
GENETICKÁ NÁCHYLNOST A OXIDAČNÍ STRES U KARCINOMU PROSTATY: INTEGROVANÝ MODEL S DOPORUČENÍMI PRO PREVENCI
Autoři:
Eric A. Klein M. D.
Působiště autorů:
Cleveland Clinic Foundation
; Glickman Urological Institute
; Cleveland Clinic Lerner College of Medicine
and Taussig Cancer Center
; Section of Urologic Oncology
Vyšlo v časopise:
Urol List 2006; 4(4): 19-24
ÚVOD
Karcinom prostaty je ve Spojených státech od roku 1984 nejčastěji se vyskytující viscerální maligní onemocnění u mužů; v současné době tvoří 1/3 všech podobných karcinomů. Odhadované riziko onemocnění během života je u bělochů 17,6 %, u afroameričanů 20,6 % a riziko úmrtí je 2,8 % u bělochů a 4,7 % u afroameričanů. Incidence karcinomu prostaty dosáhla vrcholu v roce 1992, přibližně 5 let po zavedení PSA jako screeningového testu, do roku 1995 mírně klesala a od té doby pomalu stoupá po křivce, jež se podobá křivce vzestupu před zavedením PSA. Přestože již bylo identifikováno mnoho potenciálních rizikových faktorů a přestože existuje mnoho hypotéz o příčině vzniku karcinomu prostaty, jeho etiologie zůstává stále neznámá. Máme důkazy, že na původu a vývoji tohoto onemocnění se podílí genetika i životní prostředí a studie provedené v nedávné době prokazují, že při patogenezi karcinomu hraje významnou roli zánět prostaty. V následujícím přehledu uvádíme nejnovější vědecké a klinické důkazy, které dokládají, že při genezi karcinomu prostaty je vztah mezi genetickou náchylností, predispozicí k infekci a narušeným buněčným obranným mechanizmem proti oxidačnímu stresu konvergentní, a ukazujeme, jak tato skutečnost ovlivňuje prevenci.
GENETICKÁ NÁCHYLNOST
Četné epidemiologické studie potvrzují, že na vzniku karcinomu prostaty se podílí familiární a genetická složka. První zprávy o častějším výskytu v rodinné anamnéze byly publikovány v polovině 20. století. Už tyto zprávy ukazují, že riziko vzniku karcinomu prostaty je vyšší u pacienta, jehož nejbližší příbuzný je postižen tímto onemocněním. Následné kazuistiky a skupinové studie potvrdily odhad relativního rizika (RR) vzniku karcinomu prostaty u příbuzných v první linii na 0,64–11 [1]. Studie dvojčat také prokázaly genetický prvek s vyšším procentem výskytu u jednovaječných než u dvojvaječných bratrů [2]. RR se zvyšuje v závislosti na počtu členů rodiny s karcinomem prostaty, stupni jejich příbuznosti a věku, ve kterém u nich došlo ke vzniku onemocnění (tab. 1) [3].
![Rodinná anamnéza a riziko vzniku karcinomu prostaty [7].](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image/505a6e00ced97db66ac0e5a08dcd3d83.png)
Z výzkumných důvodů lze karcinom prostaty rozdělit na 3 fenotypy: sporadický, familiární a hereditární. Sporadické karcinomy vznikají u jedinců s negativní rodinnou anamnézou. Familiární karcinom prostaty je definován jako karcinom u muže s ≥ 1 příbuzným s karcinomem prostaty. Hereditární karcinom prostaty je podmnožinou familiární formy a je definován u rodiny s ≥ 3 členy postiženými karcinomem prostaty u 3 po sobě jdoucích generací nebo u 2 postižených jedinců, u nichž je karcinom diagnostikován před dovršením 55. roku věku.[4]. Zatímco původ karcinomu prostaty je většinou polygenní, existenci skutečně hereditární formy dokazují 3 epidemiologická pozorování: 1. příbuzní pacientů mladší než 55 let mají vyšší riziko vzniku karcinomu prostaty než ti, u jejichž příbuzných došlo ke vzniku karcinomu prostaty ve vyšším věku. 2. existuje vyšší počet případů karcinomu prostaty v rodinách s časnějším nástupem karcinomu. 3. počet postižených členů rodiny a jejich věk v době vzniku karcinomu jsou mezi příbuznými nejdůležitějšími určujícími znaky rizika. Sporadické karcinomy tvoří asi 85 % všech karcinomů prostaty a asi 15 % je familiárních a/nebo hereditárních. Hereditární karcinom prostaty tvoří asi 43 % onemocnění vznikajícího v nižším věku (55 let nebo méně) ale pouze 9 % všech karcinomů vznikajících do 85. roku [4].
Z několika komplexních segregačních analýz byly získány důkazy o hlavních genech náchylnosti ke karcinomu prostaty, které se oddělují v rodinách, kdy většina vykazuje dominantní a zbytek recesivní, nebo na gen X vázané druhy dědičnosti [5]. Bylo zaznamenáno alespoň 9 kandidátů na geny náchylnosti ke karcinomu prostaty, zahrnující RNASEL/HPC1 [6], ELAC2/HPC2 [7], SR-A/MSR1 [8], CHEK2 [9], BRAC2 [10], PON1 [11], OGG1 [12], MIC-1 [13] a TLR4 [14] (tab. 2). Individuálně tvoří tyto geny pouze malý zlomek pozorované genetické predispozice ke karcinomu prostaty. Další segregační studie prokázaly existenci dalších oblastí náchylnosti na karcinom prostaty na chromozomech 1q42.2-43 (pojmenovaný PCAP) [15], 1p36 (pojmenovaný CAPB a související s tumory mozku) [16] a Xq27-28 [17], ovšem gen nebo geny vázané na tyto regiony nebyly naklonovány nebo identifikovány. Studie genomu provedené v nedávné době u větší skupiny rodin s hereditárním karcinomem prostaty identifikovaly další oblasti chromozomů spojené s karcinomem prostaty a je možné, že se počet identifikovaných genů náchylnosti zvýší.

HPC1
Ze všech známých genů náchylnosti je nejlépe charakterizován HPC1. Existence HPC1 byla prokázána pomocí studií genomu u rodin s hereditárním karcinomem [18] a později potvrzena linkage studiemi [19]. Další studie údajně identifikovala gen dekódující antivirový a proapoptický enzym RNázu L jako HPC1 [6]. RNáza L je poslední enzym systému 2-5 A, degradační dráhy RNA, jenž hraje významnou roli při zprostředkování biologického účinku IFN, zejména při odpovědi na virovou infekci. IFN typu I indukují skupinu 2-5A - -syntetáz, jež jsou aktivovány pomocí dsRNA, což vede ke konverzi ATP na skupinu krátkých oligoadenylátů vázaných na 2 a 5 (2-5A). 2-5A se váže s vysokou afinitou na RNázu L a přeměňuje ji z neaktivního monomeru na silný dimer, který degraduje single stranded (jednopramennou) RNA zabraňující replikaci virů, ovlivňující syntézu proteinů a způsobující apoptózu přenášenou kaspázou (obr. 1) [20].
![Antivirový a apoptický účinek RNázy L. Jako odpověď na interferon a virové infekce je pomocí double stranded RNA (dsRNA) aktivována skupina oligoadenylátové syntetázy (OAS), což vede k přeměně ATP na několik krátkých oligoadenylátů vázaných na (2-5A). 2-5A se váže s vysokou afinitou na RNázu L, která ji přeměňuje z neaktivního monomeru na silný dimer, jenž degraduje dsRNA a způsobuje apoptózu hostitelských buněk. Buňky karcinomu prostaty postrádající RNlázu L jsou rezistentní na apoptózu. Více podrobností v literatuře [25,31].](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image/abc3aa65e35ff4e820042328bac991ad.jpg)
Transgenní myši postrádající enzymatickou aktivitu RNázy L jsou náchylnější k virovým infekcím než myši s intaktní aktivitou RNázy L [20]. U rodin s hereditárním karcinomem prostaty byla identifikována řada neaktivujících se a mylných mutací RNASEL [21]. Na jejich základě bylo prokázáno, že nukleotidový polymorfizmus (SNP), 1385G-> A způsobující substituci argininu na glutamin (R462Q), souvisí se zvýšeným rizikem karcinomu prostaty v USA [22,23], nikoliv však u německé [24] a švédské [25] populace. Varianta R462Q snížila aktivitu RNázy L jak v systémech z volných buněk, tak v intaktních lidských buňkách [20], což může způsobovat vznik mutací způsobených oxidačním stresem, mitotické poruchy a akumulaci dalších mechanizmů způsobujících iniciaci tumoru v důsledku nedostatečné apoptózy [20,26].
Epidemiologické údaje ukazují, že mutovaný HPC1 je vzácný autozomálně dominantní gen s vysokou penetrací, což znamená, že i když není u velkého množství karcinomů prostaty přítomen, individuální nositel bude s vysokou pravděpodobností postižen karcinomem prostaty. Karcinomy vázané na HPC1 mají vyšší grade a pokročilejší stadium, přestože u sporadických karcinomů nejsou zaznamenány žádné histologické odlišnosti [27,28]. Metaanalýza provedená v současné době prokázala, že mutace R426Q jsou spojeny se závažností onemocnění v závislosti na rodinné anamnéze a rase [23]. Známá molekulární dráha aktivace RNázy L a jejich cílů by měla umožnit delineaci molekulárních strategií zacílených na překonání enzymatického defektu při prevenci i terapii postižených mužů.
DALŠÍ GENY
Biologické funkce dalších známých genů spadají do několika kategorií. SR-A/MSR a MIC-1 jsou regulátory zánětu. SR-A/MSR1 moduluje interakci hostitelských makrofágů, adhezi makrofágů, fagocytózu apoptických buněk a mikrobů a čistění a detoxifikaci mikrobiálních produktů [29]. Myši postrádající SR-A/MSR1 jsou náchylnější k bakteriálním infekcím [30]. MIC-1 je členem rodiny transformujících růstových faktorů beta (TGFβ) a reguluje aktivitu makrofágů [13]. TLR4 je ze skupiny genů nazvaných TLRs (Toll-like receptors), podobný genu Drosophila Toll, jehož mutace vede k abnormálnímu dorzoventrálnímu vývoji a predisponuje k houbovitým infekcím [31]. TLRs zprostředkovávají u myší i lidí vrozenou imunitní reakci jako odpověď na různé mikroby stimulací systému prozánětlivých cytokinů, které simultánně pomáhají vyčistit počáteční infekci a stimulovat vývoj adaptivní imunitní reakce [32]. Mutace TLRs mohou vést k nedostatečnosti mechanizmu čištění patogenů a způsobit chronickou zánětlivou reakci na přetrvávající infekci [14]. PON1 kóduje paraoxanázu, která se váže na lipoprotein o vysoké denzitě v séru a přispívá k detoxikaci organofosforových sloučenin a karcinogenních, v tucích rozpustných volných radikálů z lipidové peroxidace [11]. Bylo také prokázáno, že SR-A/MSR1 a PON1 hrají roli při zprostředkování zánětu vedoucího k ateroskleróze [33,34]. CHEK2, BRCA2 a OGG1 jsou významné při opravě DNA. CHEK 2 je aktivován a zabraňuje replikaci poškozené DNA koordinací opravy DNA, progrese buněčného cyklu a apoptózy [9]. BRCA2 podporuje opravu poškození dsDNA pomocí homologní rekombinace. OGG1 je DNA-glykosyláza/ AP-lyáza, jež opravuje oxidační poškození DNA [35]. Funkce ELAC2/HPC2 není známa.
ROLE OXIDAČNÍHO STRESU, ZÁNĚTU A INFEKCE
Chronický zánět vedoucí k buněčné hyperproliferaci nahrazující poškozenou tkáň přispívá ke vzniku karcinomů kolon, jícnu, žaludku, močového měchýře a jater spojených s infekcí [36,37]. V roce 2005 přidal US Department of Health and Human Services na seznam známých příčin způsobujících karcinom 3 infekční agens (virus hepatitidy B, virus hepatitidy C a lidský papiloma virus) [38]. Karcinomy jater, cervixu, hlavy a krku způsobené těmito viry mají stejnou patogenezi s dlouhou dobou latence po vystavení viru a zánětlivou komponentu pro promoci tumoru [38]. Jak ukázal Nelson [39], může nashromáždění epidemiologických, histologických a genetických důkazů prokázat, že podobný proces může být základem vzniku karcinomu prostaty.
Další důkazy dokládají, že karcinom prostaty může mít infekční etiologii. Metaanalýza 23 studií zaznamenala statisticky signifikantní souvislost karcinomu prostaty s anamnézou pohlavně přenosných infekcí (RR = 1,4; 95 % CI = 1,2–1,7), zahrnující RR 2,30 (95 % CI = 1,3–3,9) u syfilis a 1,34 (95 % CI = 1,2–1,6) u kapavky [40]. Podobně odhalila metaanalýza 11 studií statisticky významné riziko karcinomu prostaty (poměr šancí [OR] = 1,57, 95 % CI 1,0–2,4) u mužů s anamnézou prostatitidy [41]. Podpůrné důkazy poskytuje několik málo studií demonstrujících pozitivní souvislost protilátek proti syfilidě, lidskému papiloma viru (HPV) a lidskému herpesviru-8 (HHV-8) s karcinomem prostaty [37]. Studie také zaznamenaly vyšší koncentraci reaktantů akutní fáze a prozánětlivých cytokinů v plazmě u mužů s karcinomem prostaty. Jedná se zde o C-reaktivní protein, IL-6, IL-8, IL-1β a TNF-α, a to zejména u pokročilého nebo na hormony refrakterního onemocnění [37]. 2 studie prokázaly v tkáni lidské prostaty přítomnost virových patogenů včetně polyomaviru, lidského papilomaviru a cytomegaloviru [42,43].
V klinických vzorcích prostaty jsou často nalezeny zánětlivé infiltráty a histologické léze nazývané proliferativní zánětlivá atrofie (proliferative inflammatory atrophy - PIA) [44]. PIA představuje spektrum lézí charakterizovaných epiteliální atrofií, nízkým indexem apoptózy a zvýšeným proliferativním indexem, obvykle souvisejícím se zánětlivými infiltráty [45]. Zánět v PIA může zahrnovat mononukleární infiltráty v periglandulárním stromatu a makrofágy a/nebo neutrofily v glandulárním lumen nebo epitelu. Makrofágy aktivované IFNγ vylučují prozánětlivé cytokiny a reaktivní druhy dusíku (např. oxid dusnatý - NO). Indukovaná syntéza NO, jež katalyzuje tvorbu NO, je nadměrně vylučována v makrofázích v PIA, ovšem nikoliv v normálním epitelu [46]. Buňky PIA obvykle vykazují mnoho známek stresu včetně indukce GSTP1, GSTA1 a exprese COX-2 [46]. Důkazy dokládají, že PIA je regenerační léze, jež se objevuje jako následek infekce nebo buněčného traumatu způsobeného oxidačním poškozením, hypoxií, infekcí nebo autoimunitou, a tím, že její proliferativní stav vede ke vzniku karcinomu. PIA je často přilehlá k prostatické intraepiteliální neoplazii vyššího grade (HGPIN) nebo karcinomu v časném stadiu [45] a nashromážděné důkazy prokazují identifikovatelnou genetickou dráhu mezi PIA, HGPIN a karcinomem s progresivně častými mutacemi TP53 v centromerických sekvencích DNA na chromozomu 8 a ostrůvkovitou hypermetylaci GSTP1 CpG [39,46].
GENETICKÁ NÁCHYLNOST, INFEKCE A OXIDAČNÍ STRES: INTEGROVANÝ MODEL
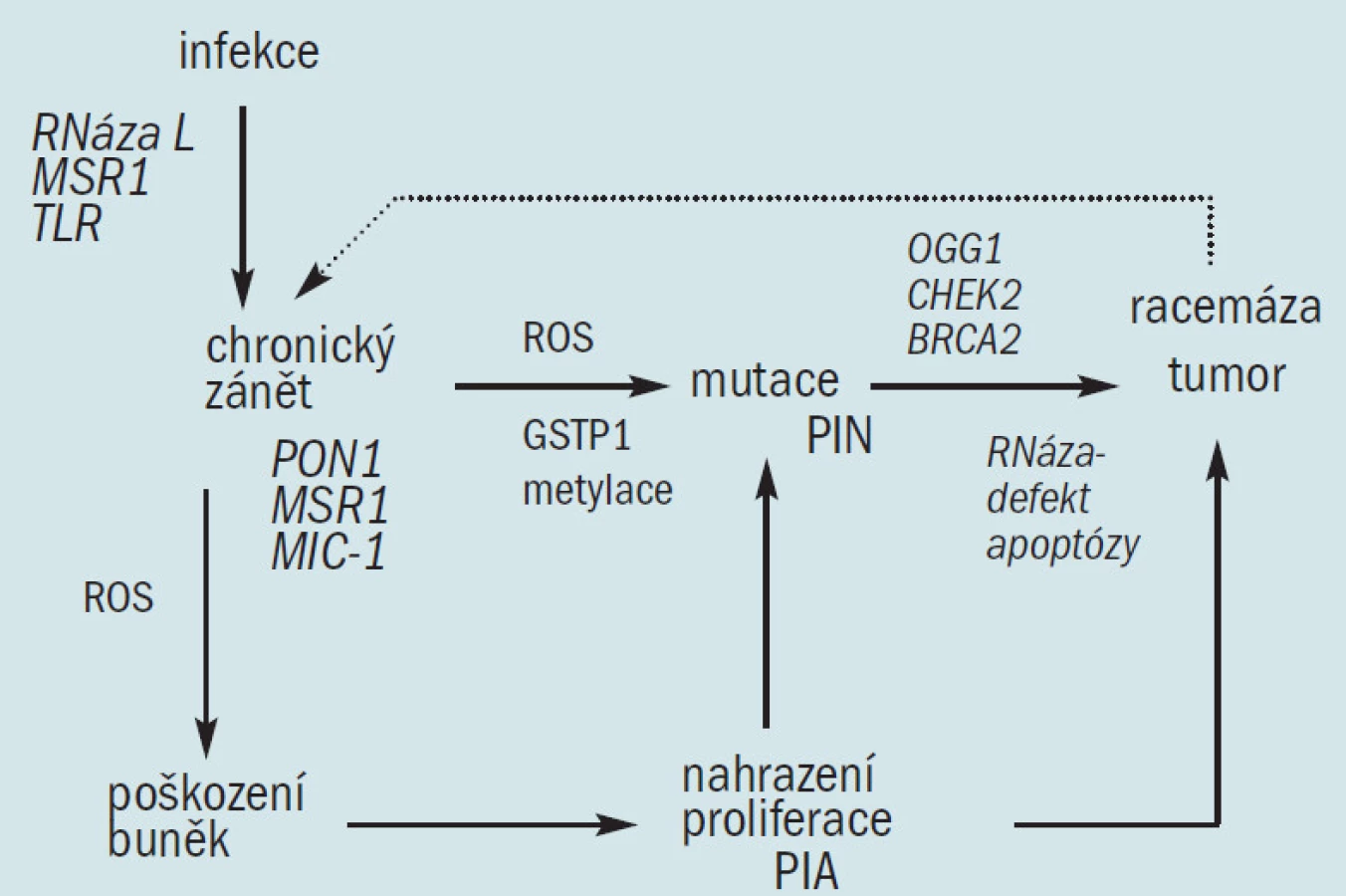
Jak bylo uvedeno výše, domníváme se, že oxidační stres chronických zánětů, který vede k maligní degeneraci, je u člověka zodpovědný za vznik řady karcinomů. Výše popsaná genetická a histologická pozorování u karcinomu prostaty prokazují, že narušená buněčná obrana proti oxidačnímu stresu může také iniciovat prostatickou karcinogenezi. Oxidační stres je zprostředkován reaktivním kyslíkem a druhy dusíku (ROS a RNS), které se váží na DNA a způsobují mutace. Důsledkem oxidačního stresu z exogenních a endogenních zdrojů je akumulace poškození DNA, které se vyskytuje spolu se stárnutím a následně způsobuje maligní onemocnění [36]. Buněčné obranné mechanizmy proti tomuto procesu zahrnují: 1. antioxidační enzymy, které ničí ROS a RNS a zabraňují vzniku mutací, 2. enzymy, které opravují mutovanou DNA a 3. schopnost podstoupit apoptózu v případě, že je DNA příliš poškozená na to, aby bylo možné ji opravit (obr. 2). Analýza funkcí známých genů náchylnosti a jiných genetických defektů u karcinomu prostaty ukazuje, že dědičné a získané defekty těchto buněčných obranných mechanizmů proti infekci a oxidačnímu stresu umožňují iniciaci prostatických tumorů. Navrhujeme model, který prokazuje, že chronická infekce vedoucí k chronickému zánětu spolu s dědičnými a získanými defekty u antioxidačních enzymů, enzymů opravujících DNA a apoptózou, vedou ke vzniku karcinomu prostaty (obr. 3), což zrcadlí proces, který se vyskytuje u dalších tumorů vznikajících jako následek zánětu. Genetická a histologická pozorování podporující tento model jsou následující: 1. defekty u 2 známých genů náchylnosti, HPC1/RNázy L a SR-A/MSR1 činí transgenní myši (a tedy možná i postižené lidské jedince) náchylné k infekci, 2. mutace TLRs mění odpověď hostitele na infekci, což může umožnit chronickou zánětlivou reakci, 3. infekce a chronický zánět podporují generaci ROS a RNS, jež způsobují buněčný oxidační stres, 4. existují četné histologické důkazy zánětlivé reakce v prostatě, zaznamenané zánětlivými infiltráty a PIA, 5. defekty vznikající různými izoformami a geny u antioxidačních enzymů PON1 a GSTP1 mohou způsobit oxidační poškození DNA, 6. defekty SR-A/MSR1 a M1C mohou umožnit neomezenou zánětlivou reakci a 7. defekty na RNASEL umožňují mutovaným buňkám uniknout apoptóze a vedou ke vzniku a šíření klonů maligních buněk. Následná exprese α-metylacyl-coA-racemázy v buňkách tumoru [47], enzymu, který okysličuje řetězce mastných kyselin z dietárních zdrojů, způsobuje tvorbu peroxidu vodíku, jenž může přispívat k oxidačnímu stresu a růstu tumoru.

DŮSLEDKY PRO PREVENCI
Tento model poskytuje základ pro další experimentální a klinické studie zabývající se rolí známých a v budoucnu eventuálně objevených genů náchylnosti, infekce, oxidačního stresu a zánětu při genezi karcinomu prostaty. Tento model také navrhuje různé strategie, které by mohly být užitečné při prevenci karcinomu prostaty; některé z nich jsou v současné době ve fázi výzkumu ve velkých klinických studiích (tab. 3). Například antioxidační látky, které ničí volné radikály, by mohly být nápomocny při zastavení mutagenních účinků ROS a RNS, zatímco protizánětlivé látky (jako NSA a inhibitory COX-2) mohou zpomalit nebo zabránit buněčnému poškození, vedoucímu k nahrazení proliferace. Existuje velké množství epidemiologických důkazů dokládajících roli obou těchto skupin látek při prevenci karcinomu prostaty a několik probíhajících klinických studií testuje jejich účinek právě v této souvislosti. Několik randomizovaných studií, zahrnujících Prostate Cancer Prevention Trial a 3 BPH studie, prokázalo statisticky signifikantní snížení incidence karcinomu prostaty u pacientů užívajících inhibitory 5α-reduktázy (finasterid nebo dutasterid) ve srovnání s pacienty užívajícími placebo [48,49]. Pokles incidence je zda pravděpodobně způsoben přímým antiandrogenním účinkem na proliferaci buněk. Je však známo, že androgeny také vyvolávají oxidační stres v prostatě [46] a alternativním nebo doplňkovým mechanizmem pro pozorované účinky těchto látek je redukce oxidačního poškození prostaty. Tato pozorování navrhují další možnou preventivní strategii, spočívající na kombinaci inhibitorů 5α-reduktázy a protizánětlivých přípravků nebo antioxidantů, které je možno užívat jako doplněk stravy. Tento model také navrhuje další, více přímočaré, strategie. Identifikace infekčního agens v etiologii karcinomu prostaty by mohla vést k vytvoření vakcíny, jež by byla aplikována před započetím sexuální aktivity. Tato strategie je příslibem při prevenci karcinomu čípku při aplikaci antiHPV-vakcíny [50].

Je třeba využít strategie genové terapie, která se zaměřuje na známé genetické defekty a snaží se obnovit funkci nedostatečných buněčných procesů, jako je například oprava DNA nebo apoptózy, nebo na vývoj látek, které obnoví funkci genů utlumených metylací nebo jiným epigenetickým fenoménem.
NEJNOVĚJŠÍ POZNATKY
I když etiologie karcinomu prostaty není známa, je zřejmé, že na jeho původu a vývoji se podílejí genetické faktory i vliv prostředí. Vědecké i klinické důkazy z poslední doby dokládají, že souvislost mezi genetickou náchylností a predispozicí k infekci může hrát při vzniku karcinomu prostaty významnou roli. Ze všech známých genů náchylnosti na karcinom prostaty je nejlépe popsán HPC1. HPC1 kóduje enzym RNázu L, antivirový gen, který hraje klíčovou roli při vrozené imunitní reakci na virové infekce. Aktivace RNázy L virovou infekcí a endogenním interferonem potlačuje virové šíření degradací RNA tím, že přinutí infikovanou hostitelskou buňku podstoupit apoptózu. Preklinické studie prokázaly, že myši s nedostatkem RNázy L jsou náchylnější k virovým infekcím.
Existují důkazy, že alely genu RNázy L mohou u lidí zvyšovat riziko vzniku karcinomu prostaty. IWe již dříve prokázala, že u heterozygotních mužů (genotyp RQ) způsobují aminokyseliny z argininu na glutamin na pozici 462 RNázy L proteinu asi o 50 % vyšší riziko karcinomu prostaty a že riziko u homozygotních na této pozici (genotyp QQ) je ve srovnání s ostatními muži dvojnásobné (genotyp RR) [1]. Pozorování zjišťující, že varianty antivirového genu jsou u mužů příčinou náchylnosti ke karcinomu prostaty, vedou náš výzkum k myšlence virové etiologie karcinomu prostaty.
Abychom ověřili tuto hypotézu, použili jsme pomůcku nazvanou ViroChip [2], vyvinutou našimi spolupracovníky J. de Reisim a Ganemen. ViroChip obsahuje vysoce konzervovaný úsek všech známých virů (téměř 1 000) z rostlinné, zvířecí i lidské říše. Hybridizace RNA z biologického vzorku (jako například sekrece dýchacích cest nebo tkáně) na čipu umožnila zjistit, jaké virové geny se ve vzorku nacházejí, identifikovat, do které skupiny virů patří a umožnila naklonování a identifikaci stejné sekvence virů. Hybridizovali jsme RNA ze vzorků periferní zóny radikální prostatektomie mužů, jejichž genotyp tvořil alely genu RNázy L. Z prvních 19 mužů jsme u 8 identifikovali nové retroviry [3]. U 7 z těchto 8 mužů s novým virem jsme zjistili, že mají genotyp QQ-genu RNázy L.
Od té doby jsme provedli screening u více než 150 mužů. Analýza údajů není kompletní, ale předběžná zjištění ukazují, že přibližně polovina mužů s mutacemi QQ je pozitivní na XMRV, zatímco ze všech mužů, kteří tuto variantu nemají, je pozitivní jen 1. Úplné vyšetření sekvencí XMRV odhalí, že velmi připomíná virus Murkin Leukemie (MuLV), totiž virus, jenž způsobuje leukemii u myší. Avšak mezi XMRV a MuLV existují významné rozdíly, včetně toho, že XMRV neinfikuje myši a má deleci v sekvenci glyko-GAG, jež napomáhá určit její virulenci.
Tkáňové studie prokázaly, že XMRV nesídlí v epitelu, ale ve fibroblastech přilehlých ke karcinomu. Existuje velké množství vědeckých důkazů, které popisují biologickou zkříženou komunikaci mezi fibroblasty souvisejícími s karcinomem a tumory epitelu. Domníváme se, že XMRV působí na tumor prostřednictvím parakrinní signalizace nebo nepřímo, poskytnutím vhodného mikroprostředí pro vznik makrofágů a bílých krvinek, což způsobuje oxidační stres. Obě tyto hypotézy jsou v současné době ve fázi výzkumu. I když je třeba prokázat přímou spojitost mezi XMRV jako příčinou karcinomu prostaty, představuje tato studie nové vzrušující poznatky možné patogeneze karcinomu prostaty.
ZÁVĚR
Epidemiologické, genetické, histologické a molekulární důkazy dokládají při genezi karcinomu prostaty integrovaný model dědičných a získaných defektů proti oxidačnímu stresu v buněčném obranném mechanizmu. Tento model je syntézou existujících důkazů a je navrhován jako základ pro další studie, které podpoří, rozšíří, vylepší nebo vyvrátí klíčové body. Tento model slouží také jako paradigma pro vývoj nových strategií při prevenci karcinomu prostaty.
Eric A. Klein, M.D.
Section of Urologic Oncology,
Glickman Urological Institute
Cleveland Clinic Lerner College of Medicine and Taussig Cancer Center,
Cleveland Clinic Foundation
Zdroje
1. Eeles RA, Dearnaley DP, Ardern-Jones A et al. Familial prostate cancer: the evidence and the Cancer Research Campaign/British Prostate Group (CRC/ BPG) UK Familial Prostate Cancer Study. Br J Urol 1997; 79(Suppl 1): 8-14.
2. Ahlbom A, Lichtenstein P, Malmstrom H et al. Cancer in twins: genetic and nongenetic familial risk factors. J Natl Cancer Inst. 1994; 89 : 287-293.
3. Bratt O. Hereditary prostate cancer: clinical aspects. J Urol 2002; 168 : 906-913.
4. Carter BS, Bova GS, Beaty TH et al. Hereditary prostate cancer: epidemiologie and clinical features. J Urol 1993; 150 : 797-802.
5. Gillanders EM, Xu J, Chang BL et al. Combined genome-wide scan for prostate cancer susceptibility genes. J Nail Cancer Inst 2004; 96 : 1240-1247.
6. Carpten J, Nupponen N, Isaacs S et al. Germline mutations in the ribonuclease L (RNase L) gene in hereditary prostate cancer 1 (HPCI)-linked families. Nat Genet 2002; 30 : 181-184.
7. Tavtigian SV, Simard J, Teng DH et al. A candidate prostate cancer susceptibility gene at chromosome 17p. Nat Genet 2001; 27 : 172-180.
8. Xu J, Zheng SL, Komiya A et al. Germline mutations and sequence variants of the macrophage scavenger receptor 1 gene are associated with prostate cancer risk. Nat Genet 2002; 32 : 321-325.
9. Dong X, Wang L, Taniguchi K et al. Mutations in CHEK2 associated with prostate cancer risk. Am J Hum Genet 2003; 72 : 270-280.
10. Edwards SM, Kote-Jarai Z, Meitz J et al. Two percent of men with early-onset prostate cancer harbor germline mutations in the BRCA2 gene. Am J Hum Genet 2003; 72 : 1-12.
11. Marchesani M, Hakkarainen A, Tuomainen TP et al. New paraoxonase 1 polymorphism 1102V and the risk of prostate cancer in Finnish men. J Nail Cancer Inst 2003; 95 : 812-818.
12. Xu J, Zheng SL, Turner A et al. Associations between hOGGI sequence variants and prostate cancer susceptibility. Cancer Res 2002; 62 : 2253-2257.
13. Lindmark F, Zheng SL, Wiklund F et al. H6D polymorphism in macrophage-inhibitory cytokine-1 gene associated with prostate cancer. J Nail Cancer Inst 2004; 96 : 1248-1254.
14. Zheng SL, Augustsson-Balter K, Chang B et al. Sequence variants of toll-like receptor 4 are associated with prostate cancer risk: results from the Cancer Prostate in Sweden Study. Cancer Res 2004; 64 : 2918-2922.
15. Berthon P, Valeri A, Cohen-Akenine A et al. Predisposing gene for early-onset prostate cancer, located on chromosome 1q42.2-43. Am J Hum Genet 1998; 62 : 1416-1420.
16. Gibbs M, Stanford JL, Mclndoe RA et al. Evidence for a rare prostate cancer susceptibility locus at chromosome 1p36. Am J Hum Genet 1999; 64 : 776-780.
17. Xu J, Meyers D, Freije D et al. Evidence for a prostate cancer susceptibility locus on the X chromosome. Nature Genet 1998; 20 : 175-178.
18. Smith JR, Freije D, Carpten JD et al. Major susceptibility locus for prostate cancer on chromosome 1 suggested by a genome-wide search. Science 1996; 274 : 1371-1374.
19. Eeles RA, Durocher F, Edwards S et al. Linkage analysis of chromosome lq markers in 136 prostate cancer families. Am J Hum Genet 1998; 62 : 653-658.
20. Silverman RH. Implications for RNase L in prostate cancer biology. Biochemistry 2003; 42 : 1805-1812.
21. Xiang Y, Wang Z, Murakami J et al. Effects of RNaseL mutations associated with prostate cancer on apoptosis induced by 2' ,5'-oligoadenylates. Cancer Res 2003; 63 : 6795-6801.
22. Casey G, Neville PJ, Plummer SJ et al. RNASEL Arg462Gln variant is implicated in up to 13% of prostate cancer cases. Nat Genet 2002; 32 : 581 - 583.
23. Rennert H, Zeigler-Johnson CM, Addya K et al. Association of susceptibility alleles in ELAC2/HPC2, RNASEL/HPC1, and MSR1 with prostate cancer severity in European American and African American men. Cancer Epidemiol Biomarkers Prev 2005; 14 : 949-957.
24. Maier C, Haeusler J, Herkommer K et al. Mutation screening and association study of RNASEL as a prostate cancer susceptibility gene. Br J Cancer 2005; 92 : 1159-1164.
25. Wiklund F, Jonsson BA, Brookes AJ et al. Genetic analysis of the RNASEL gene in hereditary, familial, and sporadic prostate cancer. Clin Cancer Res 2004; 10 : 7150-7156.
26. Malathi K, Paranjape JM, Ganapathi R, Silverman RH. HPC1/RNASEL mediates apoptosis of prostate cancer cells treated with 2',5'-oligoadenylates, topoisomerase I inhibitors, and tumor necrosis factor-related apoptosis-inducing ligand. Cancer Res 2004; 64 : 9144-9151.
27. Goode EL, Stanford JL, Peters MA et al. Clinical characteristics of prostate cancer in an analysis of linkage to four putative susceptibility loci. Clin Cancer Res 2001; 7 : 2739-2749.
28. Gronberg H, Isaacs SD, Smith JR et al. Characteristics of prostate cancer in families potentially linked to the hereditary prostate cancer 1 (HPC1) locus. JAMA 1997; 278 : 1251-1254.
29. Platt N, Gordon S. Is the class A macrophage scavenger receptor (SR-A) multifunctional? - The mouse's tale. J Clin Investig 2001; 108 : 649-654.
30. Thomas CA, Li Y, Kodama T, Suzuki H et al. Protection from lethal grampositive infection by macrophage scavenger receptor-dependent phagocytosis. J Exp Med 2000; 191 : 147-156.
31. Lemaitre B, Nicolas E, Michaut L. The dorsoventral regulatory gene cassette spatzle/Toll/cactus controls the potent antifungal response in Drosophila adults. Cell 1996; 86 : 973-983. 32. O'Neill LA, Fitzgerald KA, Bowie AG. The Toll-IL-1 receptor adaptor family grows to five members. Trends Immunol 2003; 24 : 286-290.
33. Shih DM, Gu L, Xia YR et al. Mice lacking serum paraoxonase are susceptible to organophosphate toxicity and atherosclerosis. Nature 1998; 394 : 284 - 287.
34. De Winther MP, Gijbels MJ, Van Dijk KW et al. Transgenic mouse models to study the role of the macrophage scavenger receptor class A in atherosclerosis. Int J Tissue React 2000; 22 : 85-91.
35. Boiteux S, Radicella JP. The human OGG1 gene: structure, functions, and its implication in the process of carcinogenesis. Arch Biochem Biophys 2000; 377 : 1-8.
36. Coussens LM, Werb Z. Inflammation and cancer. Nature 2002; 420 : 860-867.
37. Platz EA, De Marzo AM. Epidemiology of inflammation and prostate cancer. J Urol 2004; 171: S36-40.
38. U.S. Department of Health and Human Services, Public Health Service, National Toxicology Program, 11th Report on Carcinogens, 2005. Available at: http://ntp.niells.nih.gov/ntp/roc/tocl11.html. Accessed June 6, 2005.
39. Nelson WG, De Marzo AM, Isaacs WB. Prostate cancer. N Engl J Med 2003; 349 : 366-381.
40. Dennis LK, Dawson DV. Meta-analysis of measures of sexual activity and prostate cancer. Epidemiology 2002; 13 : 72-79.
41. Dennis LK, Lynch CF, Torner JC. Epidemiologic association between prostatitis and prostate cancer. Urology 2002; 60 : 78-83.
42. Samanta M, Harkins L, Klemm K et al. High prevalence of human cytomegalovirus in prostatic intraepithelial neoplasia and prostatic carcinoma. J Urol 2003; 170 : 998-1002.
43. Zambrano A, Kalantari M, Simoneau A et al. Detection of human polyomaviruses and papillomaviruses in prostatic tissue reveals the prostate as a habitat for multiple viral infections. Prostate 2002; 53 : 263-276.
44. DeMarzo AM, Marchi VL, Epstein JI, Nelson WG. Proliferative inflammatory atrophy of the prostate: implications for prostatic carcinogenesis. Am J Pathol 1999; 155 : 1985-1992.
45. Putzi MJ, De Marzo AM. Morphologic transitions between proliferative inflammatory atrophy and highgrade prostatic intraepithelial neoplasia. Urology 2000; 56 : 828-832.
46. Nelson WG, De Marzo AM, Isaacs WB. Prostate cancer. N Engl J Med 2003; 349 : 366-381.
47. Kumar-Sinha C, Shah RB, Laxman B et al. Elevated alpha-methylacyl-CoA racemase enzymatic activity in prostate cancer. Am J Pathol 2004; 164 : 787-793.
48. Thompson IM, Goodman PJ, Tangen CM et al. The influence of finasteride on the development of prostate cancer. N Engl J Med 2003; 349(3): 215-224.
49. Andriole GL, Roehrbom C, Schulman C et al. Effect of dutasteride on the detection of prostate cancer in men with benign prostatic hyperplasia. Urology 2004; 64 : 537-541.
50. Villa LL, Costa RL, Petta CA et al. Prophylactic quadrivalent human papillomavirus (types 6, 11, 16, and 18) LI virus-like particle vaccine in young women: a randomised double-blind placebo-controlled multicentre phase II efficacy trial. Lancet Oncol 2005; 6 : 271-278.
Štítky
Dětská urologie UrologieČlánek vyšel v časopise
Urologické listy

2006 Číslo 4
- Inkontinence jako důsledek operačního zákroku na prostatě
- Při preskripci inkontinenčních pomůcek je nezbytné hlídat limity
- Na výběru inkontinenčních pomůcek záleží − ale jak se mezi nimi neztratit?
- Inkontinence ovlivňuje duševní stav nemocného – nabízejte aktivně pomoc
- Index zdraví prostaty a jeho možné využití v klinické praxi
Nejčtenější v tomto čísle
- STRAVA A KARCINOM PROSTATY
- PSA A VČASNÁ DETEKCE KARCINOMU PROSTATY
- PÁNEVNÍ LYMFADENEKTOMIE U KARCINOMU PROSTATY A JEJÍ HRANICE
- LYMFATICKÁ DRENÁŽ PROSTATY
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy