
The Nesprin Family Member ANC-1 Regulates Synapse Formation and Axon Termination by Functioning in a Pathway with RPM-1 and β-Catenin
The molecular mechanisms that underpin synapse formation and axon termination are central to forming a functional, fully connected nervous system. The PHR proteins are important regulators of neuronal development that function in axon outgrowth and termination, as well as synapse formation. Here we describe the discovery of a novel, conserved pathway that is positively regulated by the C. elegans PHR protein, RPM-1. This pathway is composed of RPM-1, ANC-1 (a Nesprin family protein), and BAR-1 (a canonical β-catenin). Nesprins, such as ANC-1, regulate nuclear anchorage and positioning in multinuclear cells. We now show that in neurons, ANC-1 regulates neuronal development by positively regulating BAR-1. Thus, Nesprins are multi-functional proteins that act through β-catenin to regulate neuronal development, and link the nucleus to the actin cytoskeleton in order to mediate nuclear anchorage and positioning in multi-nuclear cells.
Published in the journal:
. PLoS Genet 10(7): e32767. doi:10.1371/journal.pgen.1004481
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1004481
Summary
The molecular mechanisms that underpin synapse formation and axon termination are central to forming a functional, fully connected nervous system. The PHR proteins are important regulators of neuronal development that function in axon outgrowth and termination, as well as synapse formation. Here we describe the discovery of a novel, conserved pathway that is positively regulated by the C. elegans PHR protein, RPM-1. This pathway is composed of RPM-1, ANC-1 (a Nesprin family protein), and BAR-1 (a canonical β-catenin). Nesprins, such as ANC-1, regulate nuclear anchorage and positioning in multinuclear cells. We now show that in neurons, ANC-1 regulates neuronal development by positively regulating BAR-1. Thus, Nesprins are multi-functional proteins that act through β-catenin to regulate neuronal development, and link the nucleus to the actin cytoskeleton in order to mediate nuclear anchorage and positioning in multi-nuclear cells.
Introduction
The mammalian Nuclear Envelope Spectrin repeat proteins (Nesprins) (also called Syne-1/Enaptin and Syne-2/NUANCE) mediate the anchorage of nuclei in multinucleated cells such as muscle [1], [2], and mediate nuclear movement and positioning in mononuclear cells [3], [4]. The orthologs of Nesprin-1 and 2 are called MSP-300 in Drosophila and abnormal nuclear Anchorage 1 (ANC-1) in C. elegans. MSP-300 and ANC-1 also function in nuclear anchorage, and regulate positioning of organelles including mitochondria and the endoplasmic reticulum [5], [6].
Nesprin family members are attached to the nuclear envelope by the SUN proteins (SUN 1 and 2), which together compose the Linker of the Nucleoskeleton and Cytoskeleton (LINC) complex [1], [6]. A C-terminal Klarsicht/ANC-1/Nesprin homology (KASH) domain anchors Nesprin-1 and 2 in the outer nuclear membrane by binding to SUN1 and 2, which are localized to the inner nuclear membrane. C. elegans has two SUN family proteins: Uncoordinated 84 (UNC-84) (retains ANC-1 in the nuclear membrane; expressed in most somatic cells) and SUN-1 (functions in the germ line and early embryo). Tandem calponin homology domains at the N-terminus of the Nesprins mediate binding to the actin cytoskeleton.
Nesprin-1 and 2 have functions outside of their role in nuclear anchorage. Nesprin-1 and 2 regulate centrosome orientation in migrating cells and ciliogenesis [3], [4] and regulate formation and trafficking of the Golgi [7]. Importantly, mutations in Nesprin-1 and 2 are associated with numerous diseases including: autism [8], [9], cerebellar ataxia [10], Emery Dreifuss muscular dystrophy [11], cancer [12], arthrogryposis [13], and cardiomyopathy [14]. Genome-wide association studies have also identified single nucleotide polymorphisms in Nesprin-1 that are associated with schizophrenia [15] and bipolar disorder [16], [17].
Nesprin-1 and 2 perform several functions at the neuromuscular junction (NMJ) and in the central nervous system (CNS). At the NMJ, multiple nuclei are anchored in clusters directly adjacent to the postsynaptic terminal. Nesprin-1 is enriched on these postsynaptic nuclei [18], and required for their clustering [19]. Nesprin-1 is required for axon termination of motor neurons that innervate the diaphragm [2]. Nesprin-1 and 2 are also expressed in neurons of the CNS [18], where they function in neuronal migration and neurogenesis by mediating connections between the nucleus and the cytoskeleton [4], [20]. Of particular note, Nesprin-1 shows extremely strong, broad expression in the adult murine CNS, which suggests that Nesprin-1 is likely to have an important function in neurons beyond the role it plays in neural precursor migration and neurogenesis (Allen Brain Atlas: http://mouse.brain-map.org) [21]. This is consistent with the observation that an extremely small splice variant of Nesprin-1 called Candidate Plasticity Gene 2 (CPG2) regulates synaptic plasticity [22]. At present, it remains unclear if Nesprin family members play broader roles in neuronal function and development outside of their roles in very early developmental events such as neurogenesis, and neural migration.
The role of Nesprin-1 and 2 in signal transduction has begun to be explored, but remains relatively poorly understood. In vascular smooth muscle cells, small isoforms of Nesprin-2 regulate Erk MAP kinase signaling [23]. Studies using a keratinocyte cell line showed that Nesprin-2 binds to α - and β-catenin, and regulates the nuclear localization of β-catenin [24]. While these studies demonstrate that Nesprin-2 has the potential to regulate signal transduction, the broader functional consequences of these activities remain unclear. Further, it remains unknown if Nesprin-1 and/or Nesprin-2 mediate signal transduction in neurons.
Members of the Pam/Highwire/RPM-1 (PHR) protein family are large signaling proteins that include: human Pam (also called MYCBP2), murine Phr1, zebrafish Phr1 (Esrom), Drosophila Highwire, and C. elegans Regulator of Presynaptic Morphology 1 (RPM-1) [25]. The PHR proteins are important regulators of neuronal development that function in axon outgrowth and termination [26]–[28], axon guidance [29]–[31], and synapse formation [32]–[34]. PHR proteins also function in axon regeneration [35], [36] and axon degeneration following damage [37], [38].
The PHR proteins regulate several conserved signal transduction pathways [39]–[45]. However, it is poorly understood if PHR protein activity is linked to signaling by extracellular guidance cues, morphogens, or adhesion molecules. Work in Drosophila and C. elegans has shown that Highwire negatively regulates BMP signaling [46], and RPM-1 negatively regulates Slit and Netrin signaling [29]. However, it remains unclear if PHR protein activity converges with extracellular cues on common signaling targets. It is also uncertain if the PHR proteins have the ability to positively regulate, modify or enhance signals generated by extracellular cues.
Using a proteomic approach in C. elegans, we have identified ANC-1 as an RPM-1 binding protein. Similar to rpm-1, anc-1 functions in both axon termination and synapse formation. Our analysis indicates that anc-1 functions in a genetic pathway with beta-catenin/armadillo related protein 1 (bar-1) downstream of rpm-1. Further, we identify the Wnt signaling mechanisms that regulate BAR-1 to control axon termination. Our observations provide the first evidence of a link between RPM-1 signaling and the Wnt ligands that regulate neuronal development.
Results
Identification of ANC-1 as an RPM-1 binding protein
To better understand the mechanism of how RPM-1 functions in neuronal development, we previously performed a proteomic screen to identify RPM-1 binding proteins [42]. Briefly, RPM-1 fused with Green Fluorescent Protein (GFP) was transgenically expressed using the native rpm-1 promoter. This construct was purified from whole worm lysate using an anti-GFP antibody, and RPM-1 binding proteins were identified using mass spectrometry and de novo peptide sequencing. To date, our screen has successfully identified three functional RPM-1 binding proteins: Gut Granule Loss (GLO-4, a putative Rab GEF) [42], RNA Export factor 1 (RAE-1, a microtubule binding protein) [43], and Protein Phosphatase Mg2+/Mn2+ dependent 2 (PPM-2, a PP2C phosphatase) [47]. Our screen also identified the F-box Synaptic Protein 1 (FSN-1) [42], which was previously discovered using a genetic approach [45]. Importantly, GLO-4, RAE-1, and PPM-2 are not targets of RPM-1 ubiquitin ligase activity. Thus, our proteomic screen preferentially identified RPM-1 binding proteins that are not degraded by RPM-1, and are stable interaction partners.
Another RPM-1 binding protein identified in our proteomic screen was ANC-1, a gigantic protein that is composed of 8545 amino acids and has an approximate molecular weight of 956 kDa. ANC-1 consists mostly of predicted coiled regions (including six repeats that are nearly identical at the nucleotide level), two N-terminal calponin-homology (CH) domains that bind to actin, and a C-terminal KASH domain that targets ANC-1 to the nuclear envelope (Figure 1A). Previous work showed that ANC-1 is present at the nuclear envelope and in the cytoplasm of all post-embryotic somatic cells [6].

Our proteomic analysis identified 10 peptides that covered 6.3% of the total ANC-1 protein sequence (Figure S1). The majority of peptide sequence identified was from the ANC-1 specific repeats, presumably because repeat sequence is present at 6-fold molar excess over other regions of ANC-1. To confirm the biochemical interaction between ANC-1 and RPM-1 we utilized coimmunoprecipitation (coIP) from whole worm lysates generated from transgenic animals. A prior study developed polyclonal anti-ANC-1 antibodies that recognize endogenous ANC-1, which was detected as multiple high molecular weight bands in immunoblots [6]. We used these anti-ANC-1 antibodies in coIP experiments with transgenic animals expressing RPM-1::GFP (juIs58). When RPM-1::GFP was immunoprecipitated using an anti-GFP antibody, coprecipitating ANC-1 was detected as multiple high molecular weight bands (Figure 1B, juIs58). Further examples of this coIP are shown in Figure S2. Coprecipitating bands were absent or strongly reduced in intensity in juIs58; anc-1 mutants demonstrating that these bands represent endogenous ANC-1 (Figure 1B). Coprecipitating ANC-1 was not detected in precipitates from non-transgenic animals (Figure 1B, N2). Thus, ANC-1 did not bind non-specifically to the agarose beads or the anti-GFP antibody, which demonstrates that the interaction between ANC-1 and RPM-1 is specific. These biochemical results confirm that RPM-1 binds to ANC-1, or a protein complex that contains ANC-1.
anc-1 regulates synapse formation in the GABAergic motor neurons
Previous studies have shown that rpm-1 regulates synapse formation in the GABAergic dorsal D (DD) motor neurons [32]. The DD motor neurons innervate, and inhibit the dorsal muscles of the worm (Figure 2A, schematic). The presynaptic terminals of DD neurons can be visualized in living animals using the transgene juIs1, which uses a cell specific promoter (Punc-25) to express a fusion protein of Synaptobrevin-1 and GFP (SNB-1::GFP) [48]. In wild type animals, SNB-1::GFP localized to evenly sized puncta that were uniformly positioned along the dorsal nerve cord (Figure 2A). In rpm-1(ju44) mutants, SNB-1::GFP puncta in the dorsal nerve cord were abnormally aggregated (Figure 2A, arrowheads), and there were regions of the cord lacking any puncta (Figure 2A, arrows). Quantitation showed that the number of SNB-1::GFP puncta in rpm-1 mutants was significantly lower than wild-type animals (compare 11.9±0.4 SNB-1::GFP puncta/100 µm for rpm-1 with 21.9±0.4 puncta/100 µm for wild type, Figure 2B). These findings are consistent with results from previous studies [32]. Importantly, previous electron microscopy studies showed that the defects in SNB-1::GFP puncta localization in rpm-1 mutants reflect defects in synapse formation, rather than defects in the formation of presynaptic terminals or the trafficking of synaptic vesicles [32], [39]. Subsequent studies have also shown that milder defects in organization of SNB-1::GFP puncta, such as those that occur in fsn-1 mutants, are also due to defects in both pre and postsynaptic terminals [49].
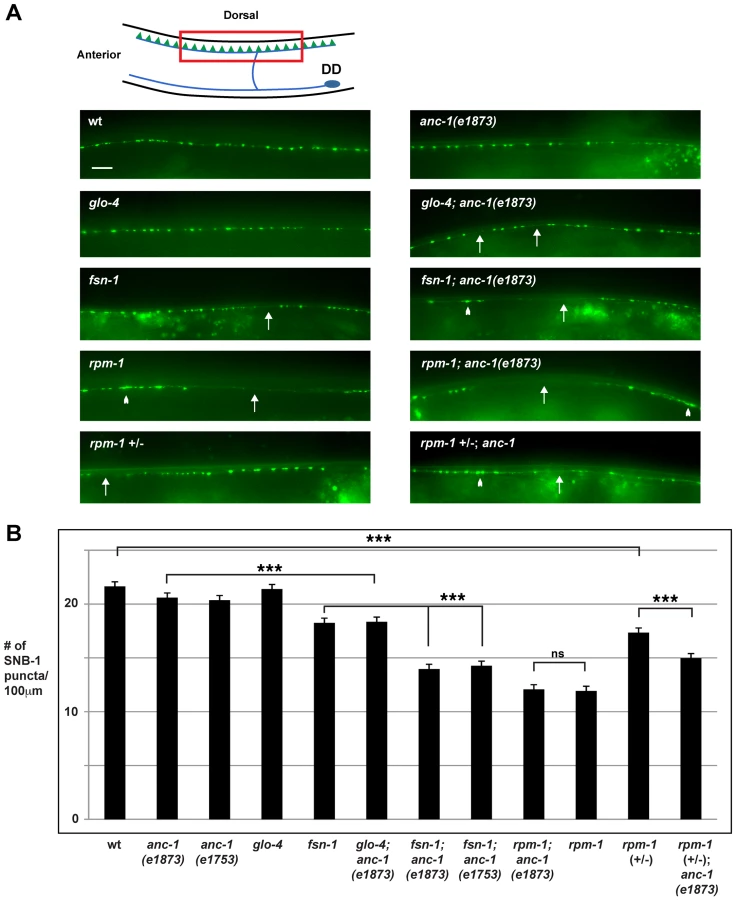
Our observation that ANC-1 binds to RPM-1 led us to hypothesize that anc-1 might function in synapse formation similar to rpm-1. To test this hypothesis, we analyzed two alleles of anc-1, e1873 and e1753. DNA sequencing confirmed that e1873 is a nonsense mutation that results in a severely truncated protein (Figure 1A) [6]. The molecular nature of the lesion in e1753 remains unknown. However, a previous study showed that antibodies raised against the repeat region of ANC-1 fail to detect all isoforms of ANC-1 greater than 175 kDa in both anc-1(e1873) and anc-1(e1753) mutants [6]. Thus, e1873 and e1753 are likely to be molecular null alleles of anc-1.
Previous studies have shown that anc-1 loss of function (lf) results in abnormal nuclear anchorage (Anc phenotype) of the nuclei in the syncytial cells that form the hypodermis of C. elegans [6]. The hypodermal nuclei can be visualized using a transgene, kuIs54, which expresses Suppressor of Activated LET-60 Ras (SUR-5)::GFP [50]. In wild-type animals, the hypodermal nuclei are anchored to the cytoskeleton, and distributed in a well organized, even pattern (Figure 1C, arrowheads). Consistent with previous results, we observed that anc-1 mutants display an Anc phenotype, in which the nuclei are no longer anchored and aggregate dramatically (Figure 1C, arrows).
With regard to synapse formation, anc-1 mutants had normal spatial distribution and number of SNB-1::GFP puncta (Figure 2A and B). We also constructed double mutants of anc-1 with two members of the RPM-1 pathway, fsn-1 and glo-4. FSN-1 is an F-box protein that binds to RPM-1 and mediates RPM-1 ubiquitin ligase activity [45]. GLO-4 binds to RPM-1, and is a putative guanine nucleotide exchange factor for a Rab pathway [42]. We found that fsn-1; anc-1 and glo-4; anc-1 double mutants had enhanced defects in synapse formation compared to single mutants (compare 14.0±0.5 puncta/100 µm for fsn-1; anc-1(e1873) with 18.3±0.3 for fsn-1, Figure 2A and B). rpm-1; anc-1 double mutants had similar defects to those observed in rpm-1 single mutants (Figure 2A and B). Additionally, we constructed rpm-1 (+/−); anc-1 animals and found enhanced defects in synapse formation compared to rpm-1 (+/−) animals (Figure 2A and B). These results are consistent with several conclusions. First, because we used null alleles, we conclude that anc-1 functions in a parallel genetic pathway to both fsn-1 and glo-4 to regulate synapse formation. Second, our observation that rpm-1; anc-1 double mutants were not enhanced, in a phenotypic assay that is not saturated [45], suggests that anc-1 functions in the same genetic pathway as rpm-1. This conclusion is further supported by our observation that rpm-1 (+/−); anc-1 animals had enhanced defects in synapse formation. Third, these results support the conclusion that ANC-1 is not negatively regulated by RPM-1 and, therefore, is unlikely to be a target of RPM-1 ubiquitin ligase activity. If this were the case, we would expect to see suppression of synapse formation defects in anc-1; rpm-1 double mutants similar to what was shown for Dual Leucine Zipper-bearing Kinase 1 (DLK-1), a known target of RPM-1 ubiquitin ligase activity [39].
The β-catenin bar-1 functions in the same genetic pathway as anc-1 to regulate synapse formation
We next sought to dissect the mechanism of how anc-1 regulates synapse formation. A previous study found that Nesprin-2 (a mammalian ortholog of ANC-1) regulates nuclear localization of β-catenin, thereby potentially regulating canonical Wnt signaling [24]. When Wnt signaling is not active, β-catenin is degraded; when Wnt signaling is activated, β-catenin accumulates, enters the nucleus, and interacts with T Cell Specific Transcription Factor (TCF)/Lymphoid Enhancer Binding Factor (LEF) family transcription factors to promote gene expression [51]. Previous work in C. elegans has shown that Wnt signaling regulates neuronal development [52]–[54]. In addition, Abnormal Cell Lineage 23 (LIN-23), an F-box protein that negatively regulates β-catenin in C. elegans, regulates the abundance of postsynaptic glutamate receptors in the ventral nerve cord [55], as well as axon termination in mechanosensory neurons [56]. Both of these developmental events are also regulated by rpm-1 [27], [57]. Based on this prior work, we hypothesized that anc-1 may function as a genetic link between rpm-1 and β-catenin signaling.
To test this hypothesis, we started by determining if β-catenin regulates synapse formation in the DD motor neurons. In C. elegans, there are four β-catenins that have diverged to perform separate functions [58], [59]. The canonical Wnt pathway operates through the β-catenin homolog BAR-1 and a single TCF homolog, Posterior Pharynx Defect 1 (POP-1) [60]. To test the role of bar-1 in synapse formation, we analyzed a null allele of bar-1, ga80 [61]. Consistent with a prior study, we observed that small, consistent sections of the dorsal cord were absent in bar-1 (lf) mutants (data not shown) [62]. However, we were able to analyze synapse formation in sections of the dorsal cord that formed normally. bar-1 mutants showed a distribution and number of SNB-1::GFP puncta that were similar to wild-type animals (Figure 3A and B). In contrast, fsn-1; bar-1 double mutants showed enhanced defects in synapse formation (compare 14.1±0.4 SNB-1::GFP puncta/100 µm for fsn-1; bar-1 with 18.3±0.3 puncta/100 µm for fsn-1, Figure 3A and B). The enhanced phenotype in fsn-1; bar-1 double mutants was similar to what was observed for fsn-1; anc-1 double mutants (Figure 3B). This result suggested that anc-1 and bar-1 may function in the same genetic pathway. To test this possibility, we constructed double and triple mutants between bar-1, anc-1 and fsn-1. anc-1; bar-1 double mutants were not enhanced compared to bar-1 and anc-1 single mutants (Figure 3A and B). Likewise, fsn-1; anc-1; bar-1 triple mutants were not enhanced compared to fsn-1; bar-1 or fsn-1; anc-1 double mutants (Figure 3A and B).

To determine if BAR-1 regulates synapse formation by acting through a canonical signaling pathway that includes the TCF transcription factor POP-1, we analyzed the role of pop-1 in synapse formation. Because null alleles of pop-1 are lethal, we opted to analyze a hypomorphic allele of pop-1, q645. The POP-1 protein produced by q645 shows reduced interaction with BAR-1 due to a mutation in the β-catenin binding domain [63]. Similar to our findings with bar-1, pop-1 (lf) animals were largely wild type, but pop-1; fsn-1 double mutants had enhanced defects in synapse formation that were of similar severity to fsn-1; bar-1 double mutants (compare 14.6±0.4 SNB-1::GFP puncta/100 µm for pop-1; fsn-1 with 18.2±0.3 puncta/100 µm for fsn-1, Figure 3A and B).
To determine if bar-1 functions in the same pathway as rpm-1, we constructed bar-1; rpm-1 double mutants. We observed no change in the severity of synapse formation defects in bar-1; rpm-1 double mutants compared to rpm-1 single mutants (Figure 3B).
As a whole, our results support several conclusions. First, anc-1, bar-1 and rpm-1 function in the same genetic pathway to regulate synapse formation. Second, the anc-1/bar-1 pathway acts in parallel to fsn-1 to regulate synapse formation. Finally, bar-1 is likely to regulate synapse formation by functioning through a canonical Wnt signaling pathway that includes the TCF transcription factor pop-1.
anc-1 regulates axon termination in the mechanosensory neurons
Previous work showed that rpm-1 regulates axon termination in the mechanosensory neurons that sense soft touch [27]. The posterior lateral microtubule (PLM) and the anterior lateral microtubule (ALM) mechanosensory neurons are an excellent system in which to study axon termination. Each PLM and ALM neuron extends a single axon that terminates extension at a precise anatomical location [64]. In addition, these neurons are easily visualized using a transgene (muIs32) that expresses GFP specifically in the mechanosensory neurons [65].
C. elegans contains two PLM neurons that sense soft touch in the posterior of the animal's body. Each PLM neuron has a single axon that terminates extension prior to the cell body of the ALM neuron (Figure 4A, schematic). In rpm-1 (lf) mutants, PLM axons fail to terminate extension properly, grow past the ALM cell body and hook towards the ventral side of the animal (Figure 4A) [27], [42]. This defect, which we refer to as a “hook”, is highly penetrant in rpm-1 mutants (87.6±1.5%, Figure 4B). Likewise, rpm-1 mutants have highly penetrant axon termination defects in the ALM neurons (Figure S3A) [27], [42].
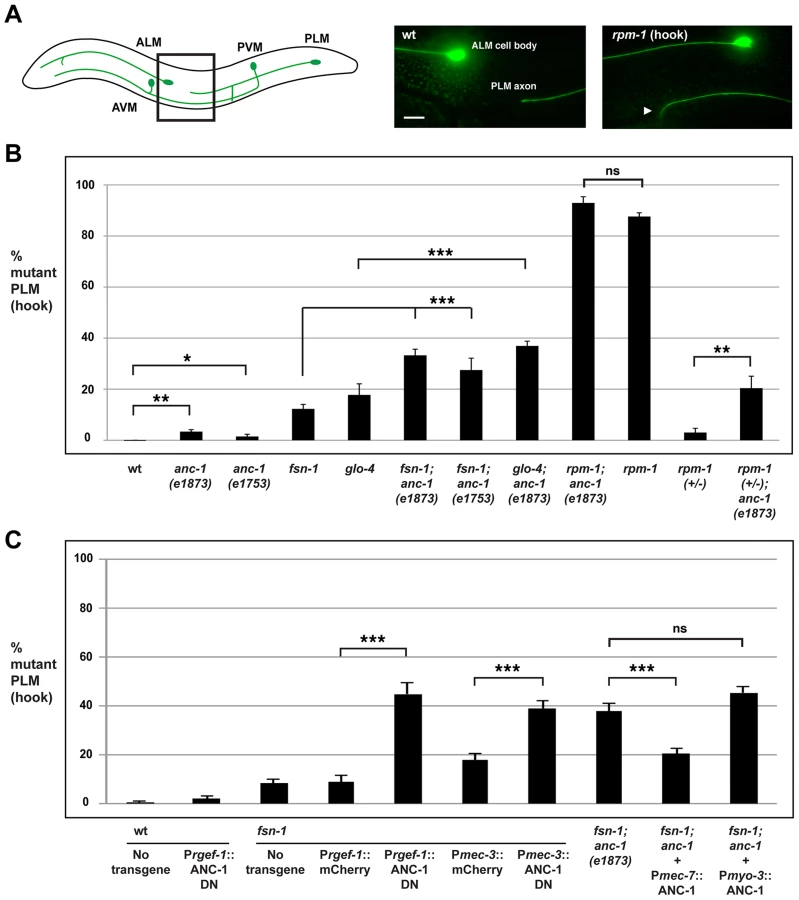
To determine whether anc-1 also functions in axon termination, we analyzed anc-1 animals as well as double mutants of anc-1 and members of the rpm-1 signaling pathway. Both anc-1(e1873) and anc-1(e1753) showed PLM hook defects that were significant compared to wild-type animals, but occurred with extremely low penetrance compared to rpm-1 mutants (3.4±0.8% for anc-1(e1873) and 1.9±0.8% for anc-1(e1753), Figure 4B). fsn-1; anc-1 double mutants showed enhanced penetrance of defects compared to single mutants (compare 33.2±2.4 for fsn-1; anc-1(e1873) and 27.5±4.6% for fsn-1; anc-1(e1753) with 9.1±1.1% for fsn-1, Figure 4B). Likewise, glo-4; anc-1 double mutants also showed enhanced penetrance of defects compared to single mutants (compare 36.9±1.9% for glo-4; anc-1(e1873) with 17.7±4.3% for glo-4, Figure 4B). We observed similar results when the function of anc-1 was analyzed with regard to axon termination in the ALM neurons (Figure S3A). These results demonstrate that anc-1 regulates axon termination by functioning in a parallel pathway to both fsn-1 and glo-4.
We also analyzed rpm-1; anc-1 double mutants, which had the same penetrance of defects as rpm-1 single mutants. This result suggests that anc-1 functions in the same genetic pathway as rpm-1 (Figure 4B). Consistent with this conclusion, we observed enhanced axon termination defects in both the PLM and the ALM neurons of rpm-1 (+/−); anc-1 animals (Figure 4B and Figure S3A).
Having established that anc-1 regulates axon termination by functioning in the same pathway as rpm-1, we wanted to test if anc-1 functions cell autonomously in the PLM neurons to regulate axon termination. Due to the enormous size of the anc-1 gene, our first approach was to transgenically overexpress a dominant negative fragment of ANC-1. Previous work by Starr and Han showed that a C-terminal fragment of ANC-1 that contains the KASH domain (see Figure 1A, C-terminal domain aa 8200–8546) acts as a dominant negative by blocking binding of endogenous ANC-1 to the SUN domain protein UNC-84, thereby preventing ANC-1 localization to the nuclear envelope [6]. We engineered fsn-1 mutants with transgenic extrachromosomal arrays that used a strong, pan-neuronal promoter (Prgef-1) to overexpress either the dominant negative ANC-1, or mCherry as a negative control. We observed enhanced penetrance of axon termination defects in fsn-1 mutants expressing the ANC-1 dominant negative (44.9±4.6%) compared to animals expressing mCherry (8.9±2.6%, Figure 4C). Transgenic overexpression of dominant negative ANC-1 in wild-type animals did not give a significant phenotype (Figure 4C). These results are consistent with ANC-1 functioning in neurons to regulate axon termination.
To more directly address cell autonomy, we used a cell specific promoter, Pmec-3, to transgenically overexpress the ANC-1 dominant negative specifically in the mechanosensory neurons. In fsn-1 mutants, when the mec-3 promoter was used to overexpress the ANC-1 dominant negative we observed enhanced penetrance of axon termination defects (38.8±3.3%) compared to fsn-1 mutants that transgenically overexpressed mCherry (17.9±2.5%, Figure 4C). Thus, transgenic overexpression of dominant negative ANC-1, specifically in the mechanosensory neurons, enhances fsn-1 (lf) to levels that are similar to what we observed in fsn-1; anc-1 double mutants (Figure 4C).
To provide further evidence that anc-1 functions cell autonomously in the mechanosensory neurons, we generated fsn-1; anc-1 double mutants that carried a transgenic extrachromosomal array containing an anc-1 mini-gene and a promoter, Pmec-7, that drives expression specifically in the mechanosensory neurons. In this case, we observed a strong, but partial rescue of the enhanced axon termination defects in fsn-1; anc-1 double mutants (compare 20.5±1.9% for fsn-1; anc-1+Pmec-7::ANC-1 with 37.6±3.2% for fsn-1; anc-1, Figure 4C). Transgenic expression of ANC-1 in the surrounding muscle cells using the myosin 3 (myo-3) promoter did not rescue defects in fsn-1; anc-1 double mutants (Figure 4C).
Our transgenic analysis supports several conclusions. First, the dominant negative and rescue experiments demonstrate that ANC-1 functions cell autonomously in the mechanosensory neurons to regulate axon termination. Second, our results with the dominant negative indicate that ANC-1 needs to be associated with the nuclear envelope via its C-terminal KASH domain in order to regulate axon termination. Finally, the lesion in anc-1 causes the enhanced axon termination defects observed in fsn-1; anc-1 double mutants.
bar-1 functions through pop-1 to regulate axon termination in the mechanosensory neurons
Our genetic analysis indicated a role for bar-1 in synapse formation, and we next sought to determine if bar-1 also functions in axon termination of the mechanosensory neurons, similar to anc-1. While axon termination defects were observed at very low penetrance in the PLM neurons of bar-1 (lf) mutants, these defects did not reach statistical significance (Figure 5A). However, the penetrance of axon termination defects was enhanced in bar-1; fsn-1 double mutants (25.8±2.5% hook) compared to fsn-1 single mutants (9.1±1.1%, Figure 5A). This level of enhancement was similar to what we observed in fsn-1; anc-1 double mutants, which suggested that bar-1 might function in the same pathway as anc-1 to regulate axon termination. To test this, we generated anc-1; bar-1 double mutants and fsn-1; anc-1; bar-1 triple mutants. As shown in Figure 5A, anc-1; bar-1 double mutants did not show enhanced PLM axon termination defects. Similarly, fsn-1; anc-1; bar-1 triple mutants were not enhanced compared to fsn-1; bar-1 double mutants and fsn-1; anc-1 double mutants (Figure 5A). Similar results were observed for bar-1 regarding axon termination in the ALM neurons (Figure S3B). Thus, bar-1 and anc-1 regulate axon termination by functioning in the same genetic pathway.

Given that BAR-1 regulates the transcription factor POP-1, we also tested if pop-1 functions in axon termination. Similar to bar-1, pop-1 (lf) mutants had axon termination defects that occurred with very low penetrance, and pop-1; fsn-1 double mutants were enhanced compared to single mutants (compare 47.7±4.3% for pop-1; fsn-1 with 9.1±1.1% for fsn-1). Importantly, pop-1; bar double mutants were not enhanced consistent with pop-1 and bar-1 functioning in the same genetic pathway (Figure 5A). Consistent with the results from PLM neurons, ALM axon termination defects were enhanced in pop-1; fsn-1 double mutants, but failed to show enhancement in pop-1; bar-1 double mutants (Figure S3B). These results are consistent with bar-1 functioning through pop-1 to regulate axon termination.
To address if bar-1 and rpm-1 function in the same genetic pathway to regulate axon termination, we generated rpm-1; bar-1 double mutants. As shown in Figure 5A, rpm-1; bar-1 double mutants had similar penetrance of hook defects to rpm-1 single mutants.
Given that ANC-1 functions cell autonomously in the mechanosensory neurons, we also wanted to determine if BAR-1 functions cell autonomously. To do so, we generated fsn-1; bar-1 double mutants that transgenically express BAR-1 using a cell specific promoter. We performed our analysis on fsn-1; bar-1 double mutants because the penetrance of axon termination defects were higher than in bar-1 single mutants. When BAR-1 was expressed using a mechanosensory neuron specific promoter, Pmec-7, the axon termination defects in fsn-1; bar-1 double mutants were significantly reduced (compare 12.1±2.0% for fsn-1; bar-1+Pmec-7::BAR-1 with 29.6±3.1% for fsn-1; bar-1, Figure 5C). In contrast, expression of BAR-1 with the myo-3 promoter (expressed in body wall muscles surrounding the PLM axon) did not rescue defects in fsn-1; bar-1 double mutants (Figure 5C).
Overall, these findings support several conclusions. First, bar-1 regulates axon termination in the mechanosensory neurons by functioning in the same genetic pathway as rpm-1, anc-1, and pop-1. Second, bar-1 functions in a parallel genetic pathway to fsn-1. Finally, bar-1 regulates axon termination by functioning cell autonomously in the mechanosensory neurons.
bar-1 functions downstream of anc-1 and rpm-1 to regulate axon termination
Our genetic analysis indicated that rpm-1, anc-1 and bar-1 function in the same pathway. We next sought to determine whether bar-1 functions up or downstream of anc-1 and rpm-1. We chose to perform our epistasis analysis using bar-1 for two reasons. First, the existing knowledge of how BAR-1 functions allowed the design and implementation of highly informative experiments. Second, a previous study in mammalian cells on Nesprin-2 and β-catenin (the orthologs of ANC-1 and BAR-1, respectively) suggested that bar-1 might function downstream of anc-1 [24]. Therefore, axon termination defects caused by loss of function in rpm-1 and enhanced axon termination defects in fsn-1; anc-1 double mutants might be due, in part, to decreased BAR-1 activity. If this model is correct, we anticipated that excess BAR-1 activity might suppress the axon termination defects caused by loss of function in rpm-1, and suppress the enhanced axon termination defects in fsn-1; anc-1 double mutants. We chose to address this question using a genetic approach that utilized a loss of function mutation in apc related 1 (apr-1), as GFP expressed by muIs32 was greatly reduced (preventing proper visualization of mechanosensory neurons) when BAR-1 was transgenically overexpressed at high levels (data not shown).
APR-1 is the C. elegans ortholog of human Adenomatous Polyposis Coli (APC) [66]. In the vertebrate canonical Wnt signaling pathway, APC forms a complex with the scaffold protein Axin and Glycogen Synthase Kinase 3β (GSK3β) to phosphorylate β-catenin and target it for destruction [51]. APR-1 interacts with the functional axin ortholog Polyray 1 (PRY-1), and has been shown to negatively regulate the β-catenin BAR-1 during vulva development and neuroblast migration [67]. Thus, APR-1 can negatively regulate BAR-1 signaling, and apr-1 mutants are likely to have increased levels of BAR-1 which is consistent with findings from other organisms on Wnt signaling and APC function.
Because axon termination defects in anc-1 mutants occur with relatively low penetrance, we chose to analyze fsn-1; anc-1 double mutants, which have enhanced penetrance of defects. As shown in Figure 5B, fsn-1; anc-1; apr-1 triple mutants had significantly reduced penetrance of hook defects (21.4±1.9%) compared to fsn-1; anc-1 double mutants (33.2±2.4%). Importantly, fsn-1; apr-1 double mutants did not show reduced penetrance of defects (Figure 5B). These results show that increased BAR-1 activity suppresses anc-1 (lf), which is consistent with bar-1 functioning downstream of anc-1.
To test if bar-1 functions downstream of rpm-1 we took a similar genetic approach using apr-1. As shown in Figure 5B, rpm-1; apr-1 double mutants showed suppressed penetrance of axon termination defects when compared to rpm-1 single mutants (compare 65.1±3.9% hook for rpm-1; apr-1 with 87.6±1.5% for rpm-1). These results demonstrate that bar-1 is likely to function downstream of rpm-1.
unc-84 regulates axon termination
Our observation that transgenic overexpression of the ANC-1 KASH domain in fsn-1 mutants results in enhanced axon termination defects suggested that ANC-1 needs to be at the nuclear envelope in order to regulate axon termination (Figure 4C). To further support this concept, we examined the role of UNC-84 in axon termination. UNC-84 is a conserved SUN domain protein that is localized to the inner nuclear membrane. UNC-84 binds to ANC-1, thereby tethering ANC-1 in the nuclear envelope and mediating formation of the LINC complex [6], [68]. Therefore, we hypothesized that if ANC-1 needs to be localized to the nuclear membrane to function in axon termination, then unc-84 should regulate axon termination similar to anc-1.
In unc-84 (lf) mutants we observed a very mild penetrance of hook defects, similar to anc-1 (lf) animals (Figure 6A). fsn-1; unc-84 double mutants had enhanced penetrance of defects compared to fsn-1 single mutants (compare 24.9±3.1% hook for fsn-1; unc-84 with 9.1±1.1% for fsn-1, Figure 6A). In contrast, anc-1; unc-84 double mutants did not show a significant increase in penetrance compared to single mutants (Figure 6A). Likewise, the penetrance of defects in fsn-1; anc-1; unc-84 triple mutants was not increased compared to fsn-1; anc-1 and fsn-1; unc-84 double mutants (Figure 6A). The unc-84 allele we used, e1410, is a hypomorph that results in loss of function in the SUN domain of UNC-84, and has nuclear anchorage defects [68]. Previous studies have shown that the SUN domain of UNC-84 is required for recruitment of ANC-1 to the nuclear envelope [6]. Thus, unc-84(e1410) is predicted to lack nuclear localization of ANC-1. This is consistent with our genetic data showing that loss of function in unc-84 does not enhance loss of function in anc-1. Thus, unc-84 regulates axon termination by functioning in the same genetic pathway as anc-1, and in a parallel pathway to fsn-1. These findings are also consistent with ANC-1 regulating axon termination by functioning at the nuclear envelope.
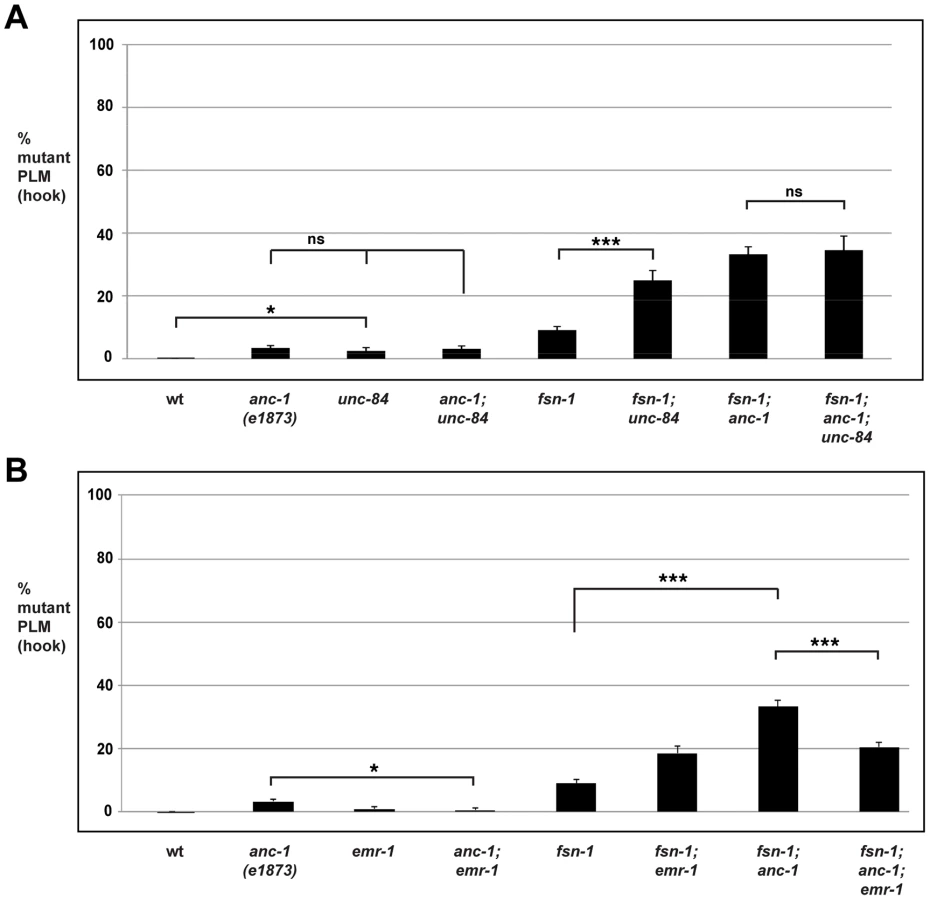
anc-1 functions through emr-1 to regulate axon termination
It was previously shown that Emerin binds to and facilitates the nuclear export of β-catenin thereby antagonizing β-catenin function [69]. Emerin also binds to Nesprin-1 and Nesprin-2 [70], [71]. These prior observations suggested that ANC-1 might regulate BAR-1 by functioning through Emerin homolog 1 (EMR-1) in C. elegans. To test this, we analyzed the genetic relationship between anc-1 and emr-1 using gk119, an allele that deletes the entire emr-1 coding sequence [72]. Although PLM axon termination defects occurred with very low penetrance in anc-1 single mutants, defects in anc-1; emr-1 double mutants were significantly suppressed (compare 3.4±0.8% hook for anc-1 with 0.6±0.6% for anc-1; emr-1, Figure 6B). Consistent with this result, we also observed that the enhanced penetrance of axon termination defects in fsn-1; anc-1 double mutants was suppressed in fsn-1; anc-1; emr-1 triple mutants (compare 33.2±2.2% hook for fsn-1; anc-1 with 20.5±1.5% for fsn-1; anc-1; emr-1, Figure 6B). Notably, fsn-1; emr-1 double mutants were mildly enhanced rather than being suppressed (Figure 6B). This result explains why fsn-1; anc-1; emr-1 triple mutants were only suppressed to the level of defect present in fsn-1; emr-1 double mutants. Our findings demonstrate that anc-1 and emr-1 function in the same genetic pathway, and that ANC-1 is a negative regulator of EMR-1.
Wnt signaling regulates PLM and ALM axon termination
Our observation that RPM-1 and ANC-1 function through BAR-1, which mediates canonical Wnt signaling, prompted us to test which Wnt ligands regulate axon termination in the ALM and PLM mechanosensory neurons. To do so, we analyzed loss of function alleles in four Wnt ligands: c. elegans wnt family 1 (cwn-1), cwn-2, egg laying defective 20 (egl-20), and lin-44. The fifth Wnt, more of ms 2 (mom-2), was not analyzed because loss of function is lethal.
With regard to the ALM neurons, loss of function in individual Wnt ligands did not result in significant defects in axon termination (Figure S3C). Analysis of double mutants with fsn-1 showed that only fsn-1; cwn-2 double mutants had enhanced axon termination defects (Figure S3C).
In the PLM neurons, the situation was more complex. cwn-1 and cwn-2 single mutants did not show defects in PLM axon termination. While fsn-1; cwn-2 double mutants failed to show enhancement, fsn-1; cwn-1 double mutants showed a small but significant enhancement (compare 13.6±1.3% hook defects for fsn-1; cwn-1 with 9.1±1.1% for fsn-1, Figure 7). In the case of egl-20, single mutants did not have a significant defect, but mild enhancer effects were observed in fsn-1; egl-20 double mutants (compare 18.8±2.3% hook defects for fsn-1; egl-20 with 9.1±1.1% for fsn-1, Figure 7). Consistent with previous work, we observed that axon polarization was abnormal in the PLM neurons of lin-44 mutants (data not shown) [54], [73]. However, axon polarization defects were not completely penetrant in lin-44 mutants, which allowed us to analyze axon termination in neurons with normal polarity. Using this approach, we observed that lin-44 mutants had significant defects in PLM axon termination (compare 18.9±4.7% hook for lin-44 with 0% defects for wild-type, Figure 7). Further, fsn-1; lin-44 double mutants were significantly enhanced compared to fsn-1 or lin-44 single mutants (compare 55.1±4.8% hook for fsn-1; lin-44 with 18.9±4.7% for lin-44, Figure 7). Consistent with LIN-44 functioning through BAR-1 to regulate axon termination, we observed no further enhancement of defects in fsn-1; lin-44; bar-1 triple mutants (Figure 7). Notably the more posterior the location of expression for a Wnt ligand the stronger the phenotypes and/or enhancer effects observed, e.g. lin-44 mutants had the strongest PLM axon termination defects and enhancer effects, and LIN-44 is the most posteriorly expressed Wnt [74].

Thus, because the ALM and the PLM neurons terminate axon extension in anatomically distinct locations, different Wnt ligands control this process for each type of neuron. Our results show that only cwn-2 regulates ALM axon termination. In contrast, a combination of cwn-1, egl-20 and lin-44 regulate PLM axon termination with lin-44 functioning most prominently.
Discussion
While the role of the Nesprins in nuclear positioning and movement is well established, mounting evidence has suggested additional roles for this protein family. High expression of Nesprin-1 in the adult brain, links to synaptic plasticity, and an association with neurological conditions suggest that the Nesprins may function in neuronal development. Here, we show that ANC-1 binds to the PHR protein RPM-1, a large signaling protein involved in numerous developmental events in neurons. Our genetic analysis indicates that ANC-1 functions cell autonomously to regulate axon termination in the mechanosensory neurons, and synapse formation in the GABAergic motor neurons. We also show that ANC-1 functions by positively regulating BAR-1, the β-catenin isoform that functions in canonical Wnt signaling (Figure 8). Our study reveals a novel role for ANC-1 in intracellular signaling and neuronal development. Importantly, we have also identified the ANC-1/BAR-1 pathway as a new mechanism by which RPM-1, and possibly other PHR proteins, regulate axon termination and synapse formation.

ANC-1 mediates RPM-1 function
Our proteomic screen for RPM-1 binding proteins identified the nuclear anchorage protein ANC-1, which was confirmed using coIP. Consistent with these findings, our genetic analysis indicated that anc-1 and rpm-1 function in the same genetic pathway to regulate synapse formation in the GABAergic motor neurons, and axon termination in the mechanosensory neurons. Transgenic analysis indicated that anc-1 functions cell autonomously in the mechanosensory neurons to regulate axon termination, similar to rpm-1. Our observation that defects in anc-1; rpm-1 double mutants were not suppressed also suggests that RPM-1 positively regulates ANC-1.
While we were unable to provide definitive evidence that anc-1 functions downstream of rpm-1, our analysis indicates that this is likely to be the case. We show that bar-1 functions downstream of both anc-1 and rpm-1, and that these three genes function in the same genetic pathway. Because similar enhancer effects are observed in fsn-1; bar-1 and fsn-1; anc-1 double mutants, and the penetrance of these defects is less than rpm-1 (lf) mutants, it is probable that anc-1, like bar-1, functions downstream of rpm-1.
The PHR protein family is highly conserved with orthologs in Drosophila, zebrafish and mice [25]. To date, all of the proteins identified in our proteomic screen for RPM-1 binding proteins (including FSN-1, GLO-4, and RAE-1) are evolutionarily conserved [42], [43]. Therefore, RPM-1 and its downstream signaling pathways are likely to function through conserved mechanisms. ANC-1 is also conserved with orthologs in Drosophila (MSP-300) and mammals (Nesprin-1 and 2). Thus, the function of ANC-1 in axon termination and synapse formation during development is also likely to be conserved. This is supported by the observation that the branches of phrenic nerves are overgrown in Nesprin-1−/− knockout mice, indicating possible axon termination defects [2]. Further addressing if the role of ANC-1 in axon and synapse development is evolutionarily conserved remains an important goal.
ANC-1 functions through the β-catenin BAR-1
Our genetic results showed that the β-catenin bar-1 functions in synapse formation in the GABAergic motor neurons, and axon termination in the mechanosensory neurons. Prior studies showed that BAR-1 regulates glutamate receptor trafficking, and axon extension in motor neurons [55], [75]. Our results show that BAR-1 plays a more expansive role in neuronal development than originally thought. We also provide significant insight into the mechanism of how BAR-1 is regulated by showing that a novel pathway containing RPM-1 and ANC-1 functions upstream of BAR-1. Our functional genetic and transgenic findings are consistent with a prior study on a keratinocyte cell line, which showed that Nesprin-2 regulates the nuclear localization of β-catenin [24]. Our results do not rule out the possibility that ANC-1 regulates the ubiquitination or phosphorylation of BAR-1. We used epifluorescent microscopy to investigate if BAR-1::GFP localization to the nucleus was altered in anc-1 mutants, but found no obvious changes (data not shown). However, it is possible that detecting such changes may require more sensitive methods.
In the canonical Wnt signaling pathway, BAR-1 activates POP-1 (the C. elegans TCF/LEF transcription factor) [58]. Our observation that defects in axon termination and synapse formation were enhanced in both fsn-1; pop-1 and fsn-1; bar-1 double mutants, and that defects were not enhanced in pop-1; bar-1 double mutants is consistent with bar-1 and pop-1 functioning in the same genetic pathway. Because fsn-1; pop-1 double mutants displayed more penetrant defects than fsn-1; bar-1 double mutants, it is possible that another β-catenin may also function through POP-1 to regulate axon termination. Humpback 2 (HMP-2) is an unlikely candidate as it functions in a cadherin-catenin complex and does not act through POP-1 [60]. Symmetrical Sister Cell and Gonad Defect 1 (SYS-1) and Worm Armadillo 1 (WRM-1) are plausible candidates as they regulate transcriptional activation and nuclear export of POP-1, respectively [76], [77]. In the case of synapse formation in the GABAergic motor neurons the situation is simpler. Phenotypes were similar in fsn-1; pop-1 and fsn-1; bar-1 double mutants suggesting that only BAR-1 is likely to function through POP-1 to regulate synapse formation in GABAergic motor neurons.
In vertebrates, β-catenin plays a broad and important role in neurons by regulating a range of processes including: neuronal differentiation [78], synaptic vesicle assembly [79], dendrite morphogenesis and plasticity [80], [81], neurite extension [82], and axon arborization and targeting in retinal ganglion cells (RGC) [83]. β-catenin also regulates the morphology of the NMJ and phrenic nerve growth [84]. Notably similar to the function of β-catenin, Phr1 regulates RGC axon arborization and targeting [31] and NMJ morphology [30], [33], and both Phr1 and Nesprin-1 regulate axon growth in phrenic nerves [2], [33]. While these studies hinted at a possible functional link between the PHR proteins, Nesprins and β-catenin, our finding that RPM-1, ANC-1 and BAR-1 function in the same pathway provides the first mechanistic explanation for these phenotypic relationships in mammals. Thus, the functional relationship between RPM-1, ANC-1 and BAR-1 is likely to be evolutionarily conserved. Further, our study outlines a signaling network that links two central and important regulators of neuronal development, the PHR proteins and β-catenin, through the function of ANC-1.
ANC-1 regulates axon termination by functioning at the nuclear envelope
ANC-1 regulates nuclear anchorage by binding to the nuclear envelope via its C-terminal KASH domain, and binding to the actin cytoskeleton via its N-terminal calponin homology domains [6]. The SUN domain protein UNC-84 mediates binding of ANC-1 to the nuclear envelope. Several of our findings demonstrate that ANC-1 needs to be anchored to the nuclear envelope in order to regulate axon termination. First, we found that transgenic overexpression of dominant negative ANC-1, which acts by inhibiting recruitment of endogenous ANC-1 to the nuclear envelope [6], enhances the axon termination defects caused by fsn-1 (lf). Second, loss of function in either anc-1 or unc-84 enhances the axon termination defects caused by fsn-1 (lf). Third, our results demonstrate that anc-1 and unc-84 function in the same genetic pathway to regulate axon termination. Finally, axon termination defects in anc-1 single mutants and fsn-1; anc-1 double mutants are suppressed by loss of function in emr-1, a nuclear envelope associated protein. Collectively, these findings support the conclusion that ANC-1 regulates axon termination by functioning at the nuclear envelope.
Previous studies showed that RPM-1 is localized to the perisynaptic zone of presynaptic terminals [32], [85]. However, a transgenically expressed fusion protein of RPM-1 and GFP that rescues rpm-1 (lf) phenotypes is also found at low levels in the cell body and excluded from the nucleus of the mechanosensory neurons (Opperman and Grill, in press) and the motor neurons (Figure S4) [32]. In addition, RPM-1 binds to RAE-1, a protein that localizes to the nuclear envelope as well as presynaptic terminals [43]. These prior observations combined with our findings here that ANC-1 binds to RPM-1, that both genes function in the same pathway, and that ANC-1 regulates axon termination by functioning at the nucleus suggest that RPM-1 may play a novel signaling function in the neuronal cell body, possibly at the nuclear envelope. This possibility is further supported by immunohistochemistry in mammals, which has shown that the rat ortholog of RPM-1 (called MYCBP2 or Pam) is present in the cell bodies of neurons in the spinal cord and throughout the brain [86], [87].
RPM-1/ANC-1 signaling and Wnt function
An emerging question is whether the PHR proteins, or the signaling pathways they control, are regulated or integrated with signals originating from outside the cell. Extracellular guidance cues, adhesion molecules and morphogens, such as Wnts, play roles in both axon guidance and synapse formation [88]. A previous study in zebrafish noted that loss of function in Phr1 and Wnt4a/Ryk causes abnormal axon stopping at the medial habenula hinting at a possible link between PHR protein function and Wnt signaling [89]. We now provide genetic evidence linking the PHR protein RPM-1 to the canonical β-catenin BAR-1 and Wnt signaling. Specifically, we show that several Wnt ligands regulate axon termination by functioning coordinately with FSN-1, a key component of RPM-1 signal transduction. In the anterior of the animal, we found that only CWN-2 regulates ALM axon termination. Our finding is consistent with previous studies showing that CWN-2 regulates axon polarization in ALM neurons, and anterior axon guidance in neurons of the nerve ring [54], [90]. Further, CWN-2 regulation of ALM axon termination is consistent with CWN-2 being the most anteriorly expressed Wnt ligand [74]. With regard to the PLM neurons in the posterior of the animal, three Wnts are involved in decreasing order of importance: LIN-44, EGL-20, and CWN-1. These findings are consistent with several previous observations. First, this same combination of Wnt ligands was shown to regulate axon polarization in the PLM neurons [54], [73]. Second, the hermaphrodite specific motor neuron (HSN) cell bodies are in roughly the same relative anterior-posterior location as the sites of PLM axon termination, and the same three Wnt ligands we have found that control PLM termination also regulate HSN migration [53].
Our genetic analysis indicates that RPM-1 and ANC-1 positively regulate BAR-1. In mammals, Nesprin-2 binds to a complex of α - and β-catenin [24]. Thus, RPM-1 may mediate formation of an ANC-1/BAR-1 complex that regulates nuclear levels of BAR-1 (Figure 8). In vitro biochemical and tissue culture experiments have shown that Nesprin-1 and Nesprin-2 bind to the nuclear membrane protein Emerin, and Emerin regulates nuclear export of β-catenin [69], [71]. We have found that loss of function in emr-1 suppressed anc-1 (lf) in the context of axon termination, which is consistent with ANC-1 inhibiting EMR-1. Thus, excess EMR-1 activity in anc-1 mutants presumably results in increased nuclear export of BAR-1, thereby mimicking loss of function in bar-1. Taken collectively, prior findings and our results here are consistent with a model in which canonical Wnt signaling controls BAR-1 protein levels and nuclear import, while RPM-1 and ANC-1 function in a linear pathway to inhibit EMR-1 and nuclear export of BAR-1 (Figure 8).
Materials and Methods
Genetics
The N2 isolate of C. elegans was propagated using standard procedures. Alleles used in this study included; anc-1(e1873), anc-1(e1753), apr-1(ok2970), pop-1(q645), fsn-1(gk429), glo-4(ok623), rpm-1(ju44), bar-1(ga80), unc-84(e1410), emr-1(gk119), cwn-1(ok546), cwn-2(ok895), lin-44(n1792), and egl-20(hu120). The strain MH1870, which contains the transgene kuIs54, and anc-1(e1873) were kind gifts from Dr. Daniel Starr. All mutants were constructed using standard procedures, and were confirmed using PCR or by the associated visible phenotypes. Heterozygous analysis with rpm-1 was done by linking rpm-1(ju44) with dpy-11(e224), and using unc-42(e270) as a balancer. Other homozygous alleles were isolated as necessary and non-dpy non-unc (rpm-1 dpy-11/unc-42) animals were scored. The transgenic strains used in this study were: muIs32 [Pmec-7GFP], juIs1 [Punc-25SNB-1::GFP], and kuIs54 [Psur-5SUR-5::GFP].
Transgenics
Transgenic animals were generated using standard microinjection procedures. Transgenes were constructed by injection of plasmid DNA or DNA generated by PCR with plasmid encoding Pttx-3RFP (50 ng/µL) and pBluescript (50 ng/µL). Dominant negative ANC-1 was cloned as a genomic fragment (bp 36523–37778). For rgef-1 promoter lines, the plasmid pBG-GY360 was amplified by PCR and injected at 10 ng/µL. For the mec-3 promoter lines, the plasmid pBG-GY370 was amplified by PCR and injected at 5 ng/µL. The anc-1 mini-gene was constructed by ligating together 4 fragments: a cDNA fragment from 1–4154 bp using an engineered ApaI site and BamHI, a BamHI to NotI genomic fragment (14,808–24,000), a NotI to KpnI genomic fragment (24,001–36,849), and a KpnI to 3′ UTR fragment containing an engineered SacII site (36,850–38,921). For anc-1 rescue analysis, a plasmid encoding Pmec-7ANC-1 (mini-gene, pBG161) was injected at 10–40 ng/µL, and a plasmid encoding Pmyo-3ANC-1 (mini-gene, pBG-183) was injected at 20 ng/µL. For bar-1 rescue analysis, a plasmid encoding Pmec-7BAR-1 (genomic clone, pBG-GY318) was injected at 1 ng/µL.
Analysis of axon termination and synapse formation
Analysis was carried out on live animals at 40× magnification using a Nikon epifluorescent microscope and a Q-imaging camera. Animals were anesthetized using 1% (v/v) 1-phenoxy-2-propanol in M9 buffer. Synapse formation defects were quantified by collecting images of juIs1 (Punc-25SNB-1::GFP) and manually scoring puncta numbers in Adobe Photoshop. Dorsal cord lengths were determined in µmeters using Q-imaging software. For each genotype 20 or more worms were analyzed from at least 3 independent experiments. Both bar-1 and pop-1 mutants displayed stereotyped, reproducible gaps in their dorsal cords (data not shown). Care was taken to avoid collecting images at these locations. Axon termination defects were visualized using muIs32 (Pmec-7GFP) and manually scored. For all genetic analysis on axon termination, averages are shown for data collected from 5–8 independent counts of 20–30 PLM neurons from young adult worms. For transgenic analysis, data shown is an average of 4 or more transgenic lines for each genotype.
Biochemistry
Proteomic analysis of RPM-1 binding proteins, including ANC-1, was described previously [42]. For biochemical analysis of RPM-1 binding to ANC-1, worms were grown in liquid culture, harvested by centrifugation, frozen in liquid N2, ground with a mortar and pestle under liquid N2, and extracted using 0.1% NP-40 lysis buffer as described previously [42]. CoIPs were performed from 40 mg of total protein extract. RPM-1::GFP was precipitated using a mouse monoclonal antibody (3E6, MP Biomedical) and protein G agarose beads (Roche). For immunoblotting, precipitates were run on an SDS-PAGE gel (3–8% Tris Acetate, Invitrogen), and proteins were wet transferred to PVDF membrane in Tris acetate transfer buffer (30 volts for 24–30 hours). Blots were blocked with part-skim milk in TBST, and probed with an anti-GFP antibody (mouse monoclonal, Roche) or purified anti-ANC-1 polyclonal antibodies that were used previously [6]. Primary antibodies were detected with secondary antibodies coupled to HRP, Supersignal FemtoWest enhanced chemiluminescent reagent (Pierce), and autoradiography.
Supporting Information
{kind=link}
Zdroje
1. StarrDA, FridolfssonHN (2010) Interactions between nuclei and the cytoskeleton are mediated by SUN-KASH nuclear-envelope bridges. Annu Rev Cell Dev Biol 26 : 421–444.
2. ZhangX, XuR, ZhuB, YangX, DingX, et al. (2007) Syne-1 and Syne-2 play crucial roles in myonuclear anchorage and motor neuron innervation. Development 134 : 901–908.
3. LuxtonGW, GomesER, FolkerES, VintinnerE, GundersenGG (2010) Linear arrays of nuclear envelope proteins harness retrograde actin flow for nuclear movement. Science 329 : 956–959.
4. ZhangX, LeiK, YuanX, WuX, ZhuangY, et al. (2009) SUN1/2 and Syne/Nesprin-1/2 complexes connect centrosome to the nucleus during neurogenesis and neuronal migration in mice. Neuron 64 : 173–187.
5. Elhanany-TamirH, YuYV, ShnayderM, JainA, WelteM, et al. (2012) Organelle positioning in muscles requires cooperation between two KASH proteins and microtubules. J Cell Biol 198 : 833–846.
6. StarrDA, HanM (2002) Role of ANC-1 in tethering nuclei to the actin cytoskeleton. Science 298 : 406–409.
7. GoughLL, BeckKA (2004) The spectrin family member Syne-1 functions in retrograde transport from Golgi to ER. Biochim Biophys Acta 1693 : 29–36.
8. YuTW, ChahrourMH, CoulterME, JiralerspongS, Okamura-IkedaK, et al. (2013) Using Whole-Exome Sequencing to Identify Inherited Causes of Autism. Neuron 77 : 259–273.
9. O'RoakBJ, DeriziotisP, LeeC, VivesL, SchwartzJJ, et al. (2011) Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat Genet 43 : 585–589.
10. Gros-LouisF, DupreN, DionP, FoxMA, LaurentS, et al. (2007) Mutations in SYNE1 lead to a newly discovered form of autosomal recessive cerebellar ataxia. Nat Genet 39 : 80–85.
11. ZhangQ, BethmannC, WorthNF, DaviesJD, WasnerC, et al. (2007) Nesprin-1 and -2 are involved in the pathogenesis of Emery Dreifuss muscular dystrophy and are critical for nuclear envelope integrity. Hum Mol Genet 16 : 2816–2833.
12. TessemaM, WillinkR, DoK, YuYY, YuW, et al. (2008) Promoter methylation of genes in and around the candidate lung cancer susceptibility locus 6q23-25. Cancer Res 68 : 1707–1714.
13. AttaliR, WarwarN, IsraelA, GurtI, McNallyE, et al. (2009) Mutation of SYNE-1, encoding an essential component of the nuclear lamina, is responsible for autosomal recessive arthrogryposis. Hum Mol Genet 18 : 3462–3469.
14. PuckelwartzMJ, KesslerEJ, KimG, DewittMM, ZhangY, et al. (2010) Nesprin-1 mutations in human and murine cardiomyopathy. J Mol Cell Cardiol 48 : 600–608.
15. AndreassenOA, ThompsonWK, SchorkAJ, RipkeS, MattingsdalM, et al. (2013) Improved detection of common variants associated with schizophrenia and bipolar disorder using pleiotropy-informed conditional false discovery rate. PLoS Genet 9: e1003455.
16. GreenEK, GrozevaD, FortyL, Gordon-SmithK, RussellE, et al. (2013) Association at SYNE1 in both bipolar disorder and recurrent major depression. Mol Psychiatry 18 : 614–617.
17. SklarP, RipkeS, ScottLJ, AndreassenOA, CichonS, CraddockN, EdenbergHJ, NurnbergerJI, RietschelM, BlackwoodD (2011) Large-scale genome-wide association analysis of bipolar disorder identifies a new susceptibility locus near ODZ4. Nat Genet 43 : 977–983.
18. ApelED, LewisRM, GradyRM, SanesJR (2000) Syne-1, a dystrophin - and Klarsicht-related protein associated with synaptic nuclei at the neuromuscular junction. J Biol Chem 275 : 31986–31995.
19. GradyRM, StarrDA, AckermanGL, SanesJR, HanM (2005) Syne proteins anchor muscle nuclei at the neuromuscular junction. Proc Natl Acad Sci U S A 102 : 4359–4364.
20. Del BeneF, WehmanAM, LinkBA, BaierH (2008) Regulation of neurogenesis by interkinetic nuclear migration through an apical-basal notch gradient. Cell 134 : 1055–1065.
21. LeinES, HawrylyczMJ, AoN, AyresM, BensingerA, et al. (2007) Genome-wide atlas of gene expression in the adult mouse brain. Nature 445 : 168–176.
22. CottrellJR, BorokE, HorvathTL, NediviE (2004) CPG2: a brain - and synapse-specific protein that regulates the endocytosis of glutamate receptors. Neuron 44 : 677–690.
23. WarrenDT, TajsicT, MelladJA, SearlesR, ZhangQ, et al. (2010) Novel nuclear nesprin-2 variants tether active extracellular signal-regulated MAPK1 and MAPK2 at promyelocytic leukemia protein nuclear bodies and act to regulate smooth muscle cell proliferation. J Biol Chem 285 : 1311–1320.
24. NeumannS, SchneiderM, DaughertyRL, GottardiCJ, EmingSA, et al. (2010) Nesprin-2 interacts with {alpha}-catenin and regulates Wnt signaling at the nuclear envelope. J Biol Chem 285 : 34932–34938.
25. PoMD, HwangC, ZhenM (2010) PHRs: bridging axon guidance, outgrowth and synapse development. Curr Opin Neurobiol 20 : 100–107.
26. LewcockJW, GenoudN, LettieriK, PfaffSL (2007) The ubiquitin ligase Phr1 regulates axon outgrowth through modulation of microtubule dynamics. Neuron 56 : 604–620.
27. SchaeferAM, HadwigerGD, NonetML (2000) rpm-1, a conserved neuronal gene that regulates targeting and synaptogenesis in C. elegans. Neuron 26 : 345–356.
28. KimJH, WangX, CoolonR, YeB (2013) Dscam expression levels determine presynaptic arbor sizes in Drosophila sensory neurons. Neuron 78 : 827–838.
29. LiH, KulkarniG, WadsworthWG (2008) RPM-1, a Caenorhabditis elegans protein that functions in presynaptic differentiation, negatively regulates axon outgrowth by controlling SAX-3/robo and UNC-5/UNC5 activity. J Neurosci 28 : 3595–3603.
30. BloomAJ, MillerBR, SanesJR, DiAntonioA (2007) The requirement for Phr1 in CNS axon tract formation reveals the corticostriatal boundary as a choice point for cortical axons. Genes Dev 21 : 2593–2606.
31. D'SouzaJ, HendricksM, Le GuyaderS, SubburajuS, GrunewaldB, et al. (2005) Formation of the retinotectal projection requires Esrom, an ortholog of PAM (protein associated with Myc). Development 132 : 247–256.
32. ZhenM, HuangX, BamberB, JinY (2000) Regulation of presynaptic terminal organization by C. elegans RPM-1, a putative guanine nucleotide exchanger with a RING-H2 finger domain. Neuron 26 : 331–343.
33. BurgessRW, PetersonKA, JohnsonMJ, RoixJJ, WelshIC, et al. (2004) Evidence for a conserved function in synapse formation reveals Phr1 as a candidate gene for respiratory failure in newborn mice. Mol Cell Biol 24 : 1096–1105.
34. WanHI, DiAntonioA, FetterRD, BergstromK, StraussR, et al. (2000) Highwire regulates synaptic growth in Drosophila. Neuron 26 : 313–329.
35. XiongX, WangX, EwanekR, BhatP, DiantonioA, et al. (2010) Protein turnover of the Wallenda/DLK kinase regulates a retrograde response to axonal injury. J Cell Biol 191 : 211–223.
36. HammarlundM, NixP, HauthL, JorgensenEM, BastianiM (2009) Axon regeneration requires a conserved MAP kinase pathway. Science 323 : 802–806.
37. XiongX, HaoY, SunK, LiJ, LiX, et al. (2012) The Highwire ubiquitin ligase promotes axonal degeneration by tuning levels of Nmnat protein. PLoS Biol 10: e1001440.
38. BabettoE, BeirowskiB, RusslerEV, MilbrandtJ, DiAntonioA (2013) The Phr1 ubiquitin ligase promotes injury-induced axon self-destruction. Cell Rep 3 : 1422–1429.
39. NakataK, AbramsB, GrillB, GoncharovA, HuangX, et al. (2005) Regulation of a DLK-1 and p38 MAP kinase pathway by the ubiquitin ligase RPM-1 is required for presynaptic development. Cell 120 : 407–420.
40. CollinsCA, WairkarYP, JohnsonSL, DiantonioA (2006) Highwire Restrains Synaptic Growth by Attenuating a MAP Kinase Signal. Neuron 51 : 57–69.
41. MurthyV, HanS, BeauchampRL, SmithN, HaddadLA, et al. (2004) Pam and its ortholog highwire interact with and may negatively regulate the TSC1.TSC2 complex. J Biol Chem 279 : 1351–1358.
42. GrillB, BienvenutWV, BrownHM, AckleyBD, QuadroniM, et al. (2007) C. elegans RPM-1 Regulates Axon Termination and Synaptogenesis through the Rab GEF GLO-4 and the Rab GTPase GLO-1. Neuron 55 : 587–601.
43. GrillB, ChenL, TulgrenED, BakerST, BienvenutW, et al. (2012) RAE-1, a Novel PHR Binding Protein, Is Required for Axon Termination and Synapse Formation in Caenorhabditis elegans. J Neurosci 32 : 2628–2636.
44. TianX, LiJ, ValakhV, DiAntonioA, WuC (2011) Drosophila Rae1 controls the abundance of the ubiquitin ligase Highwire in post-mitotic neurons. Nat Neurosci 14 : 1267–1275.
45. LiaoEH, HungW, AbramsB, ZhenM (2004) An SCF-like ubiquitin ligase complex that controls presynaptic differentiation. Nature 430 : 345–350.
46. McCabeBD, HomS, AberleH, FetterRD, MarquesG, et al. (2004) Highwire regulates presynaptic BMP signaling essential for synaptic growth. Neuron 41 : 891–905.
47. BakerST, OppermanKJ, TulgrenED, TurgeonSM, BienvenutW, et al. (2014) RPM-1 Uses Both Ubiquitin Ligase and Phosphatase-Based Mechanisms to Regulate DLK-1 during Neuronal Development. PLoS Genet 10: e1004297.
48. HallamSJ, JinY (1998) lin-14 regulates the timing of synaptic remodelling in Caenorhabditis elegans. Nature 395 : 78–82.
49. HungWL, HwangC, GaoS, LiaoEH, ChitturiJ, et al. (2013) Attenuation of insulin signalling contributes to FSN-1-mediated regulation of synapse development. EMBO J 32 : 1745–1760.
50. YochemJ, GuT, HanM (1998) A new marker for mosaic analysis in Caenorhabditis elegans indicates a fusion between hyp6 and hyp7, two major components of the hypodermis. Genetics 149 : 1323–1334.
51. CleversH, NusseR (2012) Wnt/beta-catenin signaling and disease. Cell 149 : 1192–1205.
52. KirszenblatL, PattabiramanD, HilliardMA (2011) LIN-44/Wnt directs dendrite outgrowth through LIN-17/Frizzled in C. elegans Neurons. PLoS Biol 9: e1001157.
53. PanCL, HowellJE, ClarkSG, HilliardM, CordesS, et al. (2006) Multiple Wnts and frizzled receptors regulate anteriorly directed cell and growth cone migrations in Caenorhabditis elegans. Dev Cell 10 : 367–377.
54. PrasadBC, ClarkSG (2006) Wnt signaling establishes anteroposterior neuronal polarity and requires retromer in C. elegans. Development 133 : 1757–1766.
55. DreierL, BurbeaM, KaplanJM (2005) LIN-23-mediated degradation of beta-catenin regulates the abundance of GLR-1 glutamate receptors in the ventral nerve cord of C. elegans. Neuron 46 : 51–64.
56. MehtaN, LoriaPM, HobertO (2004) A genetic screen for neurite outgrowth mutants in Caenorhabditis elegans reveals a new function for the F-box ubiquitin ligase component LIN-23. Genetics 166 : 1253–1267.
57. ParkEC, GlodowskiDR, RongoC (2009) The ubiquitin ligase RPM-1 and the p38 MAPK PMK-3 regulate AMPA receptor trafficking. PLoS One 4: e4284.
58. JacksonBM, EisenmannDM (2012) beta-catenin-dependent Wnt signaling in C. elegans: teaching an old dog a new trick. Cold Spring Harb Perspect Biol 4: a007948.
59. PhillipsBT, KimbleJ (2009) A new look at TCF and beta-catenin through the lens of a divergent C. elegans Wnt pathway. Dev Cell 17 : 27–34.
60. KorswagenHC, HermanMA, CleversHC (2000) Distinct beta-catenins mediate adhesion and signalling functions in C. elegans. Nature 406 : 527–532.
61. EisenmannDM, MaloofJN, SimskeJS, KenyonC, KimSK (1998) The beta-catenin homolog BAR-1 and LET-60 Ras coordinately regulate the Hox gene lin-39 during Caenorhabditis elegans vulval development. Development 125 : 3667–3680.
62. VashlishanAB, MadisonJM, DybbsM, BaiJ, SieburthD, et al. (2008) An RNAi screen identifies genes that regulate GABA synapses. Neuron 58 : 346–361.
63. SiegfriedKR, KimbleJ (2002) POP-1 controls axis formation during early gonadogenesis in C. elegans. Development 129 : 443–453.
64. ChalfieM, ThomsonJN (1979) Organization of neuronal microtubules in the nematode Caenorhabditis elegans. J Cell Biol 82 : 278–289.
65. Ch'ngQ, WilliamsL, LieYS, SymM, WhangboJ, et al. (2003) Identification of genes that regulate a left-right asymmetric neuronal migration in Caenorhabditis elegans. Genetics 164 : 1355–1367.
66. RocheleauCE, DownsWD, LinR, WittmannC, BeiY, et al. (1997) Wnt signaling and an APC-related gene specify endoderm in early C. elegans embryos. Cell 90 : 707–716.
67. KorswagenHC, CoudreuseDY, BetistMC, van de WaterS, ZivkovicD, et al. (2002) The Axin-like protein PRY-1 is a negative regulator of a canonical Wnt pathway in C. elegans. Genes Dev 16 : 1291–1302.
68. MaloneCJ, FixsenWD, HorvitzHR, HanM (1999) UNC-84 localizes to the nuclear envelope and is required for nuclear migration and anchoring during C. elegans development. Development 126 : 3171–3181.
69. MarkiewiczE, TilgnerK, BarkerN, van de WeteringM, CleversH, et al. (2006) The inner nuclear membrane protein emerin regulates beta-catenin activity by restricting its accumulation in the nucleus. EMBO J 25 : 3275–3285.
70. ZhangQ, RagnauthCD, SkepperJN, WorthNF, WarrenDT, et al. (2005) Nesprin-2 is a multi-isomeric protein that binds lamin and emerin at the nuclear envelope and forms a subcellular network in skeletal muscle. J Cell Sci 118 : 673–687.
71. LibotteT, ZaimH, AbrahamS, PadmakumarVC, SchneiderM, et al. (2005) Lamin A/C-dependent localization of Nesprin-2, a giant scaffolder at the nuclear envelope. Mol Biol Cell 16 : 3411–3424.
72. HaithcockE, DayaniY, NeufeldE, ZahandAJ, FeinsteinN, et al. (2005) Age-related changes of nuclear architecture in Caenorhabditis elegans. Proc Natl Acad Sci U S A 102 : 16690–16695.
73. HilliardMA, BargmannCI (2006) Wnt signals and frizzled activity orient anterior-posterior axon outgrowth in C. elegans. Dev Cell 10 : 379–390.
74. HarterinkM, KimDH, MiddelkoopTC, DoanTD, van OudenaardenA, et al. (2011) Neuroblast migration along the anteroposterior axis of C. elegans is controlled by opposing gradients of Wnts and a secreted Frizzled-related protein. Development 138 : 2915–2924.
75. MaroGS, KlassenMP, ShenK (2009) A beta-catenin-dependent Wnt pathway mediates anteroposterior axon guidance in C. elegans motor neurons. PLoS One 4: e4690.
76. KiddAR3rd, MiskowskiJA, SiegfriedKR, SawaH, KimbleJ (2005) A beta-catenin identified by functional rather than sequence criteria and its role in Wnt/MAPK signaling. Cell 121 : 761–772.
77. LoMC, GayF, OdomR, ShiY, LinR (2004) Phosphorylation by the beta-catenin/MAPK complex promotes 14-3-3-mediated nuclear export of TCF/POP-1 in signal-responsive cells in C. elegans. Cell 117 : 95–106.
78. ZechnerD, FujitaY, HulskenJ, MullerT, WaltherI, et al. (2003) beta-Catenin signals regulate cell growth and the balance between progenitor cell expansion and differentiation in the nervous system. Dev Biol 258 : 406–418.
79. BamjiSX, ShimazuK, KimesN, HuelskenJ, BirchmeierW, et al. (2003) Role of beta-catenin in synaptic vesicle localization and presynaptic assembly. Neuron 40 : 719–731.
80. YuX, MalenkaRC (2003) Beta-catenin is critical for dendritic morphogenesis. Nat Neurosci 6 : 1169–1177.
81. MuraseS, MosserE, SchumanEM (2002) Depolarization drives beta-Catenin into neuronal spines promoting changes in synaptic structure and function. Neuron 35 : 91–105.
82. VotinV, NelsonWJ, BarthAI (2005) Neurite outgrowth involves adenomatous polyposis coli protein and beta-catenin. J Cell Sci 118 : 5699–5708.
83. ElulTM, KimesNE, KohwiM, ReichardtLF (2003) N - and C-terminal domains of beta-catenin, respectively, are required to initiate and shape axon arbors of retinal ganglion cells in vivo. J Neurosci 23 : 6567–6575.
84. LiXM, DongXP, LuoSW, ZhangB, LeeDH, et al. (2008) Retrograde regulation of motoneuron differentiation by muscle beta-catenin. Nat Neurosci 11 : 262–268.
85. AbramsB, GrillB, HuangX, JinY (2008) Cellular and molecular determinants targeting the Caenorhabditis elegans PHR protein RPM-1 to perisynaptic regions. Dev Dyn 237 : 630–639.
86. EhnertC, TegederI, PierreS, BirodK, NguyenHV, et al. (2004) Protein associated with Myc (PAM) is involved in spinal nociceptive processing. J Neurochem 88 : 948–957.
87. SantosTM, HanS, BowserM, SazaniK, BeauchampRL, et al. (2006) Alternative splicing in protein associated with Myc (Pam) influences its binding to c-Myc. J Neurosci Res 83 : 222–232.
88. ShenK, CowanCW (2010) Guidance molecules in synapse formation and plasticity. Cold Spring Harb Perspect Biol 2: a001842.
89. HendricksM, MathuruAS, WangH, SilanderO, KeeMZ, et al. (2008) Disruption of Esrom and Ryk identifies the roof plate boundary as an intermediate target for commissure formation. Mol Cell Neurosci 37 : 271–283.
90. KennerdellJR, FetterRD, BargmannCI (2009) Wnt-Ror signaling to SIA and SIB neurons directs anterior axon guidance and nerve ring placement in C. elegans. Development 136 : 3801–3810.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2014 Číslo 7
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Wnt Signaling Interacts with Bmp and Edn1 to Regulate Dorsal-Ventral Patterning and Growth of the Craniofacial Skeleton
- Novel Approach Identifies SNPs in and with Evidence for Parent-of-Origin Effect on Body Mass Index
- Hypoxia Adaptations in the Grey Wolf () from Qinghai-Tibet Plateau
- DNA Topoisomerase 1α Promotes Transcriptional Silencing of Transposable Elements through DNA Methylation and Histone Lysine 9 Dimethylation in
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy