
Onemocnění asociované s imunoglobulinem IgG4. Klinické příznaky, diferenciální diagnostika a recentní mezinárodní diagnostická kritéria
IgG4-related disease. Clinical manifestation differential diagnosis and recent International Diagnostic Criteria for IgG4-related disease
Immunoglobulin G4 - related disease (IgG4-RD) is a rare systemic fibro-inflammatory disorder. Autoimmune pancreatitis is the most frequent manifestation of IgG4-RD. However, IgG4-RD can affect any organ such as salivary glands, orbits, retroperitoneum, prostate and many others. Recent research enabled a clear clinical and histopathological description of IgG4-RD and in 2019 four Clinical phenotypes of IgG4-related disease were described. Diagnosis is based on morphological examination with typical findings of lymphoplasmocellular inflammation, storiform fibrosis and obliterative phlebitis in IgG4-RD biopsies and the tissue invading plasma cells largely produce IgG4. Elevated serum IgG4 levels are found in many but not all patients. New diagnostic criteria for IgG4-RD have been published recently in 2019 and 2021. This review summarizes current knowledge on pathophysiology, clinical manifestations, diagnosis and differential diagnosis of IgG4-RD from the point of view 2022 and in next article brings overview of the IgG4-RD therapy.
Keywords:
IgG4 related disease – IgG4 immunoglobulin subclass
Autoři:
Zdeněk Adam 1; David Zeman 2; Aleš Čermák 3; Milan Dastych 4; Martina Doubková 5; Theodor Horváth 6; Šárka Skorkovská 7; Zuzana Adamová 8; Zdeněk Řehák 9; Renata Koukalová 9; Luděk Pour 1; Martin Štork 1; Marta Krejčí 1; Viera Sandecká 1; Sabina Ševčíková 10; Zdeněk Král 1
Působiště autorů:
Interní hematologická a onkologická klinika LF MU a FN Brno
1; Ústav laboratorní medicíny – Oddělení klinické biochemie, FN Brno
2; Urologická klinika LF MU a FN Brno
3; Interní gastroenterologická klinika LF MU a FN Brno
4; Klinika nemocí plicních a tuberkulózy LF MU a FN Brno
5; Chirurgická klinika LF MU a FN Brno
6; Oční klinika LF MU a FN Brno
7; Chirurgické oddělení nemocnice Frýdek Místek a Chirurgické oddělení nemocnice Vsetín
8; Oddělení nukleární medicíny, Masarykův onkologický ústav Brno
9; Ústav patologické fyziologie LF MU, Brno
10
Vyšlo v časopise:
Vnitř Lék 2022; 68(E-5): 4-19
Kategorie:
Přehledové články
doi:
https://doi.org/10.36290/vnl.2022.070
Souhrn
S imunoglobulinem IgG4 asociované onemocnění, immunoglobulin IgG4-related disease (IgG4-RD) je heterogenní porucha s multiorgánovým poškozením. Jako samostatná jednotka bylo toto onemocnění definováno teprve počátkem tohoto století. Autoimunitní pankreatitida je nejčastější a nejznámější manifestací, ale tato nemoc může postihnout prakticky kterýkoliv orgán, jako například slinné žlázy, struktury orbity, retroperitoneum ve formě retroperitoneální fibrózy a četné další. Současné znalosti umožňují tuto nemoc dobře klinicky i histologicky definovat. Diagnóza se stanovuje na základě průkazu lymfoplazmocytární infiltrace s IgG4+ plazmocyty, zánětlivými projevy, průkazu storiformní fibrózy a obliterativní flebitidy. V bioptických vzorcích jsou typicky nalézány plazmatické buňky produkující imunoglobulin typu IgG4. Zvýšené hladiny IgG4 v séru jsou nalézány u mnohých, ale zdaleka ne u všech pacientů s IgG4-RD. V roce 2019 byly rozpoznány a popsány základní 4 klinické fenotypy. Kritéria vytvořená Evropskou a Americkou revmatologickou společností byla zveřejněna v roce 2019, jsou však hodně komplikovaná, a tak v roce 2021 byla japonskými autory publikována jednodušší diagnostická kritéria. Tento přehledový článek sumarizuje současné vědomosti o patofyziologii, klinických projevech a problémech diagnostiky a diferenciální diagnostiky této nemoci z pohledu roku 2022 a následující článek bude věnován přehledu léčby IgG4-RD.
Klíčová slova:
IgG4 asociované onemocnění (IgG4-related disease) – podtřída IgG4 imunoglobulinu
Úvod
S imunoglobulinem IgG4 asociovaná choroba, anglickým termínem „immunoglobulin IgG4 related disease – IgG4-RD“, je nemaligní, chronická, imunitním systémem vyvolaná choroba.
Tato choroba může postihnout kterýkoliv orgán, od mozkových plen a CNS, přes pankreatobiliární systém a hemopoetický systém až po prostatu. A proto se s touto chorobou setkávají lékaři všech medicínských odborností, jak je patrné z citované české a slovenské odborné literatury, ale i ze složení autorského kolektivu tohoto článku. V tomto textu se na tuto chorobu podíváme ze zorného úhlu nejen internistů a hematologů, ale také urologů, plicních specialistů, očních specialistů a chirurgů. K hematologům a internistům přivádí pacienty s IgG4-RD: eozinofilie, lymfadenopatie, zvýšená hodnota celkové bílkoviny a imunoglobulinů, systémová zánětlivá reakce – synonymem B‑symptomy (1–3).
K urologům přivádí nemocné tato choroba s důsledky retroperitoneální fibrózy, hydronefróza, k nefrologům pak poškození ledvin, ke gastroenterologům poškození žlučových cest a pankreatu, často s podezřením na neoplazii pankreatu. K plicním lékařům přivede tato nemoc pacienta s projevy intersticiálního poškození plic. A tak bychom mohli pokračovat dále. Proto v textu stručně uvedeme příznaky této nemoci v jednotlivých orgánech tak, jak se s nimi setkávají lékaři všech medicínských odborností.
Pojmenování této nemoci je ustálené teprve od roku 2012, kdy byly publikovány závěry konference v Bostonu, na níž byl dohodnut mezinárodně akceptovaný název IgG4-RD a byla dohodnuta a zveřejněna první kritéria této nemoci. S postupem času se však tato první kritéria ukázala jako málo specifická, a tak byla na mezinárodním sjezdu Evropské a Americké revmatologické společnosti v roce 2019 transformována do podstatně složitějších mezinárodních kritérií. Ale i původní japonská kritéria byla v roce 2021 inovována, což dává tušit, že diagnostika a diferenciální diagnostika této nemoci je podstatně komplikovanější, než to vypadalo v roce 2012.
Ačkoliv choroba postihuje snad všechny orgány, její histologická charakteristika, až na některé výjimky, je docela uniformní a jsou pro ni typické tyto znaky:
• storiformní fibróza,
• denzní lymfoplazmocytární infiltrace se zvýšeným počtem plazmatických buněk s pozitivním imunohistochemickým průkazem IgG4+ plazmatických buněk (> 10/ zorném poli mikroskopu při vysokém rozlišení (per high power field). Četnost kolísá dle postižených orgánů. Poměr IgG4+/IgG+ plazmocytů je obvykle > 40 %,
• obliterativní flebitida,
• tkáňová eozinofilie (granulomy, neutrofilní mikroabscesy a nekrotizující vaskulitida) (1).
Odchylky od této standardní sktruktury vykazují ložiska v ledvinách a uzlinách. IgG4-RD se projevuje v kterékoliv lokalizaci tumorózními ložisky, fibrózou a infiltrátem s bohatě zastoupenými polyklonálními plazmatickými buňkami s výraznou expresí podtřídy imunoglobulinu IgG4.
V tomto článku se zaměříme na popis projevů nemoci, s nimiž se setkávají lékaři všech odbornostím, a na diagnostiku. V následujícím článku se pak budeme kontrovat na léčbu a do něj uvedeme obrazovou dokumentaci našich pacientů, která zároveň dokumentuje účinek popisované léčby.
Historie poznání IgG4‑RD
První popis pacienta, který měl zřejmě IgG4-RD, zveřejnil již před dávnými lety v roce 1892 chirurg Johann Freiherr von Mikulicz‑Radecki. Popsal pacienta se systémovým zduřením slinných žláz, které byly infiltrovány mononukleárními buňkami, aniž by ovšem tušil další souvislost. Od jeho popisu se odvíjela popisná diagnóza „Mikuliczova choroba“.
O pár let později popsal Küttner v roce 1896 pacienta s procesem v submandibulární žláze připomínající tumor, a tento nemaligní nádor byl nazván Küttnerův tumor, dnes jej nazýváme izolovaná submandibulární sklerotizující sialoadenitida. A v tom samém roce popsal Riedel „Eisenharte Struma“, nebo strumu tvrdou jako železo s fibrózní transformací štítné žlázy a přilehlých struktur. V učebnicích interny je tento typ poškození štítnice nazýván Riedelova tyreoiditida. Mnoho dalších projevů IgG4-RD (jako je retroperitoneální fibróza, sklerotizující cholangitida, sklerotizující pankreatitida) bylo v průběhu minulých let popsáno, aniž by byla zřejmá patofyziologie. Pozorným lékařům již v šedesátých letech (publikováno 1963) při popisu dvou pacientů s Riedelovou tyreoiditidou, retroperitoneální fibrózou a sklerotizující cholangitidou bylo nápadné současné postižení více tkání podobným procesem a zvažovali systémové postižení, ale ještě neměli možnost je prokázat (4).
Výrazný pokrok směřující k poznání IgG4-RD nastal až v roce 1995, kdy byl popsán pacient s chronickou pankreatitidou reagující na glukokortikoidy a byl vysloven předpoklad, že by tato pankreatitida mohla mít autoimunitní etiologii. Do té doby se netušilo, že pankreas může být postižen autoimunitní chorobou (5).
Počátek poznání této autoimunitní pankreatitidy se datuje do konce devadesátých let. Japonští vědci se snažili najít ukazatele, s jejichž pomocí by odlišili sklerotizující pankreatitidu od karcinomu pankreatu. Při analýze vzorků krve si všimli zvýšené koncentrace podtřídy (anglicky subclass) imunoglobulinu typu IgG nazvané IgG4 (6). A následovně prokázali infiltraci polyklonálními plazmatickými buňkami, které obsahovaly tuto podtřídu imunoglobulinu. K tomu bylo zapotřebí použít speciální imunohistochemické barvení na IgG4 exprimující buňky v histologických vzorcích z patologicky změněné tkáně, odebírané pacientům s autoimunitní pankreatitidou z pankreatu, z jater a případně ze žlučníku. V roce 2002 byla popsána infiltrace plazmocyty nesoucími IgG4 na svém povrchu a v roce 2003 byly potvrzeny zvýšené hodnoty IgG4 u pacientů s autoimunitní pankreatitidou a byl navržen název nové klinické jednotky „IgG4-related autoimmune disease“ (7). A také byla popsána retroperitoneální fibróza jako další komplikace sklerotizující autoimunitní pankreatitidy (8). V české literatuře se autoimunitní forma chronické pankreatitidy začíná popisovat až od roku 2010–2011 (9–11).
Jakmile byla tato jednotka laboratorně definována, tak se ukázalo, že do ní spadají choroby se zcela jinými, dříve zavedenými popisnými jmény. Tyto nálezy pak byly potvrzeny v dalších kohortách pacientů z USA a z Koreje (12, 13). A tak se objevily popisy chorob se zánětlivými a fibrotickými znaky, spojenými s vyšší hodnotou imunoglobulinu IgG4 a s infiltrací plazmocyty se zvýšenou expresí IgG4. Povědomí o této nemoci se tedy pomalu rozvíjí od roku 2000, ale název této nemoci byl poprvé dohodnut na setkání odborníků v Bostonu v roce 2011 jako „IgG4-related disease“ a závěry této konference byly publikovány v roce 2012, tedy teprve před 9 lety (14–18). Přijaté názvy nemocí na této konferenci zobrazuje tabulka 1. Bohužel k nim nebyly přiděleny kódy dle MKN-10 klasifikace.

Přijaté jméno „IgG4-related disease“ odráží univerzálnost lokalizací IgG4 + plazmocelulárních infiltrátů a také charakteristické zvýšení koncentrací podtřídy imunoglobulinu IgG4. Běžným projevem je zvětšení hlavních slinných žláz (příušní a submandibulární) a zbytnění slzných žlázek (Mickuliczova choroba), lymfadenopatie, orbitální pseudotumor, pankreatitida, sklerotizující cholangitida, retroperitoneální fibróza a tubulointersticiální nefritida. Nejčastěji postiženým orgánem je pankreas v rámci autoimunitní pankreatitidy, a proto se s tímto onemocněním setkávají gastroenterologové. Ale projevy mohou postihnout kterýkoliv orgán, jak uvádí Průcha (19).
Epidemiologie IgG4-RD
Medián věku výskytu je 6.–7. životní dekáda, poměr postižených mužů a žen je 2 : 1. Zprávy o epidemiologii se koncentrují na akutní autoimunitní pankreatitidu a pocházejí hlavně z Japonska, kde odhadují prevalenci nemoci na 6/100 000 obyvatel a incidenci 1,4 případů na 100 000 obyvatel (20).
V 8 velkých nemocnicích analyzovali v letech 2005 až 2013 celkem 235 pacientů. Medián věku při stanovení diagnózy byl 67 let a 90 % pacientů bylo ve věkovém intervalu 50–70 let. Při stanovení diagnózy byly příznaky přítomny u 70 %, ale 18 % udávalo jen nespecifické bolesti břicha. Celkem 39 % pacientů mělo diabetes mellitus a 30 % mělo buď alergickou rýmu či astma, nebo medikamentózní alergii. U mužů byla slinivka břišní nejčastěji postiženým orgánem (63 %), následována slinnými žlázami, zatímco u žen byly v 57 % postiženy slinné žlázy a na druhém místě byla slinivka břišní (20).
Počet případů popsaných u dětí je málo, ale jedna nedávná publikace popsala 25 dětí, z nichž 11 mělo orbitální formu nemoci a tři měly autoimunitní pankreatitidu (21). Údaje o incidenci či prevalenci v Evropě případně v ČR nemáme.
Patofyziologie
IgG4-RD patří do skupiny chorob způsobených patologickou imunitní reakcí, podobně jako idiopatická multicentrická Castlemanova choroba (iMCD) a jiné. Interleukin-4 (IL-4) je považován za klíčový pro rozvoj IgG4-RD, podobně jako je interleukin-6 (IL-6) klíčový pro rozvoj iMCD.
V případě IgG4-RD se k interleukinu-4 připojuje interleukin-21 (IL-21). IL-4 stimuluje rozvoj imunitní reakce Th2 typu. Četní pacienti s IgG4-RD mají atopický ekzém, alergickou rinitidu a astma. IL-4 mimo jiné také indukuje fibrotizaci přímým působením na fibroblasty. Za zdroj IL-4 jsou považovány buňky zvané „T follicular helper Tfh“, „T‑helper typ 2“, bazofily a přirozené lymfoidní buňky.
Interleukin-21 stimuluje expanzi B buněk v germinálních centrech a synergisticky s IL-4 stimuluje „IgG4 class switching“ a stimuluje diferenciaci plazmablastů, plazmocytů a Tfh buněk. Na progresi IgG4-RD se dále podílejí tyto cytokiny: IL-13, IL-10, TGF‑beta. IL-13 má podobný efekt jako IL-4.
Na patogenezi jak Ig4-RD, tak i iMCD se podílejí určité typy T‑lymfocytů. Zatím však patofyziologický mechanismus této nemoci není dopodrobna znám (22).
Přepokládá se, že neznámý podnět stimuluje iniciální TH1-immunitní odpověď, což způsobuje sekreci proinflamatorních cytokinů a aktivaci B‑buněk a plazmocytů a také eozinofilních granulocytů. To vysvětluje častou eozinofilii, stejně jako vyšší hladinu IgE protilátek u postižených pacientů. Současně tento neznámý podnět stimuluje TH2 imunitní odpověď, což vede k expanzi a tvorbě IgG4 tvořících plazmatických buněk. Zřejmě dochází také k poruše funkce regulačních T‑buněk, která má za následek perzistující TH1 a TH2 imunitní odpověď.
Poškození orgánů však rezultuje z pokračující orgánové fibrózy. Ta se dává do souvislosti s nadprodukcí TH2 typických cytokinů IL-4 a IL-13 a uvolnění IL-10 z autoregulačních T‑buněk. Výsledkem je aktivace makrofágů a produkce profibrotických cytokinů, jako jsou „transforming growth factor β1“ (TGF‑β1) a „platelet‑derived growth factor“ (PDGF).
V centru etiopatogenetického dění je klonální expanze CD8+ cytotoxických T-lymfocytů se zvýšením jejich počtu jak v periferní krvi, tak i ve fibrotických ložiscích v postižených orgánech. Podrobněji patofyziologii této nemoci popisuje Mikulenková a Král (23–25).
Od IgG4-RD se patofyziologicky zřetelně liší rovněž relativně nedávno popsaná skupina IgG4 zprostředkovaných autoimunitních onemocnění „IgG4 – related autoimmune diseases – IgG4-AID“, u kterých bývá normální koncentrace IgG4 v séru, ale jsou nacházeny IgG4 autoprotilátky proti extracelulárním autoantigenům. Tyto autoprotilátky jsou u IgG4-AID přímo patogenní, pravděpodobně blokováním fyziologických interakcí mezi různými bílkovinami. IgG4-AID nejsou provázeny zvětšením orgánů, tumoriformními lézemi ani fibrózou ani tkáňovými infiltráty IgG4+ plazmocytů (26). Mezi IgG4-AID patří myasthenia gravis s anti‑MuSK protilátkami, periferní neuropatie s protilátkami proti kontaktinu-1 (CNTN1) a neurofascinu 155 (NF155), pemfigus foliaceus (IgG4 protilátky proti desmogleinu 1), pemfigus vulgaris (IgG4 protilátky proti desmogleinu 3) a trombotická trombocytopenická purpura (IgG4 protilátky proti ADAMTS13). U dalších několika onemocnění (převážně neurologických) je patogenicita IgG4 autoprotilátek považována za pravděpodobnou. V této monografii není problematika blíže rozebírána, zájemce o podrobnější informace odkazujeme na recentní přehledový článek Koneczny 2020 a Endmayr 2022 (26, 27).
Klinické projevy
IgG4-RD postihuje téměř každý orgán. Dříve se udávalo, že vyjma synoviální tkáně, ale to již také neplatí. První případ se synovitidou byl popsán v roce 2015. Fibroinflamatorní choroba tvoří zánětlivé infiltráty a fibrotické změny predilekčně ve tkáních různých žláz. Přehled možných manifestací přináší tabulka 2. Nejčastější orgánové projevy, tak jak se manifestovaly ve dvou velkých skupinách pacientů z USA (28) a z Japonska (29), uvádí tabulka 3. Je pravděpodobné, že lépe známé projevy nemoci, jako je autoimunitní pankreatitida, orbitální postižení a postižení slinných žláz, je rozpoznáváno častěji než méně známé projevy nemoci.


K hematologovi se pacienti s IgG4-RD dostávají k diferenciální diagnostice lymfadenopatie, eozinofilie a polyklonální hypergamaglobulinemie. K chirurgovi se dostávají s podezřením na tumor pankreatu.
Zvětšení lymfatických uzlin na podkladě IgG4-RD je nejznámějším projevem manifestace IgG4-RD, postihuje 30–60 % osob s touto nemocí (28, 29). IgG4 lymfadenopatie, jak lokalizovaná, tak i generalizovaná, je někdy součástí i dalšího postižení (plic, pankreatu). Někdy je také přítomno paralelní zvětšení slzných a slinných žláz. Problém je, že postižení uzlin mohou způsobovat i další choroby (15). V následujících odstavcích stručně charakterizujeme projevy této nemoci v jednotlivých orgánech a popis začneme první rozpoznanou formou této nemoci, IgG4 autoimunitní pankreatitidou.
Pankreas
Klasickou prezentací této nemoci je autoimunitní pankreatitida typu 1, zatímco autoimunitní pankreatitida typu 2 není součástí spektra IgG4-RD, má odlišné histopatologické znaky (duktální neutrofilní abscesy a duktální poškození). Autoimunitní pankreatitida typu 1 je vzácnou příčinou chronické pankreatitidy (méně než v 5 % všech pankreatitid). Obvykle postihuje osoby středního věku. Hodnota IgG4 bývá většinou zvýšená, ale asi u 20 % bývá v normě. Obvyklým příznakem je obstruktivní žloutenka (70 % případů), zatímco bolesti nejsou pravidlem (41 % případů). Často je provázena i mimopankreatickými infiltráty a s nimi spojenými projevy. MR‑cholangiopankreatografie, endosonografie a ERCP často prokazují striktury a nepravidelný průběh. Obdobné změny mohou zároveň postihnout i žlučové cesty. CT a MR zobrazení případně prokáže fokální či difuzní zvětšení pankreatu a peripankreatický lem s kontrastním enhancementem, což se interpretuje jako peripankreatický inflamatorní proces anebo fibróza. Endosonografické vyšetření pankreatu v typických případech ukazuje difuzní zvětšení žlázy (sausage‑shape), sníženou echogenitu parenchymu, nepravidelný průběh a striktury pankreatického vývodu. Ve vzácnějších případech nacházíme obraz nespecifické chronické pankreatitidy, solidní ložiskové postižení nebo pseudotumor hlavy slinivky. V určitých případech mohou zobrazovací vyšetření a sérologické studie pomoci ke stanovení diagnózy, ale většinou je třeba opřít diagnózu o histologické vyšetření. Vzorky tkáně pankreatu jsou odebírány aspirační biopsií jehlou při endosonografickém vyšetření. Histologická verifikace je nutná především z důvodu odlišení nádoru pankreatu. V histologickém vzorku musí být prokázaná difuzní lymfoplazmocytární infiltrace a storiformní fibróza. Pozitivní plazmatické buňky IgG4+ jsou přítomny obvykle v počtu > 10 v zorném poli mikroskopu. Obliterativní flebitida je také častá, ale někdy maskovaná zánětlivým infiltrátem. V těchto případech je přínosné barvení na elastin. Bohužel hodně příznaků je společných s karcinomem pankreatu (nebolestivá žloutenka, zvětšení pankreatu) a IgG4-RD lokálním zduřením i morfologicky imituje někdy tumor hlavy pankreatu. Proto také v jedné práci, která analyzovala histologické výsledky radikální operace (pankreatoduodenektomie) byla u 2,2 % diagnostikována lymfoplazmocytární sklerotizující pankreatitida, karcinom pankreatu v 53 %, periampulární neoplazie v 38 % a cholangiokarcinom v 9 % (30). Diferenciální diagnózu je třeba tedy dělat velmi pečlivě před rozhodnutím, zda provést radikální operaci – odstranění pankreatu s tumorem, nebo zda neoperovat a léčit IgG4-RD. Nelze spoléhat na zvýšené hodnoty IgG4, které mohou být zvýšené i u karcinomu pankreatu. Rozhodování o léčbě komplikuje také skutečnost, že u pacientů s IgG4 autoimunitní pankreatitidou se může vyvinout karcinom (31–35). Proto před rozhodnutím o dalším způsobu léčby a při podezření na možnost autoimunitní pankreatitidy je ve většině případů nutná histologická verifikace.
V případě karcinomu pankreatu provázeného jistým zvýšením hladin IgG4 v séru má diagnostický přínos biopsie (intraoperativní či s pomocí endoskopické ultrasonografie) a/nebo test s krátkodobým podáním glukokortikoidu, na který reaguje IgG4-RD, ale ne tumor pankreatu. Pro diagnostiku a léčbu této formy IgG‑RD byla vytvořena samostatná doporučení (36, 37).
Žlučové cesty, žlučník a játra
Charakteristickým znakem IgG4-RD cholangitidy je obstruktivní ikterus. Typicky jsou postiženy velké žlučové cesty, často extrahepaticky ve své proximální části, zatímco postižení menších žlučových cest je variabilní. Při postižení žlučových cest je u velké části pacientů infiltrován žlučník a játra. Při endosonografickém vyšetření je typickým nálezem difuzní homogenní a ohraničené zesílení stěny žlučovodu. Elevace IgG4 není vždy přítomná, udává se u 80 % nemocných. Histologické vyšetření přináší podobné závěry jako při vyšetření jiných orgánů. Jaterní biopsie často přispěje k diagnóze, ale může přinést i nekonkluzivní výsledek. Biopsie popisuje portální infiltraci, portální sklerózu, obstrukci velkých žlučovodů, lobulární hepatitidu a kanalikulární cholestázu. Tyto formy postižení byly opakovaně popsány i českými autory (38–42). V diferenciální diagnostice je nutno především odlišit nádor žlučových cest od IgG4-RD, ale také primární sklerotizující cholangitidu, takže opravdu velmi obtížná diferenciální diagnostika, která se neobejde bez odběru vzorků pro histologické vyšetření. V případě cholangiokarcinomu na rozdíl od „IgG4 related sklerotizující cholangitidy“ se vedle obstrukčního ikteru a zvětšení pankreatu s regionální lymfadenopatií vyskytují vyšší hodnoty bilirubinu, vysoké hodnoty CA 19-9, nižší hodnota IgG4 v séru a kompletní obstrukce žlučových cest prokazatelná s pomoví ERCP. Je na gastroenterologovi, aby zvolil optimální tkáň k odebrání histologie. Obvykle jsou odebírány vzorky tkáně z choledochu z místa stenózy při ERCP nebo při cholangioskopii. Mírně zvýšené hodnoty IgG4 mohou opět provázet i tumor, takže samotná mírná elevace IgG4 při obstrukci žlučových cest diagnózu nedělá. A opět pro tento typ manifestace byla vytvořena speciální doporučení (43–46).
Štítná žláza
Riedelova tyreoiditida je od roku 2010 také součástí spektra IgG4-RD. Na rozdíl od jiných manifestací preferuje ženy. Riedelova tyreoiditida infiltruje v některých případech jen jeden lalok, v jiných celou žlázu. Specifickým nálezem je přesah fibrotických mas do okolních struktur. Nezřídka fibróza přesáhne na příštítná tělíska a způsobí tak hypoparatyreoidismus, dále na svaly, nervy a cévy. Klinickým příznakem je bolest, lokální zduření, dysfagie, chrapot a případně i zúžení trachey. Hodně pacientů s touto chorobou obvykle podstoupí tyreoidektomii pro podezření na karcinom, protože tenkojehlová biopsie je v těchto případech nekonkluzivní (47–50). A tak i pro tento typ manifestace jsou navržena diagnostická kritéria (51).
Slinné žlázy
Slinné a slzné žlázy jsou často postiženy IgG4-RD. Hlavní stížností pacientů je asymetrické či symetrické zduření slinných žláz a omezená sekrece. Bolest je výjimečná. Sialoadenitida a dakryoadenitida postihují stejně obě pohlaví. Mikroskopické vyšetření obvykle prokáže zachování lobulární architektury s denzním lymfoplazmocytárním infiltrátem a hyperplastickými lymfoidními folikuly. V diferenciální diagnostice je nutno odlišit lymfom, sialolitiázu a karcinomy. Problém je, že karcinom v některých případech má kolem sebe zánětlivý infiltrát obsahující IgG4 plazmocyty, což dále komplikuje diagnózu. Podrobné popisy těchto projevů již byly publikovány jak v české, tak ve slovenské literatuře (52–56).
V případě Sjögrenova syndromu (SS) na rozdíl od „IgG4-related SS“ jsou přítomny nízké hodnoty IgG4 v séru, případně silně pozitivní vyšetření na anti/Ro/SSA protilátky a v bioptované žláze chybí IgG4+ plazmocyty, storiformní fibróza a obliterující flebitida. A opět jsou publikována doporučení pro klinickou a morfologickou diagnostiku této nemoci (57–60).
Orbity a orbitální adnexa
Pokud se onemocnění IgG4-RD manifestuje v očnici nebo v oku, pak je označováno jako „IgG4-related ophthalmic disease (IgG4-ROD)“, které nejčastěji způsobuje nebolestivé zduření slzné žlázy nebo slzných žláz. Další stejně postižené struktury oka či orbity zahrnují okohybné svaly, očnicový tuk, infraorbitální nervy, oční víčka a odvodné slzné cesty. Diagnostická kritéria kombinují fyzikální vyšetření, zobrazovací metody a histopatologické nálezy. Zobrazovací metody usnadňují diagnózu, ale je nezbytná tkáňová biopsie k vyloučení jiných příčin onemocnění, především maligních procesů. Nejčastěji je IgG4-ROD onemocněním postižena slzná žláza (62–88 %). Další často postiženou strukturou je trojklanný nerv (9,5–39 %), okohybné svaly (19–25 %), očnicový tuk (28,6–40 %), víčka (12 %) a odvodné slzné cesty (1,5–9,5 %). IgG4-ROD na rozdíl od jiných orgánů postihuje častěji mladší skupinu pacientů (55 let), zastoupení pohlaví je stejné a může se kombinovat s postižením slinné žlázy a vyšší hladinou IgG4 v séru. Bylo také zjištěno, že IgG4-ROD může být spojeno s astmatem a alergickou rhinitidou. IgG4-ROD pacienti mají vyšší riziko non‑Hodgkinského lymfomu než pacienti s pankreatickou formou IgG4-RD onemocnění. V domácí a zahraniční literatuře jsou opět podrobné popisy těchto stavů (61–68).
Retroperitoneální fibróza a postižení velkých cév
Retroperitoneální fibróza (RPF) je vzácnou chorobou, obvykle postihuje muže středního věku a je často asociovaná s kouřením. Klinické projevy jsou poměrně variabilní, u některých pacientů se tato choroba zjistí náhodou při CT břicha provedeného z jiné indikace, jiní si stěžují na bolest v oblasti dolní páteře „low back pain“. Obvyklou komplikací je hydronefróza. Periaortitida je asymptomatická, jen výjimečně vede k tvorbě aneuryzmat a k ruptuře. Při CT zobrazení je nacházeno cirkumferenciální zesílení stěny arterií, což je způsobeno sklerotizujícím zánětem v adventicii. Postižené cévy vykazují homogenní enhancement v pozdní fázi po aplikaci kontrastu. Některé arterie mají i dilatovaný lumen. Histologie prokazuje opět typické znaky IgG4-RD, ale u dlouhotrvající nemoci začíná dominovat fibróza. V případě retroperitoneální fibrózy se vyznačuje „IgG4-related RPF“ poměrem IgG4/IgG v tkáni nad 40 %.
K urologům přivádí nemocné tato choroba důsledky retroperitoneální fibrózy, hydronefróza buďto asymptomatická, nebo s různě vyjádřenými příznaky obstrukce vývodných cest močových a poškozením ledvinných funkcí.
RPF je často provázena zánětlivými aortálními aneuryzmaty abdominální či torakální aorty. Pak se proces nazývá chronická periaortitida spojená s retroperitoneální fibrózou.
RPF se zdá být heterogenní skupinou nemocí. Někteří pacienti vykazují znaky IgG4-RD, ale jiní nemají znaky IgG4-RD, a pak se tato situace nazývá idiopatickou retroperitoneální fibrózou. Poměr idiopatických a IgG4-RD retroperitoneálních fibróz není zatím znám, protože pátrání po známkách IgG4-RD při histologickém hodnocení retroperitoneální fibrózy se provádí až v posledních letech. Zánětlivá aneuryzmata aorty mohou taktéž spadat do spektra IgG4-RD (69–82). Z dalších příčin retroperitoneální fibrózy je nutno zmínit Erdheimovu‑Chesterovu chorobu – jednu chorobu ze spektra xantogranulomu.
Onemocnění a chirurgické řešení poprvé popsal francouzský urolog Albarran v roce 1905. V roce 1948 publikoval Ormond nálezy u dvou pacientů a přinesl podklady k vyčlenění retroperitoneální fibrózy jako samostatné klinické jednotky, i když na dlouhou dobu bez znalosti skutečné etologie.
Retroperitoneální fibróza, která je charakterizována zánětlivou proliferací pojivové tkáně, vede k dislokaci a útlaku ureterů v oblasti L4-S1 a k hydronefróze. K urologovi je pacient odeslán k vyšetření a provedení diferenciální diagnostiky, vyloučení obstrukce litiázou nebo nádorovým onemocněním.
Hydronefróza může být asymptomatická, zjištěná jako náhodný nález při jiném vyšetření. Vzhledem k nevýrazným počátečním potížím je onemocnění diagnostikováno pozdě a manifestuje se až komplikacemi vzniklými na podkladě komprese vývodných cest močových.
Diagnostikován může být různý stupeň ledvinové nedostatečnosti. Až třetina pacientů přichází s afunkční ledvinou a 10 % v urémii při bilaterální hydronefróze. Někdy se retroperitoneální fibróza prezentuje jako pyelonefritida až sepse v důsledku obstrukční uropatie.
Velmi vzácné urologické manifestace
IgG4-RD zřejmě preferuje určité lokalizace, ale není jasné, zda lze definovat lokalizace, v nichž se IgG4-RD nevyskytuje, nebo zda lze pouze konstatovat, že v některých lokalizacích je tato choroba častější a v jiných vzácnější. V literatuře jsou zatím jen ojediněle popsané další urologické manifestace této nemoci.
Forma manifestující se jako tumor varlete (81).
Intersticiální cystitida je nemoc zatím neobjasněné etiologie. Zánětlivá reakce obvykle bývá přítomna a je provázena zvýšeným zastoupením mastocytů ve svalovině detruzoru močového měchýře. Do roku 2022 se zatím pouze jediná práce zaměřila na možnou souvislost chronické intersticiální cystitidy s IgG4-RD. Je to práce autorů z Houstonu (Texas). Autoři se zaměřili na možnou souvislosti s IgG4-RD, analyzovali celkem 44 pacientů a jejich bioptických vzorků. Jednalo se o 7 mužů a 37 žen. Imunohistochemické vyšetření analyzovalo přítomnost IgG a IgG4 v plazmatických buňkách. Ve 4 případech byl prokázáno signifikantně zvýšené zastoupení IgG4-pozitivních plazmatických buněk s více než 30 IgG4+plazmocyty v zorném poli (per high‑power field) a poměr IgG4/IgG byl vyšší než 0,5.
Pacienti s IgG4 pozitivní intersticiální cystitidou byli vyššího věku, měli zvýšené zánětlivé markery a sníženou kapacitu močového měchýře. Autoři této práce se domnívají, že u části pacientů je intersticiální cystitida projevem IgG4-RD a mohla by tak být i léčena (82).
Ledviny
Nejčastější manifestací v ledvinách je tubulointersticiální nefritida a membranózní glomerulonefritida. Postižení ledvin touto nemocí se projeví jako akutní či chronické renální selhání, ložisko v ledvině či obojí. V případě tubulointersticiální nefritidy způsobené Ig4-RD je při CT zobrazení s kontrastem vidět obvykle bilaterální poškození parenchymu ledvin, ale obvykle dominuje poškození kortexu ledvin ve formě kortikálních nodulů anebo difuzní poškození. Ale patologické změny mohou být nacházeny i v okolním ledvinném parenchymu ve formě lemu v měkkých tkáních kolem ledvin, případně nodularit v renálních sinusech a zesílení ledvinné pánvičky. V histologickém obraze je přítomna jak typická fibróza, tak lymfoplazmocytární infiltrát s vysokým počtem IgG4+ buněk a nález někdy doplňují depozita Ig a/nebo C3 složky komplementu v bazální membráně tubulů a nález je často doplněn různými formami glomerulárního poškození. Proto se usuzuje, že na patogenezi IgG4-RD se podílejí imunitní komplexy (83–89).
Pulmonální, mediastinální a pleurální projevy IgG4-RD
IgG4-RD postihuje také dýchací cesty, pleuru a mediastinum. Pacienti přicházejí s kašlem, dušností, s bolestí na hrudníku, ale mohou být i bez symptomů.
Plicní postižení zahrnuje tvorbu nodulů „inflammatory pseudotumor“, ale také může způsobit intersticiální plicní postižení. Na viscerální a parietální pleuře tvoří někdy IgG4-RD nodulární ložiska. Ale také bronchiální zánět, edém a stenóza bývají projevy či následky plicní formy IgG4-RD. Mediastinální fibróza je pak dalším projevem této nemoci. Nález na HRCT hrudníku je nespecifický a ukáže různé formy postižení (nodulární zastínění, konsolidace, postižení intersticia, plicní fibrózu, lymfadenopatie). Diferenciální diagnostika je velmi obtížná a poměrně široká, protože plíce mají tendenci reagovat stereotypně na různé formy poškození. Diferenciálně diagnosticky je nutné vyloučit plicní karcinom, metastázy jiného origa, intersticiální plicní procesy včetně sarkoidózy (90–98).
Lymfatické uzliny
Lokalizované či systémové postižení uzlin je u IgG4-RD časté, a tak IgG4-RD je nutno přiřadit do diferenciální diagnostiky lymfadenopatie, kam patří lymfom, metastázy karcinomů, Castlemanova choroba a jiné imunitně mediované nemoci či krevní choroby.
Horečky, noční poty čili B symptomy nejsou ale typické pro IgG4-RD.
Histologie uzlin při IgG4-RD se mírně liší od obvyklé charakteristiky, storiformní fibróza a obliterativní flebitida obvykle nejsou přítomné. A tak je nutno myslet na multicentrickou formu Castlemanovy nemoci, reaktivní folikulární hyperplazii, progresivní transformaci germinálních center. Odlišit jednotlivé formy postižení lymfatické uzliny je proto obtížné (99–101). Někdy je problém rozlišit IgG4-RD lymfadenopatii od lymfomu, neboť lymfadenopatie se může postupně transformovat do lymfomu (102).
Další orgány
Taktéž nervový systém je v některých případech poškozen IgG4-RD včetně periferních nervů. Častou lokalizací poškození jsou orbitální a spinální nervy s typickou infiltrací perineuria (perineural disease). Neurologické příznaky jsou ale řídké. Nemoc někdy způsobuje také hypofyzitidu anebo mohou vzniknout inflamatorní pseudotumory přímo v CNS. Přibližně asi jedna třetina případů s hypertrofickou pachymeningitidou je způsobena IgG4-RD (103–105). Dále se popisuje idiopatická cervikální fibróza spojená s IgG4-RD, neobvyklá sklerotizující mastitida, která je také součástí spektra IgG4-RD. U mužů byla popsána prostatitida, orchitida a paratestikulární tumory. V nečetných případech byla IgG4-RD příčinou postižení kůže, na níž byla zodpovědná za vytvoření erytematózních svědících plaků anebo podkožních nodulů. Perikard byl touto nemocí postižen ve formě konstriktivní perikarditidy. Přehled všech příznaků s německou pečlivostí rozvádí Pieringer a kol (106).
Klinické fenotypy IgG4-RD
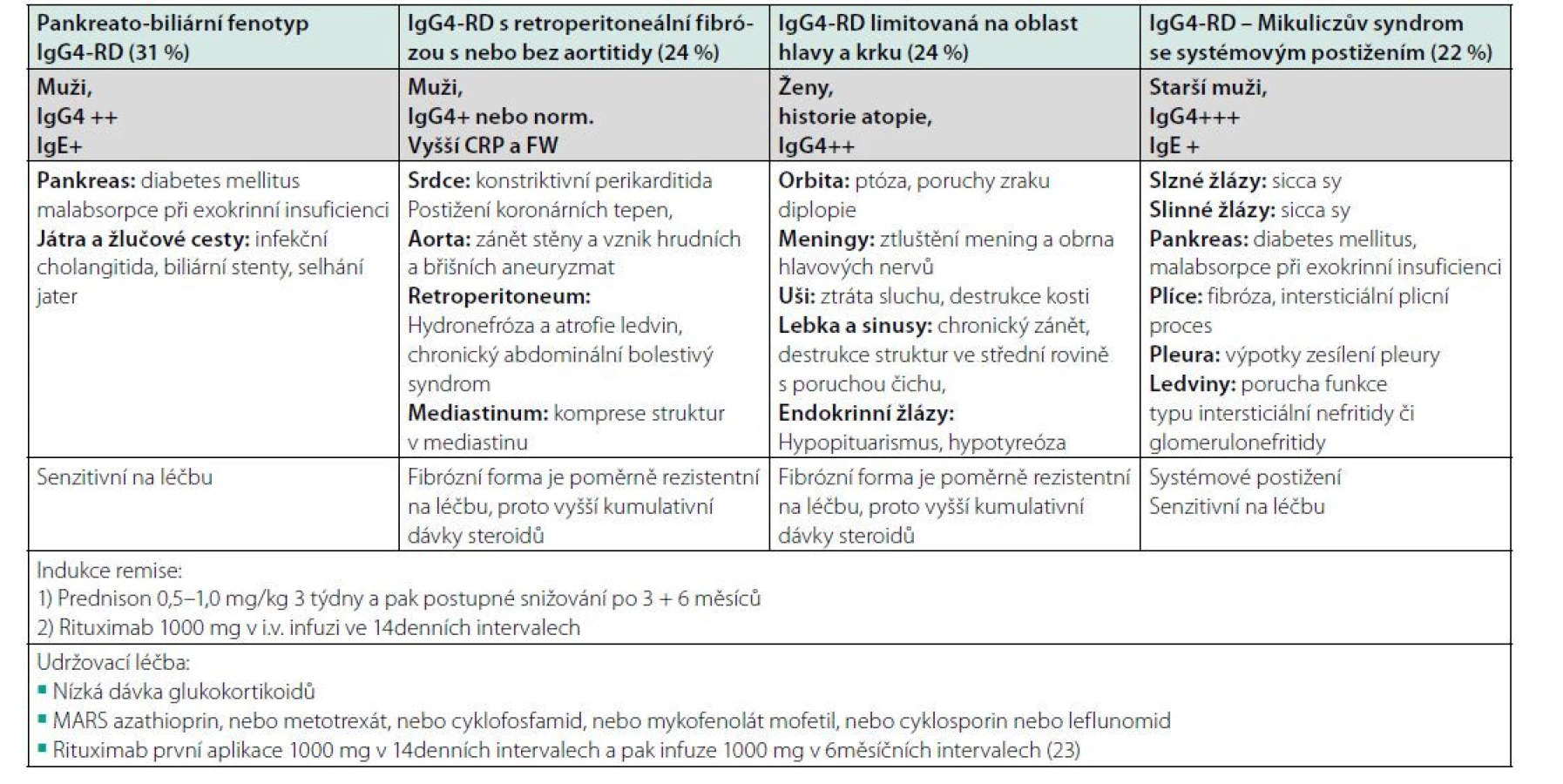
V předchozích odstavcích jsme uvedli výčet možných manifestací. Z tohoto výčtu plyne, že tato nemoc překvapuje postižením kteréhokoliv orgánu. Při analýze pacientů s touto nemocí se zjistilo, že některé formy postižení se velmi často vyskytují společně a že lze popsat čtyři základní fenotypy klinické manifestace. Toto didaktické rozdělení do několika skupin umožňuje lépe si představit, co vše se pod označením IgG4-RD skrývá.
Při analýze 800 pacientů s touto nemocí byly identifikovány čtyři homogenní fenotypy IgG4-RD:
• fenotyp pankreato-biliární (31 %),
• fenotyp retroperitoneální fibrózy s nebo bez aortitidy (24 %),
• fenotyp IgG4-RD limitovaný na oblast hlavy a krku (24 %),
• fenotyp Mikuliczova syndromu se systémovým postižením (22 %).
Uvedené fenotypy se liší nejen lokalizací postižení, ale také epidemiologickými a sérologickými nálezy. Pacienti s omezením nemoci na hlavu a krk jsou častěji ženy, ostatní typy jsou mezi ženy a muže rozloženy vyváženě. Pacienti s pankreato‑biliárním postižením jsou velmi často přijímáni do nemocnice s akutními komplikacemi. Zánětlivé parametry jsou naproti tomu nejvíce zvýšené ve třetí skupině (107). Toto relativně nové členění na čtyři fenotypy IgG4-RD uvádí tabulka 4.
Typické laboratorní nálezy
Eozinofilie
Asi 40 % pacientů s IgG4-RD má eozinofilii, často provázenou astmatem (108–110). Tedy IgG4-RD je důležitá a asi podhodnocená příčina sekundární či reaktivní eozinofilie.
Jak hypereozinofilní syndrom, tak IgG4-RD obvykle postihují kůži, plíce, zažívací trakt a lymfatické uzliny. Idiopatický hypereozinofilní syndrom a hypereozinofilie bez zjevné příčiny jsou diagnózy, které se stanoví jedině vyloučením jiných známých příčin a dle některých pramenů těchto eozinofilií bez objevené příčiny bývá až 30–50 %. Proto u pacientů s eozinofilií nejasné etiologie je nutno myslet i na IgG4-RD.
Znaky myeloidní klonální proliferace, jako je zvýšený počet blastů, abnormální karyotyp a mutace typu PDGFR‑alfa/ beta, FGFR-1 anebo PCM1-JAK2 se u pacientů s IgG4-RD nenacházejí. Ale odlišení lymfocytární varianty hypereozinofilního syndromu od IgG4-RD je obtížnější. Aberantní T‑buněčný fenotyp nalézaný v případech lymfocytární varianty hypereozinofilního syndromu zahrnuje zvýšený počet CD4+CD3−, CD3+/CD4−/ CD8 − a CD4+/CD7 − T buněk, s nebo bez T‑buněčné klonality (110, 111).
Eozinofilie způsobená IgG4-RD je obvykle mírná či střední, jen výjimečně přesahují absolutní hodnoty eozinofilů 5×109/l a mizí po kortikoidech či rituximabu (112).
Polyklonální hypergamaglobulinemie a další laboratorní nálezy
Laboratorní známky jsou často nenápadné. Zánětlivé markery, jako je sedimentace erytrocytů a CRP, jsou obvykle jen mírně zvýšené, ale mohou být i normální. Anti‑nukleární protilátky, anti-SS‑A stejně jako anti‑SS‑B protilátky jsou u většiny pacientů negativní, častější abnormalitou je snížení C3 a C4.
Snad jedině hypergamaglobulinemie je častější u této diagnózy, proto tuto nemoc uvádíme v rámci diferenciální diagnózy polyklonální hypergamaglobulinemie. Zvýšená sérová koncentrace IgE a alergie jsou přítomny asi u jedné třetiny nemocných. Vyšetření podtříd imunoglobulinů typu IgG prokáže vysoké hodnoty IgG4, ale ne u všech nemocných s IgG4-RD. Zdůrazňuje se, že koncentrace IgG4 mohou být docela zavádějící, pokud jsou použity, jako jediné kritérium pro potvrzení či vyloučení diagnózy. Na druhé straně četné jiné nemoci, jako jsou nádory, infekce a autoimunitní choroby, včetně vaskulitid, jsou spojené s vyššími hodnotami IgG4. Proto práce udávající senzitivitu zvýšené hodnoty IgG4 mají velmi divergentní výsledky od vysokých 90 % až po nízké 10 %. A záleží také na metodice vyšetření (106).
Zvýšená koncentrace IgG4 často provázená vzestupem IgG1 způsobí polyklonální hypergamaglobulinemii. Zcela výjimečně tato elevace vede k hyperviskóznímu syndromu. V současnosti není známa příčina vzestupu koncentrace IgG4, a tak je pochopitelná tendence nazývat tento jev epifenoménem. Ale zvyšují se i koncentrace volných lehkých řetězců. IgE bývá často vyšší, zvláště u pacientů s eozinofilií, zatímco IgA a IgM jsou v normě, či jen nepatrně vyšší. Diagnostika IgG4-RD má svá specifika a je nutno vyloučit klonality.
Co je příčinou hypergamaglobulinemie? V databázi PUBMED Medline jsou pouze dvě analýzy (113, 114).
Objev IgG4-RD vedl také k objevení reakcí podtříd imunoglobulinů IgG4 na různé stimuly. Například asociace hepatitidy C se zvýšením koncentrace podtřídy IgG1, asociace hypotyreózy a syndromu dráždivého trakčního se zvýšenou koncentrací podtřídy IgG2, asociace revmatoidní artritidy se zvýšenou koncentrací podtřídy IgG3 a IgG1 a také asociace celiakie se zvýšenou koncentrací podtřídy IgG4 (115). Zcela výjimečně byl popsán IgG4 myelom (7).
Vyšetření kostní dřeně v případě IgG4-RD nepřináší žádné dodatečné informace, počet plazmocytů nebývá zvýšen i při vyšší hladině imunoglobulinů třídy IgG (115).
Diferenciální diagnóza
IgG4-RD imituje četné maligní choroby v závislosti na lokalizaci. Další diferenciální diagnostický problém tvoří choroby provázené lokální či celkovou zánětlivou reakcí. Je jich tolik (116), že je zmíníme pouze v tabulce 5. Problémem je, že je spektrum chorob, které jsou provázeny vyšší hodnotou koncentrace podtřídy imunoglobulinu IgG, není malé. Hladina IgG4 bývá zvýšená u autoimunních onemocnění (ANCA – asociované vaskulitidy, revmatoidní artritida, systémový lupus erytematodes, spondyloartritidy). Taktéž některé choroby GIT (autoimunitní hepatitida, chronická pankreatitida, chronická hepatitida, jaterní cirhóza, ulcerózní proktokolitida) mohou být provázené vyšší koncentrací IgG4. A to samé bylo pozorováno v případě některých nádorových onemocnění (kolorektální karcinom, karcinom plic a zhoubná onemocnění urogenitálního traktu). Takže samotný nález vyšší hodnoty IgG4 neznamená, že se jedná o IgG4-RD.

Rozlišení IgG4-RD a idiopatické multicentrické Castlemanovy choroby (iMCD)
Problémy někdy činí rozlišení IgG4-RD od idiopatické multicentrické formy Castlemanovy choroby, protože jak pacienti s IgG4-RD, tak pacienti s iMCD mívají vyšší sérové hodnoty IgG4 a také infiltráty obsahující IgG4+ plazmocyty v ložisku nemoci (117, 118) a patolog proto svůj nález uzavře jako IgG4-RD/iMCD. Nelze spoléhat pouze na morfology, že jejich vyšetření jednoznačně odpoví na otázku, zda jde o IgG4-RD, nebo o iMCD, ale je vhodné přihlédnout i ke klinickým příznakům (119).
Japonští autoři, kteří srovnávali 45 pacientů s IgG4-RD a 33 s iMCD, popsali následující rozdíly:
• alergická onemocnění byla přítomna u 70 % pacientů s IgG4 RD a jen u 33 % s iMCD. Horečka a další projevy systémové zánětlivé reakce byly více časté u pacientů s iMCD než u pacientů s IgG4-RD,
• paravertebrální infiltráty byly jen u pacientů s IgG4-RD stejně jako postižení exokrinních žláz,
• lymfadenopatie byla přítomna vždy u iMCD, proto lze iMCD vyloučit, pokud není lymfadenopatie přítomna (120, 121).
Ale přesto je možno najít atypické případy, kdy rozlišení není dobře možné. Rozdíly mezi IgG4-RD a iMCD shrnuje tabulka 6. IgG4-RD ale postihuje starší pacienty a jen výjimečně má inflamatorní známky odpovídající systémové zánětlivé reakci, v jejímž pozadí je u Castlemanovy choroby zvýšená koncentrace IL-6 a tedy i zvýšená hodnota C‑reaktivního proteinu.

Rozlišení IgG4-RD a histiocytárních chorob
Vzácné histiocytární onemocnění, zvané Rosaiova‑Dorfmanova choroba, obsahuje také občas v infiltrátu IgG4+ buňky a má zvýšené hodnoty IgG4 v séru. Odlišení těchto dvou chorob je velký problém. Nejenže mají některé morfologické znaky stejné, vyjma fibrózy, která je typická pro IgG4-RD, ale mohou mít i některé shodné klinické projevy, jako je například pachymeningitida.
Histopatologické vyšetření v případně Rosaiovy‑Dorfmanovy choroby také vykazuje vyšší počet IgG4+ plazmatických buněk, ale obvykle spolu s CD68+ S100+ histiocyty. Rosaiova‑Dorfmanova choroba je asociovaná s morfologickým jevem zvaným emperipolesis (122–126). Diferenciální diagnostika je někdy problémem, i když se postupuje dle doporučení pro stanovení diagnózy Rosaiovy‑Dorfmanovy nemoci (127)
Klasifikace histiocytárních neoplazií doporučuje při podezření na Rosaiovu‑Dorfmanovu chorobu pátrat po zvýšeném počtu IgG4+ plazmatických buněk, ale jinak se Rosaiova‑Dorfmanova choroba nepovažuje za součást IgG4–RD (127).
Asi jedna třetina pacientů s Erdheimovou‑Chesterovou chorobou má retroperitoneální fibrózu. Ale na rozdíl od IgG4-RD více než 95 % osob s Erdheimovou‑Chesterovou chorobu má postižení skeletu, což ale nebývá u IgG4-RD. Průkaz hyperostotických změn skeletu odpovídajících Erdheimově-Chesterově nemoci dává tušit, s jakou diagnózou bude asociována retroperitoneální fibróza. Histologie u Erdheimovy‑Chesterovy choroby vykazuje CD68+S100−CD1a − histiocyty, často ve formě „foamy histiocytes“ neboli pěnitých buněk (127).
Dalším diferenciálně diagnostickým problémem je nové onemocnění, definované až v roce 2018, zvané „Immunoglobulin G4-related chronic rhinosinusitis, český ekvivalent názvu se zatím neustálil (128).
Mimoplicní sarkoidóza mívá společně rysy s IgG4-RD, včetně polyklonální hypergamaglobulinemie, lymfadenopatie plicních nodulů a sklerotizující mezenteritidu a pachymeningitidu, jak podrobněji rozvádí Doubková (97).
Pozornost byla věnována také asociaci IgG4-RD a maligních lymfomů. V asijské populaci byla častější asociace s takzvanými MALT lymfomy (mucosal‑associated lymphoid tissue lymphoma) lokalizovanými často v očnici, zatímco u bílé rasy byly popisovány divergentní typy lymfomů (difuzní velkobuněčný B‑lymfom, folikulární, lymfoplazmocytární a také MALT lymfom). Také byla popsána IgG4-RD současně probíhající s autoimunitním lymfoproliferativním syndromem (1, 101, 129). Problémy diferenciální diagnostiky shrnují tabulky 5 a 6.
Stanovení diagnózy
V rozhovoru s pacientem je třeba se zeptat na zjištěné zvětšené uzliny, které mohou svoji velikost postupně měnit (v průběhu času se zvětšovat a zmenšovat), na otoky a zduření slinných žláz, na příznaky sicca syndromu, na nevysvětlitelnou pankreatitidu a žloutenku. Zásadní je ale histologický průkaz nemoci. A pokud se podaří nemoc histologicky prokázat, tak je zásadní cílené vyšetření, pátrající po známkách poškození orgánů a po retroperitoneální fibróze. Přehled vyšetření a nálezů typických pro IgG4-RD přináší tabulka 7 (128).

Vyšetření podtřídy imunoglobulinů typu IgG (IgG1-IgG4)
Asi 70 % pacientů s IgG4-RD má zvýšené hladiny IgG4. Monoklonální imunoglobulin se musí vždy samozřejmě také vyšetřit. Zvýšená hladina IgG4 má diagnostickou senzitivitu 83–97 % a specificitu 60-85 % s tím, že za hranici se bere horní limit normálních hodnot (130). Obvykle se za horní hranici normy pro IgG4 bere 1,35 g/l, i když tato hranice vždy souvisí s použitou metodou a dle metodik se mění. Mírně zvýšené hodnoty IgG4 je možné prokázat i v jiných případech. Výrazné zvýšení koncentrace IgG4 > 5 g/l je přibližně z 90 % specifické pro IgG4-RD. Kolísání hladiny však není dáno jenom metodami stanovení IgG4, ale také v případě nemoci IgG4-RD odvisí od míry orgánového poškození. Ve studii z Bostonu mělo jen 53 ze 103 pacientů zvýšené hladiny IgG4 (28). V Japonsku ve skupině 334 pacientů mělo více než 95 % zvýšené IgG4 (29). V multietnické studii bylo prokázáno, že Asiaté měli vyšší sérové IgG4 než neasijská skupina (medián 11,2 g/l versus 2,9 g/l, p = 0,0094). Zvýšená hladina IgG4 měla 96 % senzitivitu u Asiatů a jen 67 % u ne-Asiatů (131, 132).
Poměr IgG4/IgG je typicky > 0,2 v případě IgG4-RD, ale tento poměr nezvyšuje specificitu sérové koncentrace IgG4. Proto v Evropě není měření podtřídy IgG4 vhodné pro skríning, ani pro vyloučení nemoci. V západní Evropě má asi jen 50–59 % osob s IgG4-RD zvýšené hladiny IgG4, zatímco v Asii je téměř ve 100 %. Problém je, že u některých chorob je vyšší koncentrace imunoglobulinů podtřídy IgG4, aniž by to byla IgG4-asociovaná choroba.
Zvýšené hodnoty IgG4 v séru jsou nespecifické a tento laboratorní nález se vyskytuje taktéž u multicentrické Castlemanovy choroby, alergických onemocnění včetně eozinofilní granulomatózy s polyangiitidou (syndrom Churga – Straussové), sarkoidózy.
Problém je, že také u karcinomu slinivky břišní je občas zvýšená hodnota IgG4, jak popisuje prof. Dítě a další (133, 134).
V laboratorním obraze IgG4-RD se lze setkat i s pozitivitou ANA a s pozitivitou revmatoidních faktorů, s typicky sníženou hodnotou C3 a C4 složky komplementu, se vzestupem IgE imunoglobulinu a s přítomností tkáňové depozice imunokomplexů (130).
Není ale zanedbatelný ani analytický problém a problém s interpretací výsledků. Existují dvě hlavní nefelometrické metody (Siemens a Binding Site), které se liší a které mají své interpretační problémy. A proto někteří autoři doporučují vyšetření koncentrace IgG4 hmotnostní spektrometrií (135, 136).
Plazmablasty v periferní krvi, marker aktivity nemoci
Aktivitu nemoci lze hodnotit i s pomocí průtokového cytometrického vyšetření.
Někteří autoři dávají přednost před vyšetřením hladiny IgG4 imunoglobulinu v séru podchycení obvykle dramaticky zvýšeného počtu IgG4+ plazmablastů v periferní krvi. Toto flowcytometrické vyšetření slouží nejen pro diagnostiku aktivní formy IgG4-RD, ale i pro monitoraci vývoje nemoci. Vysoký počet plazmablastů v periferní krvi lze identifikovat s pomocí vícebarevné průtokové cytometrie (CD19low, CD38+, CD20-, CD27+). Plazmablasty bývají zvýšeny i v případě normálních hladin IgG4 v séru. Vyšetření zvýšeného počtu cirkulujících plazmablastů je doporučováno jako potenciální diagnostický biomarker nemoci, využitelný i v hodnocení hloubky léčebné odezvy a odhalení progrese s potřebou zahájení opětovné léčby. Flow‑cytometrická detekce plazmablastů v krvi poskytuje více senzitivní modalitu pro diagnostiku IgG4-RD. Uvádí se senzitivita 95 % a specificita 82 % při použití hranice 900/ml (137). Tento typ flowcytometrického vyšetření vyžaduje již ale hodně výkonný mnohobarevný flowcytometr a zkušené pracovníky (137–141).
Histopatologie
Histologie se bere za základ pro stanovení diagnózy s výjimkou pro případy s typickými změnami na pankreatu odpovídajícími autoimunitní pankreatitidě. Pokud je obraz postižení pankreatu dostatečně specifický, může být ustoupeno od biopsie (52, 77). Histologický obraz IgG4-RD má tři hlavní charakteristiky:
• denzní polyklonální lymfoplazmocytární infiltrát bohatý na IgG4+ plazmatické buňky,
• storiformní fibrózu,
• obliterativní flebitidu (75).
Diagnostický počet IgG4+ plazmocytů v jednom zorném poli mikroskopu se liší dle tkáně, od hodnoty > 10/zorné pole v případě mening po > 100/zorné pole v případě kůže. Ale nehledě na absolutní počet IgG4+ plazmocytů v zorném poli, za patognomický pro IgG4 - RD se bere poměr IgG4+/IgG+ plazmatických buněk > 40 % (52, 77).
Fibróza je také požadovaná pro stanovení diagnózy IgG4-RD, buď v preparátu dominuje, nebo je prokazatelná jen fokálně. Storiformní fibróza však nemusí být zastižena v malých vzorcích, a proto je třeba vždy většího vzorku pro průkaz nemoci, aby mohla být zastižena.
Třetím charakteristickým znakem je obliterativní flebitida, žilní lumen je obliterováno inflamatorním lymfoplazmocytárním infiltrátem. Doporučuje se sledovat v histologickém preparátu průběh arterií a arteriol a pátrat, zda jsou provázeny žilními cestami, protože pokud žíly nejsou jasně zřetelné, tak mohly být nahrazeny zánětlivým infiltrátem. Barvení na elastin pomůže identifikovat kompletně obliterované žilky.
Dalším histologickým znakem je flebitida bez obliterace cévního lumen a zvýšený počet eozinofilů.
Pokud mají tito nemocní archivované vzorky z četnějších předchozích odběrů histologických vzorků vzhledem k chronickému charakteru této nemoci, tak se doporučuje zpětně v nich doplnit imunohistochemické barvení na IgG4+pozitivní buňky.
Samotný zvýšený počet IgG4+ plazmatických buněk, který je patrný ve všech vzorcích tkání, postižených IgG4-RD, není dostatečně specifický nález. Četné chronické inflamatorní procesy, jako jsou vaskulitidy, zánětlivé nemoci střeva a lymfomy, mohou vykazovat zvýšený počet IgG4+ plazmatických buněk, ale nejsou přítomné další histologické znaky, jako je storiformní fibróza, obliterující flebitida a absence granulomatózního zánětu.
Postižení kostní dřeně není u IgG4-RD běžné a obliterativní flebitida a storiformní fibróza nejsou typicky přítomné v kostní dřeni a v lymfatických uzlinách pacientů s IgG4-RD. Pro odběr histologie je tedy nutno vždy volit ten orgán a tu tkáň, které na zobrazovacích vyšetřeních vykazují známky postižení.
U pacientů s proteinurií se provádí biopsie ledvin. Postižení ledvin v případě IgG4-RD mívá jeden ze dvou histologických obrazů. V 80 % případů to bývá hypokomplementární tubulointersticiální nefritida a ve 20 % případů membranoproliferativní nefritida (142).
U pacientů, u nichž nelze postižený orgán bioptovat, se doporučuje odběr malé slinné žlázky ze rtu. Dokonce v případech bez klinického průkazu postižení velkých slinných žláz nebo sicca syndromu při minimální invazivitě někdy prokáže diagnózu. V jedné studii 66 pacientů se suspektním IgG4-RD prokázala biopsie labiální žlázy senzitivitu 55 % a specificitu 100 % (143).
Z pragmatického pohledu u pacientů, kteří mají klasické laboratorní a zobrazovací známky IgG4-RD, ale jsou křehcí na provedení biopsie nebo biopsie neprokázala jasný závěr, je možné diagnózu uzavřít jako suspektní IgG4-RD a při vyloučení jiných chorob tyto případy léčit jako IgG4-RD.
Diagnostická kritéria
U všech chorob, u nichž neexistuje jediný jasně definovaný znak, jehož přítomnost potvrzuje přítomnost nemoci a nepřítomnost vylučuje chorobou, byla, jsou a budou publikována diagnostická kritéria, která se vždy v čase vyvíjejí tak jak se vyvíjí poznání této nemoci. První diagnostická kritéria byla definována v roce 2011 a publikována v roce 2012 kolektivem japonských autorů a jsou uvedena v tabulce 8 (144). Tato kritéria pak byla upravena v roce 2020 týmem japonských odborníků a zveřejněna v roce 2021, viz tabulka 9.


Jak je vidět, tato kritéria jsou podstatně jednodušší a v praxi snáze realizovatelná než poměrně komplikovaná kritéria, která byla dohodnuta na sjezdu Evropské a Americké revmatologické společnosti. Specialitou japonských kritérií je, že akceptují mezinárodní kritéria pro jednotlivé orgány, protože obraz nemoci se v jednotlivých orgánech liší. A proto Umehara doporučuje kombinovat obecná kritéria s kritérii pro manifestaci nemoci v určitém orgánu. Dle tohoto japonského přístupu je diagnóza považována za jednoznačnou, pokud jsou splněna Umeharou uvedená kritéria anebo pokud jsou splněna kritéria IgG4-RD publikovaná pro jednotlivé orgány (145, 146) viz tabulka 10.
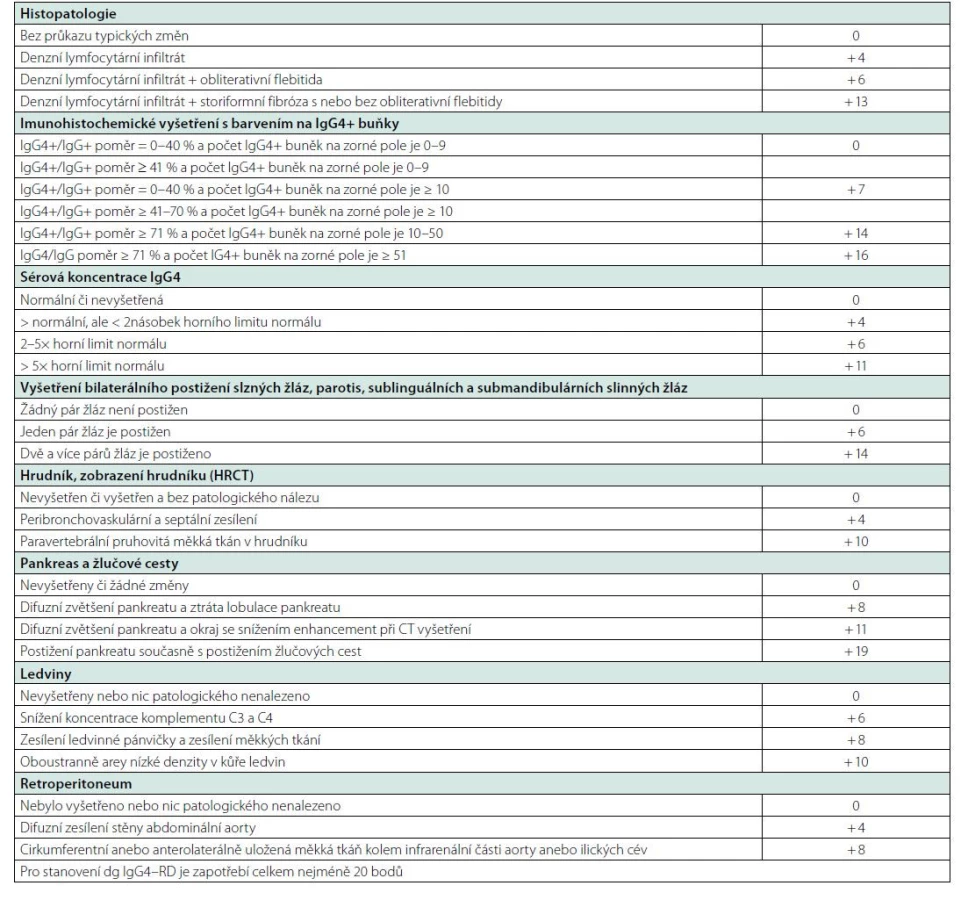
V roce 2019 byla pak ustanovena na konferenci Evropské a Americké revmatologické společnosti poměrně složitá nová mezinárodní kritéria, která byla zveřejněna v roce 2020 (147, 148), viz tabulka 10. Tato kritéria jsou nepoměrně složitější než japonská kritéria, protože dávají jednotlivým parametrům bodovou hodnotu a pro splnění diagnózy požadují dosažení 20 bodů. Na rozdíl od japonských kritérií, která považují IgG4 koncentraci za zvýšenou na hodnotě nad 135 mg/dl neboli 1,35 g/l, tak kritéria Evropské a Americké revmatologické společnosti definují zvýšení násobkem od horní fyziologické hranice.
Takže v současnosti máme k dispozici více uznávaných kritérií, dle kterých se budeme orientovat u konkrétního pacienta.
Uvedený turbulentní vývoj kritérií jen dokazuje, jak obtížné je v současnosti stanovit diagnózu této nemoci a odlišit ji od podobných chorob, které také mohou mít vyšší počet IgG4 plazmocytů anebo mohou být provázeny zvýšenou sérovou koncentrací podtřídy imunoglobulinu IgG4.
Závěr
IgG4-RD je nová choroba, která často uniká delší dobu diagnóze z několika důvodů: imunoglobulin IgG4 je stanovován pouze ve specializovaných imunologických laboratořích. Běžné laboratoře, které stanovují polyklonální a monoklonální imunoglobuliny, toto vyšetření z organizačně ekonomických důvodu v ČR neprovádějí. Provedení barvení na IgG4 pozitivní buňky není taktéž běžně prováděno, pokud není vysloveno podezření na tuto chorobu. Cílem tohoto článku je formou přehledného textu upozornit na všechny možné manifestace této nemoci, které pacienta přivedou tu s fibrózní strumou k endokrinologovi, tu s pachymeningitidou k neurologovi, tu s expanzivním procesem v orbitě k očnímu lékaři, případně s retroperitoneální fibrózou k urologovi anebo s podezřením na tumor pankreatu ke gastroenterologovi, jak také pěkně popsala Chovancová (149). Přitom se pořád jedná o jednu chorobu, i když s různými manifestacemi. Diagnostika vůbec není jednoduchá, jak vyplývá z četných pro tuto nemoc publikovaných kritérií a četnosti prací zabývajících se diferenciální diagnostikou. Prognostický index pro tuto chorobu byl vypracován relativně nedávno a je v procesu validace (150, 151), zatím není standardně používán. S prognózou a odpovědí na léčbu souvisí i přítomnost či nepřítomnost eozinofilie (152). Pacienti s eozinofilií mívají masivnější postižení a onemocnění má u nich větší tendenci recidivovat. Poznání této vzácné nemoci se rozvíjí pozvolna teprve v posledních letech, a tak každý další rok přinese další informace do mozaiky představ o této nemoci, podobně jako v roce 2015 popsali japonští autoři synovitidu provázející IgG4 related disease (153), jev, který do té doby nebyl znám.
Přehledem popsaných projevů chce multioborový tým spoluautorů přispět k časné diagnostice této nemoci. Přehled zkušeností s léčbou této nemoci uvedeme v další kapitole.
Publikace byla vytvořena na podporu těchto aktivit
MZ ČR – RVO (FNBr, 65269705)
a MOÚ: MZ ČR – RVO (MOÚ, 00209805)
KORESPONDENČNÍ ADRESA AUTORA:
prof. MUDr. Zdeněk Adam, CSc.
Interní hematologická a onkologická klinika, LF MU a FN Brno
Jihlavská 20, 625 00 Brno
Cit. zkr: Vnitř Lék. 2022;68(5):E4-E19
Článek přijat redakcí: 1. 12. 2021
Článek přijat po recenzích: 20. 5. 2022
Zdroje
1. Campr V. Monitor, aneb nemělo by vám uniknout, že co by hematopatolog měl vědět o IgG4-asociované nemoci. Česko‑slovenská patologie a Soudní lékařství. 2019;55-64 (4):200-2002.
2. Bojková M, Dítě P, Dvořáková J et al. Immunoglobulin G4, autoimmune pancreatitis and pancreatic cancer Digestive diseases (Basel. Online). 2015;33(1):86-90.
3. Hrnčíř Z. Nové imunoglobulinové biomarkery u revmatických chorob: volné lehké řetězce a IgG4. Rheumatologia (Bratislava). 2013;27(3):126-127.
4. Bartholomew LG, Cain JC, Woolner LB et al. Sclerosing cholangitis: its possible association with Riedel’s struma and fibrous retroperitonitis: report of two cases. N Engl J Med. 1963;269 : 8-12.
5. Yoshida K, Toki F, Takeuchi et al. Chronic pancreatitis caused by an autoimmune abnormality. Proposal of the concept of autoimmune pancreatitis. Dig Dis Sci. 1995;40 : 1561-1568.
6. Hamano H, Kawa S, Horiuchi A et al. High serum IgG4 concentrations in patients with sclerosing pancreatitis. N Engl J Med. 2001;344(10):732-738.
7. Kamisawa T, Funata N, Hayashi Y et al. A new clinicopathological entity of IgG4-related autoimmune disease. J Gastroenterol. 2003;38(10):982-984.
8. Hamano H, Kawa S, Ochi Y et al. Hydronephrosis associated with retroperitoneal fibrosis nand sclerosing pancreatitis. Lancet. 2002;359 : 1403-1404.
9. Dítě P. Husová L, Lukáš Z. Imunoglobulin G4 pozitivní cholangitida. Vnitř. Lék. 2010;56(8):824-826.
10. Dítě P, Novotný I, Kinkor Z. Autoimunní forma chronické pankreatitidy a IgG4 pozitivní mastitida Gastroenterologie a hepatologie. 2011;65(1):22-25.
11. Dítě P, Novotný I, Lata J et al. Autoimunitní pankreatitida a IgG pozitivní sklerotizující cholangitida. Vnitř. Lék. 2011;57(3):254-257.
12. Chari ST, Smyrk TC, Levy MJ et al. Diagnosis of autoimmune pancreatitis: the Mayo Clinic experience. Clin Gastroenterol Hepatol. 2006;4 : 1010-1016.
13. Ryu JK, Chung JB, Park SW et al. Review of 67 patients with autoimmune pancreatitis in Korea: a multicenter nationwide study. Pancreas. 2008;37 : 377-385.
14. Deshpande V, Zen Y, Chan JK et al. Consensus statement on the pathology of IgG4-related disease. Mod Pathol. 2012;25(9):1181-1192.
15. Stone JH, Khosroshahi A, Deshpande V et al. Recommendations for the nomenclature of IgG4-related disease and its individual organ system manifestations. Arthritis Rheum. 2012;64(10):3061-3067.
16. Umehara H, Okazaki K, Masaki Y et al. Research Program for Intractable Disease by Ministry of Health, Labor and Welfare (MHLW) Japan G4 team. A novel clinical entity, IgG-4-related disease (IgG4RD): general concept and details. Mod Rheumatol. 2012;22(1):1-14.doi: 10.1007/s10165-011-0508-6.
17. Deshpande V. The pathology of IgG4-related disease: critical issues and challenges.Semin Diagn Pathol. 2012;29(4):191-196. doi:10.1053/j.semdp.2012. 08. 001.
18. Stone JH. IgG4-related disease: nomenclature, clinical features, and treatment. Semin Diagn Pathol. 2012;29(4):177-190. doi: 10.1053/j.semdp.2012. 08. 002.
19. Průcha M, Sedláčková L. IgG4-related disease - a patient with multiple organ involvement Prague Medical Report. 2017;118(2-3):95-99.
20. Uchida K, Masamune A, Shimosegawa T et al. Prevalence of IgG4-Related Disease in Japan Based on Nationwide Survey in 2009. Int J Rheumatol. 2012;2012 : 358-371.
21. Karim F, Loeffen J, Bramer W et al. IgG4-related disease: a systematic review of this unrecognized disease in pediatrics. Pediatr Rheumatol Online J. 2016;14(1):18.
22. Sasaki T, Akiyama M, Kaneko Y et al - IgG4-related disease and idiopathic multicentric Castleman’s disease: confusable immune‑mediated disorders. Rheumatology (Oxford). 2021 Aug 7:keab634. doi: 10.1093/rheumatology/keab634.
23. Mikulová Š, Jílek D, Richter J. Nemoc asociovaná s IgG4. Úvod, patogeneze, diagnostika. 1. část Alergie (Praha, Print). 2015;17(1):16-24.
24. Mikulová Š, Jílek D, Richter J. Nemoc asociovaná s IgG4. Klinický obraz, orgánová postižení a terapie. 2. část. Alergie (Praha, Print). 2015;17(2):91-99.
25. Král V, Pohorská J, Stiborová I et al. Onemocnění asociovaná s IgG4 – je příčinou porucha regulace imunitní odpovědi? Klinická imunológia a alergológia. 2016;26(1):42-48.
26. Endmayr V, Tunc C, Ergin L. Anti‑neuronal IgG4 autoimmune diseases and IgG4-related diseases may not be part of the same spectrum: a comparative study. Front Immunol. 2022;12 : 78547.
27. Koneczny I. Update on IgG4-mediated autoimmune diseases: New insights and new family members. Autoimmun Rev. 2020;19 : 102646.
28. Wallace ZS, Deshpande V, Mattoo H et al. IgG4-related disease: clinical and laboratory features in one hundred twenty‑five patients. Arthritis Rheumatol. 2015;67 (9):2466–2475.
29. Yamada K, Yamamoto M, Saeki T et al. New clues to the nature of immunoglobulin G4-related disease: a retrospective Japanese multicenter study of baseline clinical features of 334 cases. Arthritis Res Ther. 2017;19(1):262-270.
30. Hardacre JM, Iacobuzio‑Donahue CA et al. Results of pancreaticoduodenectomy for lymphoplasmacytic sclerosing pancreatitis. Ann Surg. 2003;237 : 853–858.
31. Kunovský L, Dítě P, Blaho M et al. Is autoimmune pancreatitis a risk factor for pancreatic adenocarcinoma? Vnitř Lék. 2021;67(1), e9-e13.
32. Dítě P, Novotný I, Dvořáková J et al. Pancreatic Solid Focal Lesions: Differential Diagnosis between Autoimmune Pancreatitis and Pancreatic Cancer - Digestive diseases (Basel. Online). 2019;37(5):416-421.
33. Dítě P, Novotný I, Kianička B, et al. Autoimunitní pankreatitida – diagnostický konsenzus. Vnitř. Lék. 2015;61(2):114-118.
34. Peňázová P, Andrašina T, Novotný I et al. IgG4 sklerozující cholangitida – zánět imitující nádor hlavy pankreatu a cholangiokarcinom Klinická onkologie. 2019;32 (2):143-151.
35. Vaňásek J, Horrmann P, Laco J et al. IgG4 asociovaná pankreatitida a cholangoitida. Česká radiologie. 2014;68(4):294-297.
36. Takahashi M, Fujinaga Y, Notohara K et al. Working Group Members of The Research Program on Intractable Diseases from the Ministry of Labor, Welfare of Japan. Diagnostic imaging guide for autoimmune pancreatitis. Jpn J Radiol. 2020;38(7):591-612. doi: 10.1007/s11604-020-00971-z.
37. Notohara K, Kamisawa T, Fukushima N et al. Guidance for diagnosing autoimmune pancreatitis with biopsy tissues. Pathol Int. 2020;70(10):699-711. doi: 10.1111/pin.12994.
38. Novotný I, Dítě P, Trna J et al. Immunoglobulin G4-related cholangitis: a variant of IgG-4-related systemic disease. Digestive diseases (Basel. Online). 2012;30(2):216-219.
39. Dítě P, Novotný I, Kianička B et al. Imunologlobulin G4 associovaná onemocnění. Gastroenterológia pre prax. 2011;10(3):151-152.
40. Hubers LM, Beuers U. IgG4-related disease of the biliary tract and pancreas:clinical and experimental advances. Curr Opin Gastroenterol. 2017;33(4):310-314. doi: 10.1097/MOG.0000000000000362.
41. Dítě P, Trna J, Kinkor Z et al. Unusual Multiorgan Immunoglobulin G4 (IgG4) Inflammation: Autoimmune Pancreatitis, Mikulicz Syndrome, and IgG4 Mastitis. Gut and liver. 2013;7(5):621-624.
42. Honsová E, Loderová A, Kostolná E et al. Autoimunní pankreatitida s postižením žlučovodů a jater jako součást IgG4 pozitivního autoimunního onemocnění (IgG4-related autoimmune sclerosing disease). Kazuistika Česko‑slovenská patologie a Soudní lékařství. 2010;46-55(3):65-67.
43. Löhr JM, Beuers U, Vujasinovic M et al. UEG guideline working group.European Guideline on IgG4-related digestive disease - UEG and SGF evidence‑based recommendations. United European Gastroenterol J. 2020;8(6):637-666.doi: 10.1177/2050640620934911.
44. Kamisawa T, Nakazawa T, Tazuma S et al. Clinical practice guidelines for IgG4-related sclerosing cholangitis. J Hepatobiliary Pancreat Sci. 2019;26(1):9-42. doi: 10.1002/jhbp.596.
45. Chapman MH, Thorburn D, Hirschfield GM et al. British Society of Gastroenterology and UK‑PSC guidelines for the diagnosis and management of primary sclerosing cholangitis. Gut. 2019;68(8):1356-1378.doi: 10.1136/gutjnl-2018-317993.
46. Hori Y, Chari ST, Tsuji Y et al. Distinguishing IgG4-Related Sclerosing Cholangitis From Cholangiocarcinoma and Primary Sclerosing Cholangitis. Mayo Clin Proc Innov QualOutcomes. 2021;5(3):535-541. doi: 10.1016/j.mayocpiqo.2021. 03. 005.
47. Hrnčíř Z, Laco J. Klinický význam nemoci spojené s imunoglobuliny IgG4. Časopis lékařů českých. 2011;150(8):438-441.
48. Navarro‑Sánchez V, Marín‑Castañeda LA, Gallegos CA et al. IgG4-Related Fibrous Thyroiditis (Riedel’s Thyroiditis): A Case Report. Am J Case Rep. 2020;21:e928046. doi: 10.12659/AJCR.928046.
49. Rotondi M, Carbone A, Coperchini F et al. DIAGNOSIS OF ENDOCRINE DISEASE: IgG4-related thyroid autoimmune disease. Eur J Endocrinol. 2019;180(5):R175-R183. doi: 10.1530/EJE-18-1024.
50. Stan MN, Sonawane V, Sebo TJ et al. Riedel’s thyroiditis association with IgG4-related disease. Clin Endocrinol (Oxf). 2017;86(3):425-430. doi: 10.1111/cen.13238.
51. Takeshima K, Li Y, Kakudo K, et al. Proposal of diagnostic criteria for IgG4-related thyroid disease. Endocr J. 2021;68(1):1-6. doi: 10.1507/endocrj.EJ20-0557.
52. Laco J, Kamarádová K, Mottl R, et al. Plasma cell granuloma of the oral cavity: a mucosal manifestation of immunoglobulin G4-related disease or a mimic Virchows Archiv. 2015;466(3):255-63.
53. Kubíčková J, Zeleník K, Urban O et al. Manifestace IgG4 asociované nemoci v oblasti hlavy a krku. Otorinolaryngologie a foniatrie. 2015;64(3):163-167.
54. Hrnčíř Z, Laco J, Drahošová M et al. Biomarker IgG4 u Mikuliczovy choroby Rheumatologia (Bratislava). 2011;25(3)25-30.
55. Mačinga P, Jarošová J, Špičák J et al. Imonoglobulin G4 asociované onemocnění v gastroenterologii. Vnitř. Lék. 2021;67(2):76-83.
56. Laco J, Ryška A, Čelakovský P et al. Chronic sclerosing sialadenitis as one of the immunoglobulin G4-related diseases: a clinicopathological study of six cases from Central Europe. Histopathology (Oxford. Print). 2011;58(7):1157-1163.
57. Maehara T, Pillai S, Stone JH et al. Clinical features and mechanistic insights regarding IgG4-related dacryoadenitis and sialoadenitis: a review. Int J Oral Maxillofac Surg. 2019;48(7):908-916. doi: 10.1016/j.ijom.2019. 01. 006.
58. Marcus KS, Hoffman HT, Rajan Kd A et al. Not All Küttner Tumors Are IgG4-Related Disease (IgG4-RD). Head Neck Pathol. 2021. Jan 4. doi: 10.1007/s12105-020-01268-3.
59. Puxeddu I, Capecchi R, Carta F et al. Salivary Gland Pathology in IgG4-Related Disease: A Comprehensive Review. J Immunol Res. 2018;2018 : 6936727. doi: 10.1155/2018/6936727.
60. Liu Y, Xue M, Wang Z et al. Salivary gland involvement disparities in clinical characteristics of IgG4-related disease: a retrospective study of 428 patients. Rheumatology (Oxford). 2020;59(3):634-640. doi:10.1093/rheumatology/kez280.
61. Franeková L, Kozák I, Rovenský J. Onemocnění se vztahem k IgG4 Oftalmorevmatologie. První vydání. Praha, Galén 2017. 2017, 271-274.
62. Hrnčíř Z, Laco J, Slezák R et al. Mikuliczova choroba s jednostranným exoftalmem - onemocnění se vztahem k IgG4 Česká revmatologie. 2011;19(3):125-130.
63. Závorková M, Richter J, Větvička V, et al. IgG-4 asociované onemocnění v očním lékařství Česká a slovenská oftalmologie. 2017;73(3):109-112.
64. Detiger SE, Karim AF, Verdijk RM et al. The treatment outcomes in IgG4-related orbital disease: a systematic review of the literature. Acta Ophthalmol. 2019;97(5):451-459. doi: 10.1111/aos.14048.
65. Lee MJ, Planck SR, Choi D et al. Non‑specific orbital inflammation: Current understanding and unmet needs. Prog Retin Eye Res. 2021;81 : 100885. doi: 0.1016/j.preteyeres.2020.100885.
66. Lee HS, Choi W, Kim GE, Yoon KC. Case of Primary Isolated Subconjunctival IgG4-Related Disease. Cornea. 2018;37(7):926-928. doi:10.1097/ICO.0000000000001566.
67. Singh S, Selva D. Non‑infectious Dacryoadenitis. Surv Ophthalmol. 2021:S0039-6257(21)00135-1. doi: 10.1016/j.survophthal.2021. 05. 011.
68. Kubota T, Katayama M, Nishimura R et al. Long‑term outcomes of ocular adnexal lesions in IgG4-related ophthalmic disease. Br J Ophthalmol. 2020;104(3):345-349. doi: 10.1136/bjophthalmol-2018-313730.
69. Průcha M, Czinner P, Prokopová P et al. IgG4-related diseases - a rare polycystic form of Ormond’s disease. Prague Medical Report. 2016;117(2-3).124-128.
70. Průcha M, Sedláčková L. Onemocnění asociovaná s IgG4 – pacient s mnohočetným orgánovým postižením. Medicína po promoci. 2016;17(1):70-71.
71. Laco J, Podhola M, Kamarádová K et al. Idiopathic vs. secondary retroperitoneal fibrosis: a clinicopathological study of 12 cases, with emphasis to possible relationship to IgG-4-related disease. Virchows Archiv. 2013;463(5):721-30.
72. Průcha M, Kolombo I, Štádler P. Ormond’s Disease‑IgG4-related Disease. Prague Med Rep. 2015;116(3):181-92.
73. Průcha M, Šedivý P, Štádler P, et al. Aneurysma břišní aorty jako IgG4-asociované onemocnění? Anesteziologie a intenzivní medicína. 2017;28(1):53-54.
74. Bradna P, Soukup T, Tomáš J et al. Aortitis a periaortitis v rámci IgG4 – related choroby. Obtížná diagnóza s dobrou šancí léčby. Česká revmatologie. 2013;21(1):20-25.
75. Laco J, Šteiner I, Holuber T, et al., Isolated thoracic aortitis: clinicopathological and immunohistochemical study of 11 cases. Cardiovascular pathology. 2010;20(6):352-360.
76. Oyama‑Manabe N, Manabe O, Tsuneta S et al. RadioGraphics Update: IgG4-related Cardiovascular Disease from the Aorta to the Coronary Arteries. Radiographics. 2020;40(7):E29-E32. doi: 10.1148/rg.2020190219.
77. Mizushima I, Kasashima S, Fujinaga Y et al. Clinical and Pathological Characteristics of IgG4-Related Periaortitis/Periarteritis and Retroperitoneal Fibrosis Diagnosed Based on Experts‘ Diagnosis. Ann Vasc, DiS. 2019;12(4):460-472. doi: 10.3400/avd.oa.19-00085.
78. Šteiner I, Laco J, IgG4-related disease of the aortic valve. Cardiovascular pathology. 2015;24(4):264-265.
79. Lian L, Wang C, Tian JL. IgG4-related retroperitoneal fibrosis: a newly characterized disease. Int J Rheum, DiS. 2016;19(11):1049-1055. doi:10.1111/1756-185X.12863.
80. Liu Y, Zhu L, Wang Z, Zeng Q, et al. Clinical features of IgG4-related retroperitoneal fibrosis among 407 patients with IgG4-related disease: a retrospective study. Rheumatology (Oxford). 2021;60(2):767-772. doi: 10.1093/rheumatology/keaa411.
81. Wang G, Zhuo N, Luo X, et al. IgG4-Related Disease With Testicular involvement: A Case Report and Review of Literature. Front Immunol. 2021;12 : 717902. doi: 0.3389/fimmu. 2021.717902.
82. Crumley S, Ge Y, Zhou, et al. Interstitial cystitis: another IgG4-related inflammatory disease? Ann Diagn Pathol. 2013 Oct;17(5):403-7. doi: 10.1016/j.anndiagpath.2013. 03. 004.
83. Merta M. Klinický obraz onemocnění ledvin asociovaných s IgG4 Postgraduální nefrologie. 2013;11(2):26-27.
84. Tošovký M, Bradna P, Laco J et al. Case 1-2012: ANCA associated glomerulonephritis in combination with IgG4 Positive mediastinal mass in a patient with ankylosing spondylitis treated with TNF alpha inhibitors Acta Medica (Hradec Králové). 2012;55(1):42-46.
85. Kawano M, Saeki T, Nakashima H. IgG4-related kidney disease and retroperitoneal fibrosis: An update. Mod Rheumatol. 2019;29(2):231-239. doi:10.1080/14397595.2018.1554321
86. Boffa JJ, Esteve E, Buob D et al. Renal involvement in IgG4-related disease. Presse Med. 2020;49(1):104017. doi: 10.1016/j.lpm.2020.104017.
87. Kim YJ, Kim GE, Ma SK et al. IgG4-related renal disease co‑existing with retroperitoneal fibrosis. Transl Androl Urol. 2020;9(2):794-799. doi: 10.21037/tau.2020. 02. 06.
88. Tsai HC, Liao HT, Tsai CY. Retroperitoneal Fibrosis With a Damaged Kidney in IgG-4-Related Disease. J Clin Rheumatol. 2021;27(1):e1. doi:10.1097/RHU.0000000000001181.
89. Bhattad PB, Joseph DL, Peterson E. IgG4-Related Disease Manifesting as Hypocomplementemic Tubulointerstitial Nephritis: A Rare Case Report and Literature Review. J Investig Med High Impact Case Rep. 2020;8 : 2324709620952213. doi: 10.1177/2324709620952213.
90. Morales AT, Cignarella AG, Jabeen IS et al. An update on IgG4-related lung disease. Eur J Intern Med. 2019;66 : 18-24. doi:10.1016/j.ejim.2019. 06. 010.
91. Xie Y, Xiong A, Marion T et al. Lung nodules and IgG4 related disease: a single‑center based experience. BMC Pulm Med. 2020;20(1):218. doi: 10.1186/s12890-020-01250-3.
92. Moura MC, Gripaldo R, Baqir M et al. Thoracic Involvement in IgG4-Related Disease. Semin Respir Crit Care Med. 2020;41(2):202-213. doi: 10.1055/s-0039-1700995.
93. Lv X, Gao F, Liu Q et al. Clinical and pathological characteristics of IgG4-related interstitial lung disease. Exp Ther Med. 2018;15(2):1465-1473. doi: 10.3892/etm.2017.5554.
94. Matsui H, Utsumi T, Maru N, et al. A case of IgG4-related anterior mediastinal sclerosing disease coexisting with autoimmune pancreatitis. Surg Case Rep. 2020;6(1):180.doi: 10.1186/s40792-020-00939-1.
95. Corcoran JP, Culver EL, Anstey RM et al. Thoracic involvement in IgG4-related disease in a UK‑based patient cohort. Respir Med. 2017;132 : 117-121. doi: 10.1016/j.rmed.2017. 10. 005.
96. Pandita A, Wong J. IgG4-related disease in lung: a diagnostic challenge. Pathology. 2020;52(3):390-392. doi: 10.1016/j.pathol.2019. 11. 009.
97. Doubková M, Matěj R, Chovancová Z et al. Lung diseases and autoimmune hemolytic anemia associted with IgG4 disease. Vnitř Lék. 2020;66(4):47-52.
98. Terasaki Y, Ikushima S, Matsui S et al. Diffuse Lung Diseases Study Group. Comparison of clinical and pathological features of lung lesions of systemic IgG4-related disease and idiopathic multicentric Castleman’s disease. Histopathology. 2017;70(7):1114-1124. doi: 10.1111/his.13186.
99. Wick MR, O‘Malley DP. Lymphadenopathy associated with IgG4-related disease: Diagnosis & differential diagnosis. Semin Diagn Pathol. 2018;35(1):61-66. doi: 10.1053/j.semdp.2017. 11. 006.
100. Takanashi S, Kikuchi J, Sasaki T et al. Lymphadenopathy in IgG4-related disease: a phenotype of severe activity and poor prognosis, with eotaxin-3 as a new biomarker. Rheumatology (Oxford). 2021;60(2):967-975. doi: 10.1093/rheumatology/keaa648.
101. Bledsoe JR, Ferry JA, Neyaz A, et al. IgG4-related Lymphadenopathy: A Comparative Study of 41 Cases Reveals Distinctive Histopathologic Features. Am J Surg Pathol. 2021;45(2):178-192. doi: 10.1097/PAS.0000000000001579.
102. Igawa T, Hayashi T, Ishiguro K et al. IgG4-producing lymphoma arising in a patient with IgG4-related disease. Med Mol Morphol. 2016;49(4):243-249. doi: 10.1007/s00795-016-0139-2.
103. Wallace ZS, Carruthers MN, Khosroshahi A et al. IgG4-related disease and hypertrophic pachymeningitis. Medicine (Baltimore). 2013;92(4):206-216. doi:10.1097/MD.0b013e31829cce35.
104. Radotra BD, Aggarwal A, Kapoor A, et al. An orphan disease: IgG4-related spinal pachymeningitis: report of 2 cases. J Neurosurg Spine. 2016;25(6):790-794. doi: 10.3171/2016. 4. SPINE1674.
105. Sbeih I, Darwazeh R, Shehadeh M et al. Immunoglobulin G4-Related Hypertrophic Pachymeningitis of the Spine: A Case Report and Systematic Review of the Literature. World Neurosurg. 2020;143 : 445-453. doi: 10.1016/j.wneu.2020. 07. 227.
106. Pieringer H, Parzer I, Wöhrer A et al. IgG4-related disease: an orphan disease with many faces. Orphanet J Rare, DiS. 2014;9 : 110. doi: 10.1186/s13023-014-0110-z. PMID: 25026959;PMCID: PMC4223520.
107. Wallace ZS, Zhang Y, Perugino CA et al for ACR/EULAR IgG4-RD Classification Criteria Committee. Clinical phenotypes of IgG4-related disease: an analysis of two international cross‑sectional cohorts. Ann Rheum, DiS. 2019;78(3):406-412. doi: 10.1136/annrheumdis-2018-214603.
108. Luke YC Chen, Mattman A, Seidman MA et al. IgG4-related disease: what a hematologist needs to know Haematologica. 2019;104(3):444-455.
109. Della Torre E, Mattoo H, Mahajan VS et al. Prevalence of atopy, eosinophilia, and IgE elevation in IgG4-related disease. Allergy 2014;69(2):269-272.
110. Gotlib J. World Health Organization‑defined eosinophilic disorders: 2017 update on diagnosis, risk stratification, and management. Am J Hematol. 2017;92(11):1243–1259.
111. Carruthers MN, Park S, Slack GW et al. IgG4-related disease and lymphocyte‑variant hypereosinophilic syndrome: A comparative case series. Eur J Haematol. 2017;98(4):378-387. doi:10.1111/ejh.12842.
112. Zhang X, Zhang P, Li J, et al. Different clinical patterns of IgG4-RD patients with and without eosinophilia. Sci Rep. 2019;9 (1):16483. doi: 10.1038/s41598-019-52847-6.
113. Dispenzieri A, Gertz MA, Therneau TM, Kyle RA. Retrospective cohort study of 148 patients with polyclonal gammopathy. Mayo Clin Proc. 2001;76(5):476-487.
114. Zhao EJ, Carruthers MN, Li CH, et al. Conditions associated with polyclonal hypergammaglobulinemia in the IgG4-related disease era: a retrospective study from a hematology tertiary care center. Haematologica. 2020;105(3):e121-e123. doi: 10.3324/haematol. 2019.219725.
115. Engelhart S, Glynn RJ, Schur PH. Disease associations with isolated elevations of each of the four IgG subclasses. Semin Arthritis Rheum. 2017;47(2):276-280.
116. Martín‑Nares E, Hernández‑Molina G, Baenas DF et al. IgG4-Related Disease: Mimickers and Diagnostic Pitfalls. J Clin Rheumatol. 2021 Sep 17. doi: 10.1097/RHU.0000000000001787. Epub ahead of print.
117. Soto Y, Kojima M, Takata K et al. Multicentric Castleman disease with abudant IgG4 positive cells. Clinical and pathological analysis of 6 cases. J Clin Pathol. 2010, 63, 1084-1089.
118. Otani K, Inoue D, Fujikura K, et al. Idiopathic multicentric Castleman’s disease: a clinicopathologic study in comparison with IgG4-related disease. Oncotarget. 2018;9(6):6691-6706.doi: 10.18632/oncotarget.24068.
119. Sun C, Xu G, Lin J. Comparison of IgG4-Related Lymphadenopathy and Multicentric Castleman’s Disease: a Retrospective Study. Clin Lab. 2018;64(10):1671-1678. doi: 10.7754/Clin. Lab. 2018.180421.
120. Sasaki T, Akiyama M, Kaneko Y et al. Distinct features distinguishing IgG4-related disease from multicentric Castleman’s disease. RMD Open. 2017;18;3(1):e000432. doi: 10.1136/rmdopen-2017-000432.
121. Zhang X, Zhang P, Peng L et al. Clinical characteristics of a concurrent condition of IgG4-RD and Castleman’s disease. Clin Rheumatol. 2018;37(12):3387-3395. doi: 10.1007/s10067-018-4165-4.
122. Gianella P, Dulguerov N, Arnoux G et al. Thyroid Rosai‑Dorfman disease with infiltration of IgG4-bearing plasma cells associated with multiple small pulmonary cysts. BMC Pulm Med. 2019;19(1):83. doi:10.1186/s12890-019-0847-1.
123. de Jong WK, Kluin PM, Groen HM. Overlapping immunoglobulin G4-related disease and Rosai‑Dorfman disease mimicking lung cancer. Eur Respir Rev. 2012;21(126):365-7. doi: 10.1183/09059180.00001612.
124. Zhang Y, Chen H, Jiang YQ et al. Clinicopathological features of cutaneous Rosai‑Dorfman disease and its relationship to IgG4-related disease: a retrospective study. Br J Dermatol. 2019;181(4):844-845. doi: 10.1111/bjd.17939.
125. Liu L, Perry AM, Cao W et al. Relationship between Rosai‑Dorfman disease and IgG4-related disease: study of 32 cases. Am J Clin Pathol. 2013;140(3):395-402. doi: 10.1309/AJCPFH0SJ6YILXJU.
126. Zhang X, Hyjek E, Vardiman J. A subset of Rosai‑Dorfman disease exhibits features of IgG4-related disease. Am J Clin Pathol. 2013;139(5):622-632.
127. Abla O, Jacobsen E, Picarsic J et al. Consensus recommendations for the diagnosis and clinical management of Rosai‑Dorfman‑Destombes disease. Blood. 2018;131(26):2877-2890.
128. Piao Y, Zhang Y, Yue C et al. Immunoglobulin G4-related chronic rhinosinusitis: a pitfall in the differential diagnosis of granulomatosis with polyangiitis, Rosai‑Dorfman disease, and fungal rhinosinusitis. Hum Pathol. 2018;73 : 82-88. doi: 10.1016/j.humpath.2017. 12. 011.
129. Bledsoe JR, Wallace ZS, Stone JH et al. Lymphomas in IgG4-related disease: clinicopathologic features in a Western population. Virchows Arch. 2017;472(5):839-852.
130. Liu J, Yin W, Westerberg LS, Lee P et al. Immune Dysregulation in IgG<sub>4</sub>-Related Disease. Front Immunol. 2021;12 : 738540. doi: 10.3389/fimmu.2021.738540.
131. Carruthers MN, Khosroshahi A, Augustin T, Deshpande V, Stone JH. The diagnostic utility of serum IgG4 concentrations in IgG4-related disease. Ann Rheum, DiS. 2015;74(1):14-18.
132. Qi R, Chen LYC, Park S, et al. Utility of serum IgG4 levels in a multiethnic population. Am J Med Sci. 2018;355(1):61-66.
133. Dítě Z, Novotný I, Kala Z et al. Pozitivita imunoglobulinu IgG4 v krevním séru u osob s karcinomem slinivky břišní. Gastroenterologie a hepatologie. 2012;66(3):187-190.
134. Slavíčková J Lašťovička J. Elevace sérového imunoglobulinu G4 a diagnostika IgG4-asociované nemoci., Alergie (Praha, Print). 2018;20(2):98-102.
135. van der Gugten G, DeMarco ML, Chen LYC et al. Resolution of spurious immunonephelometric IgG subclass measurement discrepancies by LC‑MS/MS. Clin Chem. 2018;64(4):735-742.
136. Kawa S, Skold M, Ramsden DB, Serum IgG4 Concentration in IgG4-Related Disease. Clin Lab. 2017;63(9):1323-1337.
137. Wallace ZS, Mattoo H, Carruthers M et al. Plasmablasts as a biomarker for IgG4-related disease, independent of serum IgG4 concentrations. Ann Rheum, DiS. 2015;74(1):190-195.
138. Mizushima I, Yamada K, Harada K et al. Diagnostic sensitivity of cutoff values of IgG4-positive plasma cell number and IgG4-positive/CD138-positive cell ratio in typical multiple lesions of patients with IgG4-related disease. Mod Rheumatol. 2018;28(2):293-299. doi: 10.1080/14397595.2017.1332540.
139. Akiyama M, Suzuki K, Yamaoka K et al. Number of Circulating Follicular Helper 2 T Cells Correlates With IgG4 and Interleukin-4 Levels and Plasmablast Numbers in IgG4-Related Disease. Arthritis R heumatol. 2015;67(9):2476-81.
140. Lanzillotta M, Della‑Torre E, Stone JH. Roles of Plasmablasts and B Cells in IgG4-Related Disease: Implications for Therapy and Early Treatment Outcomes. Curr Top Microbiol Immunol. 2017;401 : 85-92. doi: 10.1007/82_2016_58.
141. Lin W, Zhang P, Chen H, Lipsky PE. Circulating plasmablasts/plasma cells: a potential biomarker for IgG4-related disease. Arthritis Res Ther. 2017;19(1):25. doi: 10.1186/s13075-017-1231-2. PMID: 28183334;PMCID: PMC5301376.
142. Seidman MA, Barbour SJ, Levin A, Carruthers M, Chen LY. Recognizing IgG4-related tubulointerstitial nephritis. Can J Kidney Health, DiS. 2016;3 : 34.
143. Moriyama M, Ohta M, Furukawa S et al. The diagnostic utility of labial salivary gland biopsy in IgG4-related disease. Mod Rheumatol. 2016;26(5):725-729.
144. Umehara H, Okazaki K, Masaki Y et al. Comprehensive diagnostic criteria for IgG4-related disease (IgG4-RD). Mod Rheumatol. 2012;22 : 21-30.
145. Umehara H, Okazaki K, Kawa S et al. Research Program for Intractable Disease by the Ministry of Health, Labor and Welfare (MHLW) Japan. The 2020 revised comprehensive diagnostic (RCD) criteria for IgG4-RD. Mod Rheumatol. 2021;31(3):529-533. doi: 10.1080/14397595.2020.1859710,
146. Umehara H, Okazaki K, Nakamura T et al. Current approach to the diagnosis of IgG-4-related disease - Combination of comprehensive diagnostic and organ‑specific criteria. Mod Rheumatol. 2017;27(3):381-391.
147. Wallace ZS, Naden RP, Chari S et al for IgG4-RD Classification Criteria Working Group. The 2019 American College of Rheumatology/European League Against Rheumatism classification criteria for IgG4-related disease. Ann Rheum Dis 2020;79(1):77-87.
148. Wallace ZS, Naden RP, Chari S et al IgG4-Related Disease Classification Criteria Working Group. The 2019 American College of Rheumatology/European League Against Rheumatism Classification Criteria for IgG4-Related Disease. Arthritis Rheumatol. 2020;72(1):7-19.
149. Chovancová Z, Filipenský P, Rotnáglová S et al. Podtřída imunoglobulinů IgG4 a s ní související patologické stavy aneb jak účinně imitovat nádorové onemocnění Klinická onkologie. 2022;35(1):20-31.
150. Wallace ZS, Khosroshahi A, Carruthers MD et al. An International Multispecialty Validation Study of the IgG4-Related Disease Responder Index. Arthritis Care Res (Hoboken). 2018;70(11):1671-1678. doi: 10.1002/acr.23543.
151. Fernández‑Codina A, Pinilla B et al. Spanish Registry of IgG4 Related Disease (REERIGG4) investigators;Autoimmune Diseases Group (GEAS);Spanish Internal Medicine Society (SEMI). Treatment and outcomes in patients with IgG4-related disease using the IgG4 responder index. Joint Bone Spine. 2018;85(6):721-726. doi: 10.1016/j.jbspin.2018. 01. 014.
152. Zhang X, Zhang P, Li J et al. Different clinical patterns of IgG4-RD patients with and without eosinophilia. Sci Rep. 2019;9(1):16483. doi: 10.1038/s41598-019-52847-6.
153. Tomiyama F, Watanabe R, Fujii H et al. Synovitis in a Patient with IgG4-related Disease. Intern Med. 2015;54(11):1427-32. doi: 0.2169/internalmedicine.54.4320.
Štítky
Diabetologie Endokrinologie Interní lékařstvíČlánek vyšel v časopise
Vnitřní lékařství

2022 Číslo E-5
- Inovace v hojení ran: krytí Zetuvit Plus Silicone Border pro optimální management exsudátu z ran
- Ivabradin zlepšuje kvalitu života starších pacientů se srdečním selháním
- Fixní kombinace kandesartan/amlodipin v terapii arteriální hypertenze
- Clopidogrel je v prevenci kardiovaskulárních příhod přínosnější než kyselina acetylsalicylová
Nejčtenější v tomto čísle
- Onemocnění asociované s imunoglobulinem IgG4. Klinické příznaky, diferenciální diagnostika a recentní mezinárodní diagnostická kritéria
- Střednědobá úspěšnost single stage hybridní ablace perzistující a dlouhodobě perzistující fibrilace síní
- Critical Appraisal of Guidelines in Internal Medicine Working Group
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy