivých forem a jejich názvů jsme uvedli v článku Lokalizovaná (unicentrická) forma Castlemanovy nemoci. Klinické projevy, diagnostika a léčba dle mezinárodních doporučení z roku 2020 (2), v němž jsou základní fakta o vývoji poznání celé skupiny nemocí, které patří pod zastřešující název „Castlemanova choroba“. V tomto článku se budeme věnovat příznakům multicentrické formy Castlemanovy choroby (multicentric Castleman disease – MCD), diagnostice a léčbě a uvedeme nejdůležitější fakta z posledních mezinárodních doporučení pro tuto chorobu. Pro často se vyskytující choroby máme k dispozici národní doporučené postupy pro diagnostiku a léčbu, vypracované jednotlivými odbornými společnostmi. Pro velmi vzácné choroby však národní doporučení nejsou připravována, a tak v praxi je nutno přihlížet k mezinárodním doporučením.
Etiologie nemoci a role viru Kaposiho sarkomu (HHV-8)
Multicentrická forma Castlemanovy nemoci je termín pro heterogenní skupinu patologických stavů, spojených s proliferací morfologicky benigních lymfocytů. Příčinou jsou výrazně zvýšené hladiny proinflamatorních cytokinů, dominantně interleukinu-6 (Il-6), s jehož koncentrací koreluje agresivita nemoci. Il-6 je multifunkční cytokin, který indukuje plazmocytózu v kostní dřeni (3), hypergamaglobulinemii, zvýšenou tvorbu vaskulárního endoteliálního růstového faktoru (VEGF), trombocytózu, tvorbu proteinů akutního zánětu v játrech (tedy i CRP), aktivaci makrofágů a T‑buněk. Il-6 je zodpovědný i za autoimunitní projevy, které Castlemanovu nemoc občas provázejí.
Na rozdíl od plazmocelulární varianty Castlemanovy nemoci se v případě pacientů s POEMS syndromem (což je akronym pro Polyneuropathy, Organomegaly, Endokcinopathy, Monoclonal gammopathy, Skin changes) předpokládá zvýšení VEGF (vascular endothelial growth factor) nezávislé na Il-6 (3).
Určitou roli v patofyziologii hraje také interleukin-1 (Il-1), neboť byli popsáni pacienti, kteří nereagovali na léčbu protilátkami proti Il-6, ale zareagovali na protilátky proti interleukinu-1 (3), a dále byli popsáni nemocní, kteří při podávání inhibitoru receptoru pro interleukin - 1 (anakinra) dosáhli remise (3). Il-6 je tedy zřejmě hlavním vyvolávajícím faktorem, ale ne vždy a ne jediným!
Zatím je pouze známo, že humánní herpesvirus 8 (HHV-8) indukuje tvorbu virového homologu Il-6, a tím zvýšení všech prozánětlivých cytokinů. Nejasnou otázkou zůstává, co je příčinou zvýšených hladin prozánětlivých cytokinů u HHV-8 negativních osob.
Z pohledu vyvolávajícího činitele lze rozdělit multicentrickou formu Castlemanovy nemoci na nemoc způsobenou infekcí HHV-8 a na idiopatickou formu s neznámou vyvolávající příčinou (3). V roce 2021 se člení multicentrická forma této nemoci dle výsledků virologických vyšetření na:
idiopatickou multicentrickou Castlemanovu chorobu bez pozitivity HHV-8 a bez průkazu viru HIV,
multicentrickou Castlemanovu chorobu s průkazem viru HHV-8 obvykle s pozitivitou HIV. Přítomnost viru HIV byla prokázána jen u menšiny pacientů (u 15 %), přítomnost viru HHV-8 byla prokázána jen u 17 % nemocných (4).
Ačkoliv je vyšetření HHV-8 součástí diagnostiky Castlemanovy choroby a je zakotveno i v indikaci pro siltuximab v dokumentech SÚKL, tak v současné době neexistují obecná doporučení pro výběr diagnostické metody, biologického materiálu ani standardizované laboratorní postupy. Monitorování HHV-8 infekce je možné pomocí polymerázové řetězové reakce (PCR) v periferní krvi, ale pro stanovení diagnózy HHV-8 asociované Castlemanovy choroby je třeba průkaz latentního nukleárního antigenu LANA-1 pomocí imunohistochemického barvení.
Příznaky idiopatické multicentrické Castlemanovy nemoci
Choroba má jako všechny nemoci individuální průběh, od chronicky probíhajících jen mírně agresivních až po značně agresivní formy. Pro označení mírné formy se používá termín „flu‑like“. Pro značně agresivní případy, odpovídající cytokinové bouři, pak termín „sepsis‑like“. Těchto agresivních průběhů je však méně než průběhů neagresivních. Velmi agresivní případy jsou spojené s anasarkou s multiorgánovým selháním a vedou často ke smrti. Klinické příznaky lze edukačně rozdělit na příznaky pravidelně se vyskytující a příznaky vyskytující se nepravidelně. A tyto nepravidelně se vyskytující pak na příznaky imunitně mediované a příznaky nejasné patogeneze.
Pravidelně se vyskytující příznaky a laboratorní nálezy
Nadprodukce interleukinu-6 ve svém důsledku zvýší produkci hepcidinu, který blokuje jak vstřebávání železa z trávicího traktu, tak jeho uvolňování z depotních forem v makrofázích. Proto nemoc provází pravidelně anémie chronických chorob. Interleukin-6 dále snižuje tvorbu albuminu v játrech a způsobuje tak hypoalbuminemii. Zvýšená tvorba interleukinu-6 zvyšuje tvorbu vaskulárního endoteliálního růstového faktoru (VEGF), což stimuluje angioneogenezi a zvýšenou vaskulární permeabilitu. K pravidelně vyskytujícím se příznakům patří systémová zánětlivá reakci organismu, která se projeví:
neinfekčními subfebriliemi či febriliemi,
úbytkem hmotnosti,
nočním pocením,
patologickou únavou.
Tyto příznaky provázejí následujícími laboratorní nálezy:
zvýšené hodnoty zánětlivých markerů: CRP, sedimentace erytrocytů, ferritin,
vzestup počtu trombocytů, které reagují také jako reaktant akutní fáze a vzestup koncentrace fibrinogenu,
anémie s rysy anémie chronických chorob,
klesající hodnoty albuminu,
zvýšená hodnota celkové bílkoviny,
hraniční zmnožení plazmocytů v kostní dřeni občas, které jsou polyklonální,
zvýšená koncentrace polyklonálních imunoglobulinů, hlavně imunoglobulinů třídy IgG.
Tyto příznaky jsou způsobeny dominantně interleukinem-6 (IL-6) a VEGF, byť tyto interleukiny nejsou běžně vyšetřitelné.
Nepravidelně se vyskytující příznaky imunitní etiologie
Multicentrická Castlemanova choroba je často asociována s různými autoimunitními poruchami, nejčastější jsou:
autoimunní anémie či trombocytopenie, méně často je cytopenie způsobena hemofagocytózou,
vaskulitidy, které mohou být příčinou cévní mozkové příhody,
paraneoplastický pemphigus,
Sjögrenův syndrom,
myasthenia gravis,
jiné formy autoimunitního poškození organismu.
Četné imunitně mediované projevy MCD jsou formou popisů případů dokumentovány i v české a slovenské odborné literatuře, které citujeme. Z popsaných imunitně mediovaných případů je zřejmé, že v této oblasti může mít nemoc opravdu velmi různorodé příznaky (5, 6).
V zahraniční literatuře je několik analýz četnosti autoimunitních projevů. Poslední analýza z roku 2021 obsahuje soubor 40 pacientů, z nichž 9 (22,5 %) mělo prokázány autoimunitní projevy. Jednalo se o paraneoplastický pemphigus u 4 pacientů s UCD a u 1 pacienta s iMCD, dále o 1 případ Sjögrenova syndromu, 1 případ autoimunitní hemolytické anémie, 1 případ myasthenia gravis a 1 případ psoriasis. Autoimunitní poškození u některých nemocných předcházelo stanovení diagnózy Castlemanovy choroby (CD), někdy probíhalo v době stanovení diagnózy a někdy byla autoimunita diagnostikována po ukončení léčby CD. Pacienti s CD a autoimunitou měli častěji než pacienti s CD bez autoimunity kožní a/nebo mukózní komplikace a také plicní komplikace. Pacienti s CD a autoimunitou vykazovali deregulaci imunitní odpovědi s nižšími počty CD3+ T buněk a zvýšeným počtem NK buněk (7). Proto je u každé nové diagnózy CD nutno provést základní vyšetření na autoimunitní choroby a počítat i s jejich pozdějším výskytem při sledování.
Nepravidelně se vyskytující příznaky nejasné etiologie
Do této kategorie lze zařadit následující komplikace:
poškození ledvin: endoteliózou v 60 %, AA‑amyloidózou ve 20 %; mírné poškození funkce ledvin je popisováno relativně častěji, zatímco poškození funkce ledvin s retencí dusíkatých látek, vedoucí až k úplné anurii, je u Castlemanovy nemoci výjimečné (8, 9),
souběh s POEMS syndromem, (periferní neuropatie, osteosklerotické změny skeletu); POEMS syndrom zmíníme podrobněji v dalším odstavci,
nekardiální dušnost a retence tekutin – ascites, plicní výpotek, plicní abnormality typu infiltrátů, restriktivní plicní poruchy, intersticiální pneumonitidy či dokonce bronchiolitis obliterans neboli rysy TAFRO syndromu, který je zmíněn dále,
kožní změny včetně hyperpigmentace, kožní raš, tvorba kožních hemangiomů,
zcela výjimečně průjmy a hmotnostní úbytek,
dysfunkce jater,
osteosklerotické změny na skeletu.
Podrobně o těchto typech poškození se lze dočíst v odpovídajících popisech případů a přehledových článcích (9, 10, 11)
Příznaky odpovídající POEMS syndromu
POEMS syndrom je akronym pro polyneuropatii, organomegalii, endokrinopatii, monoklonální gamapatii a kožní změny (skin changes) (12, 13).
Tato jednotka může existovat samostatně, ale může se také prolínat s Castlemanovou chorobou. Při POEMS syndromu bývá přítomen edém papily, nevýrazná neuropatie a při zobrazení skeletu jsou zřetelné osteosklerotické změny. Protože příznaky POEMS syndromu a Castlemanovy choroby se mohou překrývat, může být problém, jak nemoc nazvat.
Dle autorů z Mayo Clinic by se termín POEMS syndrom měl používat pouze pro nemocné s periferní neuropatií a dalšími znaky POEMS syndromu, kteří mají prokázanou klonální expanzi plazmocytů v kostní dřeni. Pro nemocné s Castlemanovou chorobou, kteří sice mají některé známky POEMS syndromu, ale nemají ani neuropatii a ani klonální plazmocyty v kostní dřeni, doporučují používat termín „variantní forma Castlemanovy choroby se znaky POEMS syndromu“.
V souboru 113 pacientů s multicentrickou formou Castlemanovy choroby byly u 34 % zjištěny příznaky POEMS syndromu (9).
K projevům POEMS syndromu u MCD patří tvorba kožních hemangiomů, které lze považovat za jeden ze znaků (12–16). Výskyt jednotlivých symptomů dokumentuje tabulka 1.

Stanovení diagnózy dle mezinárodních kritérií Castlemanovy nemoci
K diagnóze se obvykle dojde v rámci vyšetřování febrilií či subfebrilií nejasného původu (fever of uknown origin – FUO). Doporučení pro diagnostiku FUO dnes obsahují v rámci širšího pátrání i provedení FDG‑PET/ T vyšetření. Toto vyšetření zobrazí nejen velikost lymfatických uzlin, ale stanoví také míru akumulace FDG v lymfatických uzlinách. A pro histologickou diagnostiku se doporučuje odběr právě uzlin s nejintenzivnější akumulací FDG. FDG‑PET/ CT může detekovat mimo lymfadenopatie také hepatomegalii a splenomegalii a osteosklerotická kostní ložiska či plicní infiltráty, které u této nemoci také mohou být (17–19). Pro stanovení diagnózy jsou přínosná vyšetření uvedená v tabulce 2. Může být prokázána i retence tekutin. K diferenciální diagnóze, zda je retence kardiálního či nekardiálního původu, může pomoci vyšetření mozkového natriuretického faktoru (NTProBNP či BNP), který je zvýšený u kardiální etiologie a ECHO srdce.
Zásadní význam má histomorfologické stanovení diagnózy. Morfologové stále rozlišují jen dva základní typy a další dva typy jako odvozené varianty. V souboru asi 198 HIV negativních případů multicentrické formy Castlemanovy nemoci byly stanoveny tyto histologické typy:
hyalinně‑vaskulární typ 17–49 %,
plazmocelulární typ 46–77 %,
smíšený typ 4–20 %,
vzácný plazmablastický typ, který se ale vyskytuje jedině při HHV-8 infekci.
Zde je možná transformace v plazmablastické lymfomy.
V přehledu se morfologii věnuje Fajgenbaum a další (20, 21).
Bioptická diagnostika Castlemanovy choroby je obtížná. Zejména u plazmocelulární varianty jsou morfologické znaky značně nespecifické. Existuje celá řada nádorových, autoimunitních a infekčních onemocnění, která mohou imitovat obraz Castlemanovy choroby. Na druhou stranu jsou málokdy všechny morfologické znaky plně vyjádřené a odlišení od reaktivně změněné lymfatické uzliny může být problematické. Histopatologickou diagnostiku ztěžuje fakt, že není definován žádný diagnostický marker této nemoci, a proto je nutná diskuze mezi patologem a klinikem. Japonští autoři udávají ve svém souboru interval od prvních symptomů nemoci do stanovení diagnózy 27,5 měsíce, což jen dokresluje problémy při morfologickém stanovení diagnózy. Doporučený rozsah vyšetření dle mezinárodního doporučení uvádí tabulka 2.

V roce 2017 byla definována a publikována diagnostická kritéria této nemoci uvedená v tabulce 3 (20). Takže diagnózu stanovuje společně patolog a klinický lékař konfrontací morfologických, laboratorních a klinických nálezů s požadovanými kritérii.

V roce 2013 vyšel popis nové varianty Castlemanovy nemoci, kterou japonští autoři nazvali TAFRO syndrom. TAFRO syndrom je akronym pro trombocytopenii, ascites (anasarku), fibrotické změny v kostní dřeni s mikrocytární anémií, renální insuficienci, organomegalii a histologický nález Castlemanovy nemoci – obvykle smíšeného nebo hyalinně vaskulárního typu. TAFRO varianta Castlemanovy nemoci nebývá provázena polyklonálním zmnožením imunoglobulinů a v kostní dřeni nebývá proliferace polyklonálních plazmocytů, jak bývá u klasické plazmocelulární formy Castlemanovy nemoci, ale spíše megakaryocytární hyperplazie (22–24). Po prvních popisech TAFRO syndromu se věřilo, že tato choroba je vázaná jen na japonskou rasu. V roce 2015 se objevil první popis této nemoci u rozené obyvatelky Itálie (25) a následně i další zprávy o rozšíření v evropské populaci. V případě TAFRO syndromu je zřetelné výrazná vaskularizace lymfatických uzlin a bylo popsáno odlišné cytokinové spektrum se zvýšeným vaskulárním endoteliálním faktorem (VEGF), ale nižší elevací IL-6 a byla navržena stratifikace dle závažnosti a léčba (26). Kritéria TAFRO uvádí tabulka 4.

Průběh idiopatické multicentrické formy Castlemanovy nemoci
Idiopatická multicentrická forma Castlemanovy nemoci se typicky objevuje ve 4. a v 5. dekádě života, častěji u mužů než u žen. Z popisu případů je však zřetelné, že u většiny nemocných má choroba dlouholetý průběh, pacientům sice nebere rychle život, ale výrazně zhoršuje jeho kvalitu. Projevy případné vaskulitidy mohou trvale invalidizovat (cévní mozkové příhody) a komplikují další léčbu i ošetřování. Vzhledem k narušení imunity se zvyšuje u těchto nemocných výskyt dalších maligních chorob ve srovnání s průměrnou populací. V podstatě se popisují následné 4 formy klinických průběhů:
opakované relapsy a remise (flu‑like),
stabilní perzistující choroba,
progredující fatální choroba (sepsis‑like),
transformace v maligní lymfom.
Mezinárodní kritéria pro léčbu rozdělují léčebné postupy při „flu‑like“ průběhu a „sepsis ‑ like“ průběhu a definují stavy „sepsis ‑ like“ (Tab. 5). Mezi oběma formami existuje kontinuální vývoj. A tak se inovace léčebných doporučení možná bude v dalších letech odvíjet od prognostického indexu, který byl publikován v roce 2020 a je uveden v tabulce 6 (27, 28). Pro účely klinických studií jsou definovány i léčebné odpovědi (20).

Přehled léků používaných pro léčbu multicentrické formy Castlemanovy nemoci
Glukokortikoidy
Glukokortikoidy jsou široce účinkující imunosupresivní léky, které tlumí jak akutní, tak i chronickou zánětlivou reakci. Snižují tvorbu proinflamatorních cytokinů a chemokinů a klíčových enzymů zánětlivé reakce, tedy Il-2, Il-6, TNF alfa. Pro odstranění příznaků nemoci je však třeba vysokých dávek glukokortikoidů, přičemž tyto vysoké dávky nelze podávat dlouhodobě. Příznaky nemoci se rychle obnovují po ukončení kortikoterapie či při snížení jejich dávky. Proto jsou dnes používány jako součást kombinační léčby (29–31).
Klasická chemoterapie
V letech před příchodem biologických léků do klinické praxe (antiCD20 protilátky a thalidomidu a lenalidomidu a siltuximabu) byly testovány klasické chemoterapeutické kombinace používané pro léčbu maligních lymfomů, tedy kombinace CHOP (cyklofosfamid, vinkristin, adriamycin a prednison), CVAD (cyklofosfamid, vinkristin, adriamycin a dexametazon), případně režimy s etoposidem, monoterapie etoposidem podaným denně či intermitentně, aplikace kladribinu, chlorambucil v monoterapii a podobné režimy v té době považované za moderní léčbu maligních lymfomů (29–31).
V době, kdy byly tyto klasické režimy používány bez přidání nových léků, byla diagnostika této nemoci méně přesná než dnes a byly publikovány jen zkušenosti v malých skupinách pacientů. V roce 1998 Herrada popsal 27 % dlouhodobých remisí po klasické chemoterapii (32). Chronowsky (2001) popsal progression free interval 23–119 měsíců u 4 z 9 pacientů léčených klasickou chemoterapií (33).
Zhu (2013) popsal 10 pacientů léčených kombinací CHOP anebo COP. U jednoho pacienta bylo dosaženo kompletní remise, u 6 parciální remise, při mediánu sledování 34 měsíce zůstávali všichni pacienti naživu (34).
Liu v retrospektivním přehledu z roku 2016 uvádí, že cytostatická terapie navodila kompletní remisi u 19 ze 43 pacientů s mediánem intervalu do selhání léčby (time to treatment failure) 6 měsíců (35, 36).
Léčba založená na kombinaci cytostatik v některých případech vedla k vymizení symptomů nemoci – tedy k léčebné odpovědi u 36 % pacientů (11, 30).
Mezinárodní doporučení pro léčbu Castlemanovy nemoci z roku 2018 uvádí, že kombinované chemoterapeutické režimy je vhodné ponechat pro ty pacienty, jejichž nemoc progreduje při použití nových účinných léků, jako je rituximab, siltuximab či tocilizumab, případně IMiDs (29–31). Tento názor je založen na mínění odborníků, kteří se touto léčbou zabývají, protože pro nízkou incidenci této nemoci nebyly prováděny srovnávací klinické studie vyjma klinické studie se siltuximabem, zmíněné níže.
Anti‑CD20 protilátka (rituximab)
Rituximab je monoklonální protilátka proti povrchovému antigenu CD20, který je lokalizovaný na fyziologických i transformovaných B‑lymfocytech. Po navázání dochází k destrukci těchto lymfocytů. A protože polyklonální B‑lymfocyty tvoří převážnou část uzlin postižených Castlemanovou chorobou, je racionální použít cílené destrukce těchto buněk pomocí anti‑CD20 protilátky. Rituximab je u této choroby účinný i v monoterapii, jak vyplývá z četných citovaných popisů případů, ale je možné také účinek rituximabu potencovat kombinací s dalšími léky, které se používají při léčbě maligních lymfomů, tedy s glukokortikoidy a dále s alkylačními či antracyklinovými cytostatiky. Rituximab má potenciál navodit léta trvající kompletní remisi a podstatné je, že účinnost této léčby byla potvrzena velkým počtem publikací, dominantě sice u HHV-8 pozitivních případů, ale také u HHV-8 negativních nemocných. Takže na rituximab v této indikaci lze nahlížet jako na velmi účinnou léčbu (35–38).
Gerald (2007) popsal 92 % léčebných odpovědí při kombinaci rituximabu s etoposidem, přičemž léčebné odpovědi měly dlouhodobý charakter, 5leté OS bylo 92 % a PFS 82 % (39).
Léčba rituximabem snižuje dle některých pozorování riziko pozdější transformace v maligní lymfom (40). Před podáním rituximabu se doporučuje vyšetřit virové hepatitidy, protože léčba rituximabem může vést k reaktivaci těchto infekcí.
Rituximab je také účinný v léčbě HHV-8 pozitivní formy nemoci, protože eradikuje lymfocyty, v nichž HHV-8 virus žije, a tím sníží tvorbu IL-6 (29–31).
Na našem pracovišti podáváme pacientům s Castlemanovou chorobou rituximab v kombinaci s cyklofosfamidem a dexametazonem. Používáme tedy stejný režim, jaký podáváme pacientům s Waldenströmovou makroglobulinemií.
Jen v jednom případě jsme použili rituximab pouze v kombinaci s dexametazonem. Šlo o mladou ženu toužící mít vlastní děti, a tak jsme s pacientkou prodiskutovali vliv všech dostupných léků na zárodečné buňky a dohodli se na léčbě rituximabem a kortikoidy. Efekt této léčby přetrval však pouze 3 roky, nyní nemoc u pacientky pomalu dle laboratorních parametrů i FDG‑PET/ CT progreduje, ale zatím asymptomaticky, a tak další léčbu odkládáme. U pacientů, kteří měli kombinaci rituximabu, cyklofosfamidu a dexametazonu, jsou remise delší, ale obdobné je to například u pacientů s Waldenströmovou makroglobulinemií, kteří jsou léčeni rituximabem a dexametazonem anebo rituximabem, dexametazonem a cyklofosfamidem (anebo bendamustinem). Přidání cytostatika k rituximabu a dexametazonu prodlužuje také u Waldenströmovy makroglobulinemie trvání remise.
V literatuře je pouze jedna publikace, která se snaží srovnávat siltuximab a rituximab. Pro srovnání nepoužívá klasický ukazatel OS, ale používá progression free survival – PFS. Liu ve svém přehledu porovnal výsledky léčby rituximabem a siltuximabem a dle výsledků v tomto článku je léčba siltuximabem spojena s delším PFS než při aplikaci rituximabu (36).
IMiDs – imunomodulační léky
Thalidomid byl jako první zařazen do skupiny IMiDs – immunomodulatory drugs. V lidském organismu má velmi pestré účinky. Mimo jiné potlačuje tvorbu některých cytokinů, mezi nimi IL-1, IL-6, IL-12 a VEGF. Stimuluje naopak T buňky cestou cereblonové inhibice.
A právě tato jeho vlastnost, potlačování tvorby prozánětlivých cytokinů, jej činí účinným při léčbě Castlemanovy nemoci, jak dokládají četné popisy případů (41–47). Podání imidů vede k poklesu tvorby Il-6, a to má za následek pokles hodnoty CRP.
V literatuře lze najít několik prací, které popisují synergický účinek podání rituximabu a thalidomidu. Thalidomid, imunomodulační, anti‑inflamatorní a anti‑angiogenní lék, má silnou aktivitu proti této nemoci i v monoterapii. Proto je doporučována kombinovaná léčba rituximab a thalidomid (41–47). Ale z vlastních klinických zkušeností víme, že tolerance thalidomidu není ideální, způsobuje neuropatii, dělá útlum a patologickou únavu, takže léčbu delší než 6 měsíců pacienti obtížně tolerují i při dávce jen 100 mg denně. V případně Castlemanovy nemoci jsme používali stejné schéma jako u mnohočetného myelomu, kombinaci thalidomidu, cyklofosfamidu a dexametazonu.
Lenalidomid má podstatně lepší toleranci než thalidomid. V jednom případě jsme jej s úspěchem použili, pacient byl rok léčen lenalidomidem s následující 6 let trvající remisí a nyní znovu nemoc recidivovala a vyžaduje další léčbu (48, 49). Naše pozitivní zkušenost s léčbou iMCD lenalidomidem byla později potvrzena i dalšími publikovanými zkušenostmi na zahraničních pracovištích (50, 51).
Pokud by bylo možné použít kterýkoliv lék ze skupiny IMiDs, tak jistě lépe tolerován než thalidomid bude lenalidomid a nejlepší toleranci z těchto léků má pomalidomid, jak vidíme u pacientů s mnohočetným myelomem. K datu léto 2021 nebylo použití pomalidomidu u MCD popsáno.
Bortezomib
Bortezomib je selektivní inhibitor proteazomu, preferenčně působící na plazmatické buňky, v nichž mimo jiné snižuje tvorbu Il-6 a také inhibuje NFkappaB. Úspěchy bortezomibu u mnohočetného myelomu vedly k testování bortezomibu u plazmocelulární varianty Castlemanovy nemoci. A dle tří publikací, které popisují tuto léčbu, bylo podání bortezomibu spojeno s dosažením kompletní remise. Bortezomib je tedy po rituximabu dalším novým účinným lékem pro tyto nemocné (52–57). Na našem pracovišti jsme tuto léčbu použili jednou pro recidivu nemoci ve stejném režimu, jaký podáváme pacientům s mnohočetným myelomem, tedy bortezomib, cyklofosfamid a v tomto případě prednison místo dexametazonu. A otázkou je, zda by byl účinný i carfilzomib, lék ze skupiny proteazomových inhibitorů, podobně jako bortezomib.
Protilátka proti interleukinu-6 a jeho receptoru
V literatuře lze nalézt popisy případů s multicentrickou formou Castlemanovy nemoci úspěšně léčených protilátkou proti receptoru interleukinu-6, zvanou tocilizumab. Tato látka je používána hlavně v Asii a Japonsku, jak plyne z citované literatury (58, 59).
Tocilizumab je humanizovaný antagonista receptoru interleukinu - 6, je schopen zablokovat transmembránovou signalizaci, kterou by se normálně aktivoval interleukin-6. Tocilizumab snižuje zánětlivou reakci mediovanou signální kaskádou navázanou na receptor interleukinu-6. Tocilizumab je registrován pro léčbu idiopatické multicentrické Castlemanovy nemoci v Japonsku, v jiných zemích je registrován pro léčbu revmatoidní artritidy.
Po léčbě tocilizumabem byla také popsána regrese orgánového poškození při Castlemanově nemoci, ústup kardiomyopatie anebo zlepšení funkce ledvin (58, 59).
V Evropské unii je od května roku 2014 registrována pro léčbu multicentrické formy Castlemanovy choroby protilátka proti interleukinu-6 zvaná siltuximab (SYLVANT).
Siltuximab je chimérická IgG1 protilátka, která tvoří komplexy s interleukinem - 6, takže brání jeho vazbě na solubilní a membránové receptory. Siltuximab má registraci pro léčbu idiopatické multicentrické formy Castlemanovy nemoci v USA a v Evropě. Podmínkou pro jeho podání je diagnóza idiopatické formy Castlemanovy nemoci, tedy nepřítomnost infekce HHV-8 a nepřítomnosti infekce HIV. Siltuximab tvoří stabilní komplexy s bioaktivními formami interleukinu-6 a brání jeho vazbě na rozpustné i membránové vazebné receptory. Siltuximab se podává v doporučené dávce 11 mg/kg formou intravenózní infuze 1x za tři týdny až do případného selhání léčby (anebo nepřijatelné toxicity). Ošetřující lékař si musí být vědom, že přípravek siltuximab může maskovat známky akutního zánětu, včetně potlačení horečky a reaktantů akutní fáze, a naopak může vést ke zvýšení hodnoty triglyceridů a cholesterolu v séru. Proto se požaduje, aby pacienti vždy před podáním siltuximabu měli počet neutrofilů vyšší než 1,0 x 109/l a počet trombocytů vyšší než 50 x 109/1, a při závažné infekci by se léčba siltuximabem měla vždy přerušit.
V první studii fáze I bylo léčeno 67 pacientů, z nich 29 bylo léčeno déle než 1 rok. Z 37 hodnocených mělo 32 (86 %) pacientů zlepšen alespoň jeden ze sledovaných parametrů. Celkem 12 z 36 hodnocených mělo léčebnou odpověď dokumentovanou na zobrazovacích vyšetřeních. Kompletní remise (CR) byla zaznamenána v 1 případě, parciální remise (PR) v 11 případech. Tato studie byla zaměřena na toleranci a bylo použitorůzné dávkování, takže počty léčebných odpovědí z této studie nejsou relevantní pro vytvoření představy o účinnosti léku. Závěry této studie fáze I lze interpretovat, že lék byl pro část pacientů účinnou léčbou a že je celkem dobře tolerován (60).
V následující fázi II klinického zkoušení bylo 79 pacientů randomizováno v poměru 2 : 1 do skupiny (53 pacientů) dostávající 11 mg/kg siltuximabu 1x za tři týdny, nebo do skupiny dostávající placebo (26 pacientů). Celkem 18 z 53 (34 %) pacientů dosáhlo trvalé léčebné odpovědi (1x CR a 17x PR), zatímco ve skupině s placebem nedošlo k žádné léčebné odpovědi, které by odpovídala CR nebo PR. Parciální remise (PR) byla definována nejen vymizením symptomů, ale také zmenšením lymfadenopatie nejméně o 50 %. U dalších 30 pacientů (57 %) byl pozorován ústup symptomů beze změny velikosti lymfadenopatie či hepatosplenomegalie při hodnocení nemoci pomocí zobrazovacích vyšetření (61).
Do prodloužené léčby pak bylo zařazeno 19 pacientů, kteří dostávali siltuximab dlouhodobě až 7 let. Tato studie prokázala, že pokud dojde k léčebnému efektu po siltuximabu, tak tento léčebný efekt dlouhodobě přetrvává při aplikaci siltuximabu. Intervaly mezi jednotlivými aplikacemi byly v této studii při dobrém efektu prodlužovány někdy až na 6 týdnů. Nežádoucí účinky byly hodnoceny jako tolerovatelné (62). Nejčastější nežádoucí účinky byly pruritus (42 %), infekce horního dýchacího traktu (36 %), fatigue (34 %), makulopapulární raš (34 %) a periferní edémy (32 %) (62).
Randomizované srovnání léčby siltuximabu ve srovnání s placebem prokázalo u 53 hodnotitelných pacientů léčených siltuximabem celkem jen 34 % léčebných odpovědí definovaných jako CR či PR, i když ke zlepšení stavu došlo u většího počtu léčených (61). Siltuximab jako jediný z výše vyjmenovaných léků má registraci pro léčbu Castlemanovy nemoci, ale léčebnou odpověď, definovanou jako CR a PR, přinesl pouze třetině nemocných, i když ústup symptomů byl pozorován u více pacientů. Srovnání účinnosti siltuximabu a rituximabu v rámci klinické studie nebylo provedeno k dispozici je pouze srovnání publikovaných dat, které v roce 2016 zveřejnil Liu a uvedl, že PFS bylo při léčbě siltuximabem delší než při léčbě rituximabem (36). Se siltuximabem zatím vlastní zkušenosti nemáme.
Další klinické studie prokázaly, že siltuximab u dříve léčených pacientů jinými léky anebo u nově léčených dosahuje shodný počet odpovědí (63).
Menší studie probíhaly i v Evropě, k dispozici je hodnocení italské (9 pacientů) a polské studie (11 pacientů) léčebná odpověď byla dosažena u 33 % pacientů, tedy podobně jako v registrační studii (64, 65).
Správný lék správnému pacientovi je heslo posledních let, a tak je pochopitelné, že na registrační studie navazují další, které se snaží vytipovat prediktivní markery léčebné odpovědi po podání siltuximabu. Zatím to vypadá, že výrazná systémová zánětlivá reakce se svými markery (vysoké hodnoty CRP a fibrinogenu, snížená koncentrace hemoglobinu) budou signalizovat vysokou pravděpodobnost léčebné odpovědi na siltuximab.
Léčebná odpověď se dostavila častěji u pacientů s iMDC při zvýšení laboratorních markerů korelujících se systémovou zánětlivou reakcí (pokles hemoglobinu, vzestup hodnoty fibrinogenu, CRP a imunoglobulinů) než u pacientů bez laboratorních známek systémové zánětlivé reakce. Byly dosaženy následující výsledky:
15 z 18 (83 %) mělo léčebnou odpověď a mělo pravděpodobnost léčebné odpovědi > 50 %
19 z 22 (86 %) nemělo léčebnou odpověd a mělo pravděpodobnost léčebné odpovědi < 50 %.
V této studii se podařilo dosáhnout 86% pravděpodobnost rozlišení pacientů reagujících a nereagujících na siltuximab (66, 67). Tato první pozorování zřejmě brzy bude časem následovat přesnější definování podmínek, kdy siltuximab bude dosahovat podstatně vyššího počtu léčebných odpovědí, než bylo v registračních studiích fáze I a II.
Při léčbě siltuximabem proběhla i další sledování, která mimo jiné prokázala zlepšení depresivní nálady a ústup anhedonie (neschopnosti se radovat) vlivem léčby siltuximabem (68).
Anakinra
Anakinra je monoklonální protilátka proti receptoru pro interleukin - 1 (Il-1 receptor antagonist). Její biologický poločas je ale jen 4–6 hodin. Anakinra je schválená agenturou FDA pro léčbu revmatoidní artritidy, ale v průběhu posledních let se prokázal účinek tohoto léku u četných dalších chorob včetně Stillovy nemoci, juvenilní artritidy a také u četných autoinflamatorních chorob s manifestací v dětství, a také u syndromu Schnitzlerové. Podává se obvykle denně v dávce 100 mg 1x denně. Její léčebné použití u Castlemanovy nemoci je založeno na faktu, že interleukin-1 stimuluje tvorbu interleukinu-6, který stimuluje projevy Castlemanovy nemoci. Siltuximab tlumí chorobu blokádou účinku interleukinu-6. Anakinra tlumí chorobu blokádou aktivity interleukinu-1, takže jednu úroveň níže než siltuximab. V odborné literatuře není zatím žádná randomizovaná klinická studie analyzující účinek anakinry u Castlemanovy nemoci. K dispozici jsou tři publikace popisující zásadní zlepšení po podání anakinry u pacientů s Castlemanovou chorobou, často již značně předléčených. První popis léčby Castlemanovy nemoci anakinrou je z roku 2008. U tohoto pacienta předcházela léčba chemoterapií a rituximabem, která však nedosáhla léčebné odpovědi. Až podání anakinry vedlo k vymizení příznaků nemoci (69).
V dalším případě byla anakinra s úspěchem použita u ještě více předléčeného pacienta, u něhož předcházela léčba kladribinem, rituximabem, steroidy, etanerceptem (protilátka proti TNF) a protilátkou proti interleukinu-6, přičemž nemoc byla na vše uvedené refrakterní. Takže klasické léčebné možnosti byly u této ženy, léčené v MD Anderson Cancer, vyčerpány. Proto byla použita anakinra (100 mg denně), do týdne příznaky vymizely a na kontrolních FDG‑PET/ CT zobrazeních vymizela lymfadenopatie a vymizela zvýšená akumulace FDG v kostní dřeni (70).
Anakinru použili také francouzští autoři u případu Castlemanovy nemoci s anasarkou a trombotickou trombocytopenickou purpurou, odpovídající TAFRO syndromu (71).
Anakinra tedy představuje léčebnou alternativu, ověřenou více autory, která může pomoci, když selžou jiné léčebné postupy, ale zase není účinná u všech případů (72–75). Výhodou anakinry je excelentní tolerance nemocnými. Anakinru používáme pro léčbu syndromu Schnitzlerové a Stillovy nemoci, pro léčbu Castlemanovy nemoci jsme ji zatím nepoužili.
Cyklosporin, sirolimus a takrolimus
Kalcineurinové inhibitory cyklosporin a sirolimus působí na T‑buňky. A protože u Castlemanovy nemoci dochází k jejich aktivaci, byly tyto léky také testovány v léčbě Castlemanovy nemoci. Klinické zkušenosti v indikaci Castlemanovy nemoci jsou zatím omezené, cyklosporin navodil remisi u pacientů s TAFRO syndromem (76, 77). V současnosti s těmito léky probíhají další studie (78, 79). S těmito léky zatím nemáme vlastní zkušenosti. Jsou však velmi doporučovány pro léčbu případu s rysy TAFRO syndromu.
Vysokodávkovaná chemoterapie s autologní transplantací krvetvorných buněk
V literární databázi PubMed‑Medline jsou k dispozici jen tři zprávy o použití vysokodávkované chemoterapie s transplantací autologních krvetvorných buněk, 2× melfalanu v dávce 200 mg/m2 (80, 81) a 1x kombinace etoposid thiotepa, cytarabin cyklofosfamid a melfalanu s podporou autologní transplantace krvetvorných buněk (82). Všichni autoři popisují dosažení kompletní remise. Pouze jedna zpráva popisuje provedení alogenní transplantace (83). Takže i vysokodávkovaná chemoterapie s autologní transplantací je alternativou, kterou možno zvážit při neúspěchu klasické léčby.
Doporučení pro léčbu dle mezinárodního doporučení pro léčbu idiopatické multicentrické Castlemanovy nemoci 2018
Léčba méně závažné formy (flu‑like) idiopatické multicentrické formy Castlemanovy nemoci dle mezinárodního doporučení
Vzhledem k tomu, že jediná léčba testovaná v rámci klinické studie byl siltuximab, případně příbuzný tocilizumab, tak v obou případech sepsis‑like i flu‑like průběhu citované mezinárodní doporučení (29–31) začíná léčbu siltuximabem či tocilizumabem, při neúčinnosti této léčby pak doporučuje rituximab (375 mg/m2 4–8 dávek) v kombinaci s glukokortikoidy a případně s dalšími imunomodulačními (thalidomid, lenalidomid) a imunosupresivními léky (cyklosporin A, sirolimus, takrolimus), dále bortezomib, IL-1β receptor antagonista – anakinra, retinoidy a případně interferon ‑ α (31), jak uvádí schéma 1 a tabulka 7. Cyklosporin A byl používán hlavně pro iMCD‑TAFRO částečně s cílem zlepšit trombocytopenii. Anakinra, blokující e IL-1β receptor a zřejmě i NF‑kB signalizaci, byla s úspěchem podávána pacientům refrakterním na siltuximab.


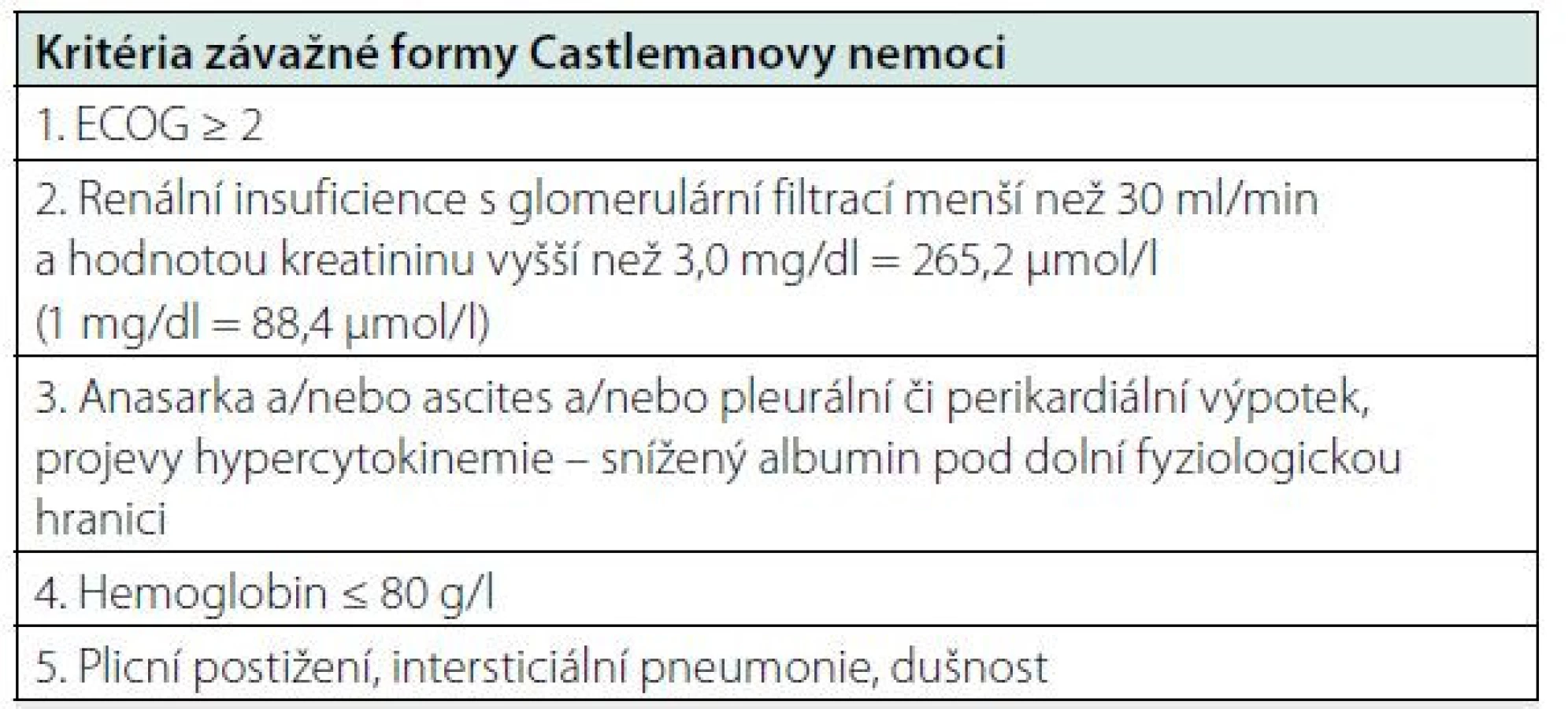
Léčba závažné formy (sepsis‑like) idiopatické multicentrické formy Castlemanovy nemoci
Přibližně 10–20 % pacientů s idiopatickou Castlemanovou nemocí splňuje kritéria závažné formy nemoci s orgánovou dysfunkcí, špatným celkovým stavem a potřebuje velmi intenzivní léčbu. Pro tuto podskupinu pacientů se doporučuje zahájit léčbu vysokými dávkami glukokortikoidů (metylprednisolon 500 mg denně) spolu se siltuximabem. V tomto případě se doporučuje podávání siltuximabu 1x týdně po dobu jednoho měsíce.
U pacientů, kteří na tuto léčbu reagují, se má po měsíci přejít na aplikaci v třítýdenních intervalech a postupně snižovat glukokortikoidy.
V případě, že v prvním týdnu nedojde ke zlepšení, doporučuje se použít polychemoterapeutické režimy používané pro B ‑ lymfomy, tedy režimy obsahující antiCD20 protilátku rituximab, nebo režimy obsahující etoposid a cyklofosfamid, které se používají pro léčbu hemofagocytující lymfohistiocytózy, tedy režimy které mohou potlačit cytokinovou bouři (29–31). Citované doporučení uvádí následující polychemoterapeutické režimy: R‑CHOP (rituximab, cyklofosfamid, doxorubicin, vinkristin, prednison), CVAD (cyklofosfamid, vinkristin, doxorubicin, dexametazon), nebo CVP (cyklofosfamid, vinkristin, prednison), léčebné režimy používané pro myelom nebo etoposid/cyklofosfamid obsahující režimy ve stejném složení, v jakém je používán pro léčbu hemofagocytující lymfohistiocytózy. Chemoterapeutické kombinace jsou vhodné pro pacienty s velmi závažným průběhem nemoci a mohou dosáhnout 78 % léčených odpovědí, ale s odpovídající toxicitou. Pokud ani tyto polychemoterapeutické režimy nepřinesou očekávanou léčebnou odpověď, tak již další léčebné kroky nejsou přesně definovány. Je možné po utlumení prvotní cytokinové bouře opět použít anti ‑ IL-6 léčbu s nadějí, že nyní po utlumení choroby již bude přínosem, nebo kterýkoliv z léků s popsanou účinností u této nemoci, pokud tuto léčbu pacient toleruje (IMiDs, imunosupresiva typu cyklosporinu A, sirolimu, nebo léky používané pro léčbu mnohočetného myelomu VTD (bortezomib, thalidomid, dexametazon)).
Autologní a alogenní transplantace byla použita pouze v ojedinělých případech a představuje poslední možnost.
Závažné průběhy iMCD se často prezentují jako TAFRO subtyp. Dle analýzy 49 případů iMCD‑TAFRO jsou účinné kortikosteroidy, anti - -IL-6 protilátky, cytotoxická léčba a cyklosporin A. U pacientů s TAFRO syndromem je cyklosporin A považován za lék volby, může zmenšit ascites a trombocytopenii. Japonští autoři doporučují pro nemocné s TAFRO syndromem kombinaci vysokých dávek steroidů, tocilizumab a cyklosporin A. Analýza ukázala, že u těchto pacientů je často upregulována dráha mTOR. V souboru 69 pacientů po neúspěchu kortikosteroidů testovali tři způsoby léčby druhé linie, 21 pacientů dostalo tocilizumab, 14 cyklosporin a 8 rituximab ke kortikoidům. Léčbu hodnotili dle intervalu do další léčby či smrti (time to next treatment or death – TTNT). Medián TTNT ve skupině s tocilizumabem byl 2,8 měsíce, s cyklosporinem 9,2 měsíce a v skupině s rituximabem nebylo mediánu v době publikace dosaženo. Tato zkušenost zveřejněná v roce 2021 podtrhuje roli cyklosporinu a rituximabu u agresivní formy nemoci zvané TAFRO syndrom (84).
Prognóza
Na Mayo Clinic byla popsána série 113 pacientů. Celkové pětileté přežití OS bylo jen 65 % u pacientů s multicentrickou formou nemoci a 91 % s unicentrickou formou nemoci při mediánu sledování 5,8 roku. V průběhu sledování zemřelo 37 pacientů. Nejvíce indolentní průběh byl u pacientů, kteří měli současně i POEMS syndrom s osteosklerotickými ložisky. Tato podskupina měla 5leté OS 90 % (1, 3, 4).
Závěry pro praxi
Vyšetření celkové bílkoviny a albuminu by mělo být součástí každého skríningového biochemického vyšetření. Zvýšená koncentrace celkové bílkoviny informuje o zvýšené koncentraci imunoglobulinů, a pak se postupuje dle popsané diferenciální diagnostiky hypergamaglobulinemie (85) a může se dojít k diagnóze Castlemanovy nemoci.
PET‑CT zobrazení je užitečné pro nasměrování odběru uzliny u místa nejvyšší akumulace FDG, ale také pro zjištění rozsahu nemoci i pro hodnocení léčebného efektu.
Základními léky pro multicentrickou formu Castlemanovy choroby jsou: monoklonální protilátka proti interleukinu-6 (siltuximab), monoklonální protilátka anti‑CD20 (rituximab), léky ze skupiny IMiDs (thalidomid, lenalidomid) a protilátka inhibující účinek interleukinu-6 (tocilizumab).
Ve formě popisů případů byla potvrzena účinnost protilátky proti receptoru interleukinu-1 (anakinra). Hypotetické zdůvodnění účinnosti anakinry u této nemoci je blokáda stimulace tvorby interleukinu-6 vlivem blokády interleukinu-1.
V případě, že nemoc nereaguje na jednu z výše uvedených alternativ, je třeba otestovat další ze jmenovaných léků, protože nemoc nereagující na jeden typ léčby může kompletně ustoupit po jiné léčbě. U některých pacientů nemoc lépe ustupuje po léčbě obsahující IMiDs, zatímco u jiných po léčbě obsahující rituximab a praxe vždy ukáže, který z výše jmenovaných léků je pro konkrétního pacienta optimální.
Publikace z posledních let často při neúspěchu léčby první linie kombinují léky s prokázanou účinností. Které kombinace jsou nejúčinnější, nelze říci, ale v případě TAFRO příznaků jsou doporučovány kombinace obsahující cyklosporin A nebo sirolimus.
Článek vznikl v souvislosti s Institucionální podporou MOÚ: MZ ČR – RVO (MOÚ, 00209805)
KORESPONDENČNÍ ADRESA AUTORA:
prof. MUDr. Zdeněk Adam, CSc.
Interní hematologická a onkologická klinika, LF MU a FN Brno Jihlavská 20, 625 00 Brno
Cit. zkr: Vnitř Lék 2022;68(1):41-53
Článek přijat redakcí: 17. 9. 2021
Článek přijat po recenzích: 9. 12. 2021
Zdroje
1. Munshi N, Mehra M. van de Velde H, Desai A et al. Use of a claims database to characterize and estimate the incidence rate for Castleman disease. Leuk Lymphoma 2015; 56(5):1252-1260.
2. Adam Z, Pour L, Krejčí M et al. Lokalizovaná (unicentrická) forma Castlemanovy nemoci. Klinické projevy, diagnostika a léčba dle mezinárodních doporučení z roku 2020. Vnitř Lék 2021;67(8):462–471.
3. Fajgenbaum DC, vanRhee F, Nabel ChS. HHV-8 negative idiopathic multicentric Castleman disease: novel insight into biology pathogenesis and therapy. Blood 2014;123 (19):2924-2933.
4. Bower M, Pria AD, Coyle C et al. Diagnostic criteria schemes for multicentric Castleman disease in 75 cases. J Acquir Immune Defic Syndr. 2014;65(2):e80–e82.
5. Jakubíková M, Piťha J, Latta J, et al. Myasthenia gravis, Castleman disease, pemphigus, and anti‑phospholipid syndrome Muscle and nerve. 2013;47(3):447-451.
6. Cibičková Ľ, Soukup T, Bradna P et al. Asociace revmatoidní artritidy a Castlemanovy choroby. Česká revmatologie. 2005;13(3):106-109.
7. Sun DP, Chen WM, Wang L et al. Clinical characteristics and immunological abnormalities of Castleman disease complicated with autoimmune diseases. J Cancer Res Clin Oncol. 2021;147(7):2107-2115.
8. El Karoui K, Vuiblet V, Dion D et al. Renal involvement in Castleman disease. Nephrol Dial Transplant 2011;26(2):599-609.
9. Sydor A, Madura M, Wagrowska‑Danilewicz M. Amyloid a amyloidosis and renal failure in a course of Castleman disease. Nephrology (Carlton). 2007;12(6):620-621.
10. Dispenzieri A, Armitage J O, Loes M J et al. The clinical spectrum of Castleman’s disease. Amer J Hematol 2012;87 : 997-1002.
11. Dispenzieri A, Fajgenbaum DC. Overview of Castleman disease. Blood. 2020;135(16):1353 - 1364.
12.Dispenzieri A. POEMS syndrome: 2014 update on diagnosis, risk‑stratification, and management. Am J Hematol 2014;89(2):214-223.
13. Fajgenbaum DC, Rosenbach M, van Rhee F, et al. Eruptive cherry hemangiomatosis associated with multicentric Castleman disease: a case report and diagnostic clue. JAMA Dermatol. 2013 Feb;149(2):204-208.
14. Garcia T, Dafer R, Hocker S, Schneck M et al. Recurrent strokes in two patients with POEMS syndrome and Castleman’s disease. J Stroke Cerebrovasc, DiS. 2007;16(6):278-284.
15. Huang J, Wang L, Zhou W, Jin J. Hyaline vascular Castleman disease associated with POEMS syndrome and cerebral infarction. Ann Hematol 86(1):59-61.
16. Szalat R, Munshi NC. Diagnosis of Castleman Disease. Hematol Oncol Clin North Am. 2018;32(1):53-64. doi: 10.1016/j.hoc.2017. 09. 005.
17. Ferda J, Ferdová E, Záhlava J et al. Fever of unknown origin: a value of (18)F‑FDG‑PET/ CT with integrated full diagnostic isotropic CT imaging.European journal of radiology. 2010;73(3):518-525.
18. Koa B et al. Emerging role of 18 F‑FDG PET/CT in Castleman disease: a review Insights Imaging.2021;12 : 35.
19. Koukalová R, Selingerová I, Řehák Z. FDG‑PET/ CT v diagnostice a hodnocení léčebné odpovědi Castlemanovy choroby – retrospektivní studie 29 případů z jednoho centra. Klinická onkologie. 2021;34(2):120-127.
20. Fajgenbaum DC, Uldrick TS, Bagg A et al. International, evidence‑based consensus diagnostic criteria for HHV-8-negative/idiopathic multicentric Castleman disease. Blood 2017;129(12):1646-1657.
21. Cronin DM, Warnke RA. Castleman disease: an update on classification and the spectrum of associated lesions. Adv Anat Pathol. 2009;16 : 236-246.
22. Masaki Y, Nakajima A, Iwao H et al. Japanese variant of multicentric castleman’s disease associated with serositis and thrombocytopenia--a report of two cases: is TAFRO syndrome (Castleman - Kojima disease) a distinct clinicopathological entity? J Clin Exp Hematop 2013;53(1):79-85.
23. Kawabata H, Takai K, Kojima M et al. Castleman‑Kojima disease (TAFRO syndrome): a novel systemic inflammatory disease characterized by a constellation of symptoms, namely, thrombocytopenia, ascites (anasarca), microcytic anemia, myelofibrosis, renal dysfunction, and organomegaly: J Clin Exp Hematop 2013;53(1):57-61.
24. Inoue M, Ankou M, Hua J et al. Complete resolution of TAFRO syndrome (thrombocytopenia, anasarca, fever, reticulin fibrosis and organomegaly) after immunosuppressive therapies using corticosteroids and cyclosporin A: a case report. J Clin Exp Hematop. 2013;53(1):95-99.
25. Tedesco S, Postacchini L, Manfredi L et al. Successful treatment of a Caucasian case of multifocal Castleman’s disease with TAFRO syndrome with pathophysiology targeted therapy - case report. Exp Hematol Oncol 2015;4(1):3-10.
26. Masaki Y, Kawabata H, Takai K et al. Proposed diagnostic criteria, disease severity classification and treatment stratedy for Tafro syndrom, 2015 version. Int. J. Hematol 2016;103 : 686-692.
27. Yu L, Shi M, Cai Q et al. A Novel Predictive Model for Idiopathic Multicentric Castleman Disease: The International Castleman Disease Consortium Study. Oncologist. 2020;25(11):963-973.
28. Fang X, Sun Z, Xu‑Monette ZY et al. Model for Idiopathic Multicentric Castleman Disease Supporting Treatment Decisions. Oncologist. 2021;26(1):4–6.
29. Wu D, Lim MS, Jaffe ES. Pathology of Castleman Disease. Hematol Oncol Clin North Am. 2018;32(1):37-52.
30. van Rhee F, Greenway A, Stone K. Treatment of Idiopathic Castleman Disease. Hematol Oncol Clin North Am. 2018;32(1):89–106. doi: 10.1016/j.hoc.2017. 09. 008.
31. Van Rhee F, Voorhees P, Dispenzieri A et al al. International evidence based consensus treatment guidelines for idiopahtic multicentric Castleman disease. Blood 2018;132 (20):2115-2124.
Další literatura u autora a na www.casopisvnitrnilekarstvi.cz
Štítky
Diabetologie Endokrinologie Interní lékařstvíČlánek vyšel v časopise
Vnitřní lékařství

2022 Číslo 1
- Inovace v hojení ran: krytí Zetuvit Plus Silicone Border pro optimální management exsudátu z ran
- Ivabradin zlepšuje kvalitu života starších pacientů se srdečním selháním
- Fixní kombinace kandesartan/amlodipin v terapii arteriální hypertenze
- Clopidogrel je v prevenci kardiovaskulárních příhod přínosnější než kyselina acetylsalicylová
Nejčtenější v tomto čísle
- Pacient s jaterní cirhózou na interním oddělení
- Měření cholesterolu a současná doporučení
- Diferenciální diagnostika zvětšení hypofýzy
- Levotyroxin
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy