
Diagnostická patológia non-Langerhansových histiocytóz
Pathology of histiocytoses of non-Langerhans cell type
The paper offers a review on pathology of histiocytoses of non-Langerhans cell type, especially focused on neoplastic forms of the histiocytic disorders according to the WHO classification of tumours of hematopoietic and lymphoid tissues (2008). The review is based on lineage - specific histogenetical typing of histiocytic and dendritic cells distinguishing histiocytes (makrophages), dendritic cells of myeloid origin and dendritic cells of the stromal type (of mesenchymal origin). The tumours are classified as follows: histiocytic sarcoma, sarcoma of interdigitating, indetermined, dermal (intersticial) and plasmacytoid dendritic cellss and sarcomas (tumours) of follicular dendritic and fibroblastic reticular cells. Within each of the tumor categories, the histogenesis, clinical manifestation and prognosis is discussed and the histomorphology and phenotypic profile of the tumour is shortly described. Besides that also the relations of all these tumours to fibrohistiocytic tumours of the soft tissue category leading to classification conceptual problems are discussed.
Key words:
histiocyte – dendritic cells – non-Langerhans cell histiocytoses – histiocytic sarcoma – dendritic cell sarcomas and tumours – fibrohistiocytic soft tissue tumours
Autoři:
L. Plank 1,2
Působiště autorů:
Ústav patologickej anatómie a Konzultačné centrum bioptickej diagnostiky ochorení krvotvorby Jesseniovej lekárskej fakulty UK a UN Martin, Slovenská republika, prednosta prof. MU Dr. Lukáš Plank, CSc.
1; Martinské bioptické centrum, s. r. o., Martin, Slovenská republika
2
Vyšlo v časopise:
Vnitř Lék 2010; 56(Supplementum 2): 39-63
Kategorie:
Histiocytóza z Langerhansových buněk a některá další vzácná hematologická onemocnění
Souhrn
Autor podáva prehľad o patológii non-Langerhansových histiocytóz, so zameraním na nádorové formy histiocytových ochorení podľa klasifikácie nádorov krvotvorných a lymfatických tkanív podľa SZO z roku 2008. Prehľad sa opiera o líniovo - špecifickú histogenetickú typizáciu histiocytových a dendritických buniek s rozlíšením histiocytov (makrofágov), myeloidných dendritických buniek a dendritických buniek stromálneho typu (mezenchýmového pôvodu). Nádory z týchto buniek sú rozdelené na histiocytový sarkóm, na sarkómy z interdigitujúcich, neurčených, dermálnych (intersticiálnych) a plazmocytoidných dendritických buniek a na sarkómy, resp. nádory z folikulových dendritických buniek a fibroblastových retikulárnych buniek. V každej kategórii sa diskutuje o histogenéze, klinickej manifestácii a prognóze a stručne sa komentuje histomorfologický obraz a fenotypový profil nádoru. Okrem toho sa diskutuje aj o vzťahu uvedených nádorov ku fibrohistiocytovým nádorom mäkkých tkanív a o s tým súvisiacich klasifikačných problémoch.
Kľúčové slová:
histiocyt – dendritické bunky – non-Langerhansove histiocytózy – histiocytový sarkóm – sarkómy dendritických buniek – fibrohistiocytové nádory
Úvod
Problematika diagnostickej patológie „histiocytóz“ a „non-Langerhansových histiocytóz“ obzvlášť patrí medzi najzložitejšie a súčasne najmenej známe oblasti hematopatológie, a to aj pre nasledovné dôvody:
- a) pretrvávajúce problémy súvisiace s meniacou sa definíciou „histiocytózy“ a definíciou základného bunkového elementu tohoto procesu – „histiocytu“,
- b) terminológia „histiocytóz“ je nejednotná. Aj preto pre ciele supplementa, ktorého súčasťou je aj táto práca, bol akceptovaný názov „histiocytóza“. V prvých ucelenejších a pomerne recentných textoch na túto tému v československom písomníctve použili Adam et al [1,3] názov „histiocytární choroby“, kam logicky zaradili tak „malígne histiocytární onemocnění“, ako aj „reaktivní zmnožení“ histiocytov. Ani pojem „non-Langerhansove histiocytózy“ (ďalej non-LCH - histiocytózy), použitý aj v názve predloženého článku, nie je oficiálnym termínom klasifikácie krvotvorných a lymfatických tkanív podľa SZO z roku 2008 [82]. Tento v odbornej literatúre často používaný názov bol editormi tohto supplementa použitý pre vymedzenie „histiocytóz iných, než sú histiocytózy Langerhansových buniek“ (podobne ako v klasifikácii malígnych lymfómov) a v tomto zmysle ho používame aj v predloženom texte,
- c) zriedkavosť „histiocytóz“ vše-obecne a osobitne aj „non-LCH histiocytóz“. V mnohých prípadoch ide o chorobné jednotky s celosvetovým výskytom v rádovo desiatkach až stovkách prípadov. S tým súvisí nedostatok skúseností s diagnostikou a liečbou týchto ochorení, ako aj nedostatočnosť a niekedy aj neprehľadnosť literárnych údajov,
- d) dlhoročná absencia jednotnej a celosvetovo akceptovanej klasifikačnej schémy histiocytóz, ktorá by bola klinicky relevantná a súčasne zrozumiteľná pre patológa, ako aj pre príslušného klinického pracovníka. Výnimku predstavovali 2 zdroje: klasifikácia SZO z roku 2001 [45] a klasifikácia podľa International Histiocyte Society [28,39], ktoré však nepoužívali jednotnú terminológiu ani klasifikačné východiská. Medzitým je k dispozícii nová klasifikácia nádorov krvotvorného a lymfatického tkaniva podľa SZO z roku 2008 [71,82], ktorú pre zjednodušenie tu budeme nazývať len klasifikácia SZO z roku 2008,
- e) zložitosť bioptickej diagnostiky (a diferenciálnej diagnostiky) non-Langerhansových histiocytóz z hľadiska ich morfologických čŕt, fenotypu, genotypu a klinických prejavov, pri empiricky známej častej nestabilite až nekonštantnosti týchto prejavov.
V predloženom texte sa pokúsime, pochopiteľne z hľadiska patológa, sumarizovať známe a aj vlastné poznatky z oblasti histiocytóz a ich bioptickej diagnostiky, pričom dôraz kladieme na „nádorové histiocytózy“ (klonálne nádorové ochorenia histiocytov) a ostatné, reaktívne, zmieňujeme len názvom a pre objasnenie súvislostí.
„Histiocytóza“ a „histiocyty“ – úvod a definície
Histiocytóza je pojem pomerne často a niekedy aj nesprávne používaný, ktorý v širšom slova zmysle znamená akékoľvek nahromadenie histiocytov v tkanive. Tento jav môže byť súčasťou celého radu patologických udalostí, takže z hľadiska patogenézy možno histiocytózy v uvedenom slova zmysle rozdeliť na sekundárne a primárne (tab. 1 a 2).
![Klasifikácia histiocytóz (ochorení histiocytov – kompilácia podľa viacerých literárnych zdrojov, najmä [12,45]).](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image/ec4aa1dc3c87583ac9cebcccbb21a33f.jpg)
![Klasifikácia primárnych histiocytóz (histiocytových proliferácií) podľa Histiocyte Society [28,45,68].](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image/ff7622df19338e462cdd1574bd25e24a.jpg)
Najviac známe a v praxi aj najčastejšie sú zápalové a imunitné reakcie rôznych tkanív, pri ktorých v dôsledku známeho primárneho patologického podnetu dochádza k sekundárnemu (sekundárne antigénom, infekčným agens a pod.) indukovanému nahromadeniu histiocytov, t.j. buniek, ktoré sú súčasťou celulárnych mechanizmov vrodenej imunitnej reakcie. Tak napr. súčasťou veľmi častej a bežnej reakcie lymfatického tkaniva lymfatickej uzliny na antigénové podnety rôznej povahy je tzv. sínusová „histiocytóza“, t.j. zmnoženie a sekundárna aktivácia „histiocytových“ buniek vystielajúcich sínusy uzliny. Ďalšími príčinami sekundárneho hromadenia histiocytových buniek môžu byť vrodené funkčné defekty týchto elementov alebo ich systémové aktivácie (tab. 1).
Preto v predloženej práci používaný pojem „histiocytóza“ by mal ostať obmedzený na skupinu ochorení nazývaných aj histiocytové proliferácie, ktorých podstatou je primárne nahromadenie alebo proliferácia „histiocytových elementov“, ktoré:
- a) utláčajú (efekt nahromadenia „masy“ histiocytov) okolité tkanivo zväčša v zmysle expanzie lézie a útlaku či tlakovej atrofie okolitých štruktúr (napr. v CNS a i.),
- b) a/ alebo infiltrujú postihnuté tkanivá a orgány s ich prípadnou deštrukciou a s tým súvisiacim potenciálom metastatického rozsevu a disseminácie. V oboch prípadoch proliferácia „histiocytov“ spôsobuje následné poškodenie štruktúry a funkcie postihnutých tkanív a orgánov s príslušnou klinickou prezentáciou.
Podobne ako termín histiocytóza, aj termín „histiocyt“ vyžaduje terminologický formálny výklad a obsahové upresnenie. Formálne „histiocyt“ znamená „tkanivová bunka“, pričom z hľadiska obsahu uvedeného pojmu je vhodné si uvedomiť, že v posledných rokoch došlo vplyvom akumulácie nových poznatkov z oblasti morfológie, biológie a imunológie k značnému posunu názorov [20,71,72]. Tzv. histiocytové proliferácie v dnešnom ponímaní [17,45,82] zahŕňajú proliferácie 2 skupín „histiocytov“:
- histiocyty, alebo mononukleárne fagocyty, t.j. makrofágy,
- antigén prezentujúce dendritické bunky (viď ďalej definície histiocytových elementov, obr. 1, 2 a tab. 2, 3).
![Klasifikácia nádorov histiocytových a dendritických buniek podľa SZO klasifikácie z roku 2008 [82].](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image/7c00f59a4d00d1f31a41897f1cecd182.jpg)
Histiocytové elementy tak predstavujú početnú skupinu morfologicky a funkčne odlišných buniek, ktoré možno podľa ich príslušnosti k diferenciačným líniám rozdeliť z pohľadu patológa podľa dvoch relevantných kritérií:
-
a) podľa ich pôvodu na:
- elementy odvodené od kmeňovej hemopoetickej bunky kostnej drene, ktoré po vzniku v dreni cirkulujú ako prechodné formy – monocyty v periférnej krvi a potom vstupujú do jednotlivých tkanív a
- elementy odvodené od kmeňovej mezenchýmovej bunky, bez účasti cirkulujúceho poolu periférnej krvi.
- b) podľa ich príslušnosti k jednotlivým bunkovým líniám na (viď aj obr. 1):
b.1) makrofágy, t.j. histiocyty monocytového - histiocytového, resp. makrofagocytového radu – sú odvodené kmeňovej hemopoetickej bunky kostnej drene. Diferenciácia a vyzrievanie elementov tohto radu v kostnej dreni vedie, ako sme uviedli, k produkcii monocytov, ktoré sa vyplavujú do periférnej krvi, a z nej sa dostávajú do tkanív, t.j. väčšina poolu tkanivových histiocytov má pôvod v monocytoch periférnej krvi. Tieto histiocyty sú schopné aktivácie a premeny buď na makrofágy (alebo na epiteloidné bunky), pričom súčasťou ich aktivácie môže byť aj tvorba obrovských viacjadrových buniek.
Z hľadiska ortologicko-štrukturálneho možno hovoriť o tkanivo - špecifických makrofágoch lymfatického tkaniva [uzlín, sleziny a pod. – sú v sínusoch, dreňových povrazcoch, folikulových zárodočných centrách, v parakortexe (T - zóne)] a iných tkanivo - špecifických makrofágoch [70]. Sú to napr. pľúcne alveolárne makrofágy, histiocyty kože a spojivového tkaniva, makrofágy seróznych blán, kostné osteoklasty, mozgová mikroglia, pečeňové Kupferove bunky, glomerulové mezangiové bunky v obličke, Hofbauerove bunky placenty a pod.
Z hľadiska funkcie makrofágy sú fagocytujúce elementy nešpecifickej (vrodenej) imunity, ktorých fyziologickou funkciou v tkanivách je fagocytovať (a ďalej rozkladať a inaktivovať, resp. ničiť) patogénny, ako sú cudzorodé látky, infekčné agens, ako aj tkanivovú a bunkovú debris a odstránením týchto substancií vytvoriť predpoklady pre obnovu poškodeného tkaniva. Pritom spolupracujú s ostatnými bunkami (dendritickými, leukocytami, B - a T-lymfocytami), s pro - a protizápalovými mediátormi lokálneho mikroprostredia, podieľajú sa na indukcii špecifickej získanej imunity (bunkovej aj humorálnej) a pod. [45].
b.2) histiocyty dendritického radu, nazývané dendritické bunky (ďalej DCs) sú predstaviteľmi tzv. akcesórnych antigén - prezentujúcich buniek. Od makrofágov sa tak líšia nielen inou funkciou a odlišnou morfológiou, ale aj s tým súvisiacou bunkovou výbavou, ako je slabá až nulová lyzozómová enzýmová aktivita a pod. [3]. Tvoria komplexný systém, ktorý sa skladá z niekoľkých odlišných bunkových populácií (obr. 1), ktoré osídľujú štruktúry lymfoidných a nelymfoidných orgánov a majú schopnosť migrácie špecifickými cestami. V každom mieste ich výskytu sa vyznačujú inými morfologickými, imunofenotypovými a funkčnými charakteristikami. Podobne ako makrofágy sú tkanivovo - špecifické: sú umiestnené napr. v koži, mukokutánnych spojeniach a pľúcach (Langerhansove bunky), v spojivovom tkanive (dermálne a intersticiálne DCs), v LU, slezine, týmuse a iných lymfatických tkanivách (interdigitujúce a folikulové DCs, fibroblastové retikulárne bunky) a pod. V týchto tkanivách sídlia ako strážne bunky, ktoré vychytávajú signály z vonkajšieho alebo vnútorného prostredia. Po signalizácii, zväčša sprostredkovanej cytokínami, sa aktivujú, vyzrievajú a menia fenotyp z bunky schopnej prijať a spracovať antigén (mechanizmami pinocytózy, fagocytózy a i.) na bunky antigén prezentujúce [45], čo si vyžaduje buď schopnosť migrácie do štruktúr imúnneho systému (bunky ďalej uvedené ako b.2.1), alebo byť v stálom kontakte s lymfatickým systémom (bunky ďalej uvedené ako b.2.2). DCs sa vyskytujú na rôznych miestach v rôznom štádiu aktivácie, a sú preto veľmi rôznorodé (a aj preto nemajú jeden spoločný imunohistochemický marker). Po prezentácii zachytených antigénov elementom špecifickej imunity sa iniciujú mechanizmy aktivácie B bunkami a T bunkami sprostredkovanej imunity. Okrem toho niektoré druhy DCs sú zrejme aj sami sekrečne aktívne v zmysle produkcie intereferónu, cytokínov a pod. (viď ďalej).
Podľa pôvodu možno DCs rozdeliť na [45,82]:
b.2.1) DCs, ktoré majú pôvod v kmeňovej hemopoetickej bunke kostnej drene, t.j. DCs myeloidného pôvodu, čiže tzv. myeloidné DCs, ktorých prekurzorové bunky cirkulujú v poole monocytov v periférnej krvi a ktoré po osídlení v tkanive sú schopné migrácie. Sem patria bunky 5 nasledovných skupín:
- Langerhansove bunky: sú dobre známe a morfologicky, fenotypove a ultraštrukturálne dobre definované špecializované DCs bariérových štruktúr kože a slizníc, ktoré po aktivácii prezentujú antigén T-lymfocytom, a preto sú schopné migrovať cez lymfatické cievy do T zóny lymfatických uzlín, resp. lymfatického tkaniva,
- interdigitujúce dendritické bunky: sú špecializované DCs T zóny lymfatických uzlín/ lymfatického tkaniva, resp. periarteriolárnych T bunkových oblastí sleziny, ktoré do značnej miery majú spoločný pôvod v línii Langerhansových buniek. Ich názov odráža skutočnosť, že ultraštrukturálne tieto bunky vykazujú komplexy vzájomne prepojených (interdigitujúcich) cytoplazmatických výbežkov, bez tvorby dezmozómov [45,68],
- indeterminované – neurčené (seu záhadné – „veiled“) DCs predstavujú nie celkom známu populáciu kožných DCs, sú schopné migrovať do regionálneho lymfatického tkaniva,
- dermálne/ intersticiálne dendritické bunky sú DCs dermis a mäkkotkanivových štruktúr, mnohí ich považujú za tkanivové DCs analogické ku kožným Langerhansovým bunkám, ich úlohou je aktivovať sa a množiť v prípade miestnej zápalovej reakcie,
- plazmocytoidné dendritické bunky (predtým aj plazmocytoidné monocyty) majú neistý pôvod (patria zrejme k myeloidnej/ monocytovej línii) a neistú funkciu (najmä pokiaľ ide o vzťah k prezentácii antigénu). Je však známe, že na rozdiel od iných antigén prezentujúcich DCs sú schopné produkovať interferón [45,88]. Ide o populáciu pomerne málo známych a konfúzne vnímaných buniek. Ich terminológia sa totiž postupne menila v hierarchii od pôvodného názvu Lennerta a Remmeleho [58] „lymfoblasty v zhlukoch“, resp. neskoršieho názvu Lennerta et al [59] T asociované plazmocytoidné plazmocyty cez plazmocytoidné monocyty až po súčasný názov plazmocytoidné dendritické bunky. Presnejšie ide o tzv. plazmocytoidné DCs2 bunky s fenotypom CD4+, CD56+, CD45RA+, CD123+ [11], ktoré sa fenotypove líšia od iných myeloidných dendritických buniek (DCs1 buniek, ktoré majú fenotyp CD11c+, CD86+, CD40+, CD80+, CD86+ [46]). Ich „normálny“ ortologický výskyt nie je známy, ale skúsený hematopatológ ich môže niekedy zastihnúť ako náhodný nález v malej časti biopticky vyšetrovaných lymfatických uzlín s obrazom „chronickej nešpecifickej lymfadenitídy“, zväčša v podobe málo rozsiahlych a nenápadných ložísk v parakortexe uzliny. Podobné nálezy možno pozorovať aj pri Castlemanovej chorobe, granulomatóznych lymfadenitídach a pod. [31]. Biologický význam tohto fenoménu nie je jasný. Okrem toho je známa ich výrazná a často rozsiahla proliferácia v obraze tzv. Kikuchiho (Kikuchiho - Fujimotovej) histiocytárne - nekrotizujúcej lymfadenitídy [3,70]. Ich proliferácia je tu zrejme súčasťou hyperimúnnej reakcie v rámci parainfekčných, resp. postinfekčných (postviróznych) stavov, prípadne systémových imunopatologických procesov a proces nekrobiózy v štádiu nekrotizácie môže súvisieť s ich sekrečnou aktivitou.
b.2.2) DCs stromálneho typu, tzv. dendritické stromálne bunky, nepatria do línie odvodenej od kmeňovej hemopoetickej bunky, lebo majú pôvod v kmeňovej mezenchýmovej bunke. Súčasne tak majú úzky vzťah k stromálnym progenitorom kostnej drene s črtami (myo - )fibroblastov. Na rozdiel od predošlých patria medzi tzv. stabilné nemigrujúce elementy.
Delia sa na 2 skupiny:
- folikulové dendritické bunky, predstavujú dobre morfologicky a fenotypove definované elementy, ktoré sa de norma vyskytujú v zárodočných centrách lymfatických folikulov. V nich tvoria stabilnú sieť folikulu, pričom ich zosieťovanie sprostredkovávajú dezmozomálne prepojenia. V centre lymfatického folikulu zachytávajú antigény a prezentujú ich B bunkám, čím sa aktivuje B bunková mašinéria zárodočných centier folikulov [16,82],
- fibroblastové retikulárne bunky, ktoré sa de norma nachádzajú v lymfatickom tkanive uzlín. Predstavujú snáď medzi najmenej známu populáciu DCs. Patria k tzv. fixným alebo nemigrujúcim bunkám, ktoré s folikulovými DCs spája nielen spoločný mezenchýmový pôvod [obe sú odvodené zo stromálnych kmeňových (progenitorových) buniek kostnej drene], ale aj podobnosť s myofibroblastami (aj v ultraštrukturálnom obraze). Fibroblastové retikulárne bunky aj obsahujú v cytoplazme intermediárne cytoskeletónové filamentá hladkosvalového aktínu. Preto ich možno pri ich nenápadnosti v histologickom obraze dobre znázorniť pomocou pozitívnej imunohistochemickej reakcie na dôkaz hladkosvalového antigénu a identifikovať ako podporné elementy opúzdrujúce postkapilárne venuly. Sú viditeľné napr. v parafolikulárnej oblasti kôry LU alebo v extrafolikulovej časti sleziny a tonzily. Sú jednou zo základných zložiek tkanivového mikroprostredia, udržujú integritu lymfatického tkaniva, vytvárajú sieť ako podklad pre signalizáciu imunitných mechanizmov a v rámci ich homeostatického pôsobenia sú schopné produkovať a transportovať cytokíny [13,49]. Imunohistochemickým dôkazom cytokeratínov (CK) možno rozlíšiť CK - negatívne a CK-pozitívne fibroblastové retikulárne bunky [24]. Je známe aj to, že okrem homeostatickej funkcie možno hyperpláziu fibroblastových retikulárnych buniek pozorovať v LU pacientov so zápalovými a autoimúnnymi ochoreniami (indukovaná hyperplázia?), rovnako však aj pri Castlemanovej chorobe [36].
Výskyt histiocytóz
„Histiocytózy“ všeobecne patria medzi zriedkavé, neraz veľmi zriedkavé až raritné ochorenia. Preto aj hematologicky, hematoonkologicky či na iné príslušné segmenty patológie orientovaní a špecializovaní patológovia (podobne ako ich klinickí partneri) majú zväčša nedostatok skúseností s ich diagnostikou, resp. stretávajú sa obyčajne s len relatívne častejšími histiocytózami typu histiocytózy Langerhansových buniek (ďalej LCH). Aj preto obyčajne máme my, patológovia, nedostatočný prehľad či skúsenosti s celým spektrom histiocytových proliferácií. Navyše, s rozvojom nášho poznania v posledných 2 desaťročiach došlo v celej hematopatológii, a v oblasti patológie „histiocytóz“ vôbec, k celému radu zmien, neraz aj koncepčného charakteru. To viedlo k „zániku“ niektorých tradičných kategórií histiocytových proliferácií, ktorých dnes už známa podstata ich radí medzi iné skupiny ochorení, napr. do kategórie malígnych lymfómov. Naopak, rozvoj poznatkov umožnil napriek absencii typických fenotypových a genotypových markerov spoznať a definovať niektoré nové typy a druhy ochorení. Napriek pokroku situáciu v oblasti tak diagnostickej patológie, ako aj v rovine klasifikačných prístupov sťažuje aj nepresnosť hraníc jednotlivých typov histiocytóz a isté prekrývanie sa niektorých jednotiek, vrátane neostrých až nejasných hraníc medzi reaktívnymi (nenádorovými) a nádorovými histiocytózami.
Klasifikácia histiocytóz
Pre klasifikáciu histiocytóz možno v princípe použiť viaceré východiskové princípy. Z hľadiska patológie a patológa možno deliť histiocytózy podľa:
- a) histogenetického princípu,
- b) ich biologickej povahy (biologických vlastností),
- c) rozsahu procesu histiocytózy a
- d) prítomnosti čŕt fagocytózy.
a) Histogenetický princíp v klasifikácii histiocytóz znamená rozdeliť histiocytózy podľa východiskového elementu procesu, resp. v prípade nádorových histiocytóz podľa príslušnosti nádorových buniek k danej bunkovej „histiocytovej“ línii. Uplatnenie histogenetického princípu vyžaduje spoľahlivo identifikovať tie histiocytové bunky, ktoré sú podstatou lézie, resp. tvoria jej väčšinu, a to morfologickou, fenotypovou alebo ultraštrukturálnou analýzou. Situáciu tu totiž môže komplikovať skutočnosť, že morfológia a biologická povaha proliferujúcich histiocytov sa môže v priebehu ochorenia meniť (tzv. plasticita – viď ďalej), histiocyty môžu vyzrievať a meniť sa aj vplyvom terapie, vplyvom imunitnej reakcie hostiteľa, vplyvom cytokínov a fibroprodukcie v rámci interakcie histiocyt – mikroprostredie, a i., čo môže prekryť prejavy samotnej histiocytózy.
Poznanie morfológie, fenotypu a funkcie „normálnych histiocytov“ je dôležité aj preto, že pri štúdiu nádorových histiocytóz sa zdá byť oprávnené aplikovať princíp porovnávania buniek histiocytózy s ich proťajškami v bunkovom a vývojovom cykle „normálneho“ histiocytu. Nádory odvodené od histiocytov, resp. makrofágov a dendritických buniek vykazujú morfologické, imunohistochemické a iné črty podobné črtám ich nenádorových proťajškov [17]. Súčasne však je potrebné zvážiť, že v systéme neoplasticky transformovaných a aktivovaných histiocytových a dendritických buniek (a celkove aj v systéme krvotvorných a lymfatických tkanív vôbec – viď aj ďalej) existuje značná plasticita. Táto plasticita bola recentne podrobne opísaná v myšacom modeli „histiocytózy“, v ktorom vznikajú bunky s hybridným fenotypom medzi makrofágom a DCs [57]. Preto neprekvapuje, že aj pri vzniku humánnych histiocytóz DCs môžu napr. „konvertovať“ na makrofágy. Zrejme preto existujú aj histiocytózy s fenotypom „sivej zóny“, kedy v „histiocyte“ nemožno spoľahlivo odlíšiť fenotyp makrofágu od fenotypu DCs [45,73]. Tak je tomu napr. v nasledovných prípadoch:
- bunky sínusovej histiocytózy s masívnou lymfadenopatiou (Rosaiova - Dorfmanova choroba) vykazujú fenotypové črty makrofágov (CD14+, CD68+, CD163+), ale aj DCs (S - 100 proteín+ a fascín+),
- bunky skupiny juvenilného xantogranulómu, ktoré vykazujú črty makrofágov (CD14+, CD68+, CD163+), ale aj dermálnych a intersticiálnych DCS (F XIIIa +, fascín+).
Ďalšie dôležité momenty prispievajúce k plasticite histiocytového systému sú nasledovné:
- mnohé histiocytové lézie často obsahujú zmes histiocytov, makrofágov a DCs, takže môže byť obtiažne identifikovať podstatnú bunkovú proliferujúcu populáciu,
- fúzia makrofágov vyúsťuje do tvorby viacjadrových obrovských buniek,
- pod vplyvom mikroprostredia a produkcie napr. interleukínov sa histiocyty menia na epiteloidné bunky a pod.
Histogenetický princíp sa dnes, v súlade s princípmi klasifikácie nádorov krvotvorného a lymfatického systému podľa SZO z roku 2008, plne uplatňuje najmä v klasifikácii nádorových histiocytóz (tab. 3 a obr. 2).

Na tomto mieste je ešte vhodné pripomenúť, že východiskovým princípom klasifikácie SZO z roku 2008 je, že v rámci jednotlivých bunkových línií sa vždy rozlišujú nádory z prekurzorových a nádory z periférnych (zrelých) buniek. V tomto kontexte väčšina nádorov histiocytov a DCs patrí medzi nádory z periférnych buniek, výnimku tvoria nádory z plazmocytoidných dendritických buniek (viď aj ďalej).
b) Podľa biologickej povahy procesu, resp. podľa biologických vlastností proliferujúcich histiocytových buniek, by sa primárne histiocytózy mali rozdelovať na:
- nenádorové (reaktívne) histiocytózy,
- na nádorové histiocytózy a tie by sa mali, analogicky k iným nádorovým ochoreniam, rozdeľovať na benígne a malígne.
V hematoonkológii je však dobre známe, že pre viaceré zhubné nádorové ochorenia krvotvorného a lymfatického systému nepoznáme ich identifikovateľný „benígny“ proťajšok. Keďže biologický priebeh ochorenia typu „histiocytózy“ je značne variabilný, tak v klinike sa všeobecne akceptovalo rozdelenie histiocytóz podľa povahy ochorenia na:
- jednoznačne benígne procesy, bez histologických a klinických známok infiltratívno - deštruktívneho rastu a bez potenciálu metastazovať,
- jednoznačne malígne „histiocytózy“, s pomerne dobre definovanými kritériami určenia ich malígnej povahy na základe ich bioptického vyšetrenia a
- histiocytózy neistého biologického priebehu s individuálnou variabilitou prejavov, pri diagnostike ktorých absentujú jasné histopatologické kritériá predpovede ich biologickej povahy.
c) Podľa rozsahu procesu možno histiocytózy deliť na:
- lokalizované a generalizované, resp. systémové histiocytózy, alebo, prihliadajúc na počet postihnutých orgánov na
- formy monoorgánové (unifokálne, resp. multifokálne) a na formy multiorgánové.
Pri tomto druhom prístupe sa multiorgánové histiocytózy ešte podľa časového priebehu ochorenia v minulosti delili na akútne, subakútne a chronické.
d) Podľa prítomnosti či absencie fenoménu fagocytózy, resp. tezaurizácie fagocytovanej substancie, možno rozlišovať:
- histiocytózy s fagocytózou (obyčajne s hemofagocytózou) a/ alebo tezaurizujúce histiocytózy [3,12,45] a
- histiocytózy bez prejavov fagocytovej aktivity.
Okrem tu vymenovaných prístupov možno samozrejme použiť aj iné, vrátane častosti ich výskytu. Tak napr. pri konštrukcii seminára, na ktorom bola prednesená aj táto prezentácia, sa použil (v súlade s prístupmi tzv. histiocytovej spoločnosti [39]) jednoduchý, ale klinicky dobre zrozumiteľný prístup rozdelenia histiocytóz na:
- častejšie „Langerhansove histiocytózy“ a
- zriedkavejšie „non-Langerhansove histiocytózy“.
V klinickej praxi sa však najčastejšie používa kombinovaný klasifikačný prístup, ktorý zohľadňuje tak histogenetické, ako aj biologické kritériá. Klasifikácie podľa tu diskutovaných prístupov a podľa rôznych autorít, resp. literárnych zdrojov, sumarizujeme v tab. 1 – 3 a na obr. 2.
Jednotlivé nádorové non-LCH histiocytózy
Nádory histiocytov (makrofágov)
Na tomto mieste je žiadúce najprv objasniť viacero pojmov a definovať východiskové kritériá. Nádorová histiocytóza makrofágov vzniká klonovou neoplastickou proliferáciou buniek, ktoré histogeneticky a fenotypove zodpovedajú zrelým bunkám typu histiocytov, resp. makrofágov, t.j. buniek odvodených od monocytového - histiocytového radu. Podľa klasifikácie SZO z roku 2008 sem patrí len histiocytový sarkóm (tab. 3), ale nie leukémie monocytového radu, ktoré nespĺňajú uvedenú definíciu (viď aj ďalej). Mnohé morfologické aj fenotypové črty makrofágov vykazuje aj disseminovaný juvenilný xantogranulóm, ale jeho histogenetický pôvod je zrejme odlišný (viď ďalej), a je preto podľa citovanej klasifikácie SZO aj v tejto práci radený samostatne.
Okrem toho existuje celý rad lézií, aj nádorových, v názvosloví ktorých sa v rôznom tvare objavuje slovo histiocyt a pri ktorých sa diskutuje o ich histiocytovom pôvode. Ide tu najmä o spektrum procesov mäkkých tkanív, ktoré sa tradične radili medzi tzv. fibrohistiocytové nádory ako nádory s črtami histiocytov a fibrocytov, resp. fibroblastov [93]. Tieto sa rozdeľujú na benígne, nádory stredného (intermediárneho) stupňa malignity (majú signifikantné riziko lokálnej rekurencie, ale nízke riziko regionálneho alebo vzdialeného metastazovania) a na malígne (agresívne) nádory (tab. 4). Konceptuálne ide o chaotické usporiadanie, lebo:
- nie všetky lézie týchto skupín sú pravými nádormi,
- nie všetky lézie majú skutočne histiocytový, ale skôr fibroblastový alebo možno aj iný pôvod, čo platí aj pre malígny fibrózny histiocytóm, takže názvoslovie je tu do značnej miery zavádzajúce,
- alebo vykazujú zmiešaný histiocytovo - fibroblastový fenotyp
- a napokon v skupine agresívnych je zaradená „povrchová“ kožná forma malígneho fibrózneho histiocytómu (sarkómu), ktorá nie je biologicky agresívna.
![Klasifikácia tzv. fibrohistiocytových nádorov mäkkých tkanív [93].](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image/bc36d29a9eb45d324b31bab4653f9baf.jpg)
Ide však o praktický klinickou praxou akceptovaný zoznam nádorov a nádorom podobných lézií. Z hľadiska nami predloženého prehľadu non-Langerhansových histiocytóz boli tieto nádory tu uvedené len veľmi stručne pre orientáciu čitateľa vzťahov v celej problematike, pre detaily odkazujeme na špecializovanú literatúru nádorov mäkkých tkanív.
Pre ich stručný komentár na tomto mieste boli aj ďalšie 2 dôvody:
- a) niektoré z lézií skupiny „fibrohistiocytových nádorov“ sa do istej miery prekrývajú s tu diskutovanými nádormi histiocytov (napr. xantóm, ktorý je nepravým nádorom, pseudotumorom a xantogranulóm, ktorý zrejme zahŕňa spektrum procesov medzi hyperpláziou histiocytov a pravým histiocytovým nádorom),
- b) podľa klasifikácie histiocytovej spoločnosti (tab. 2) medzi histiocytózy makrofágov patrí aj retikulohistiocytóm a multicentrická retikulohistiocytóza (viď ďalej), ktoré sú radené do spektra benígnych fibrohistiocytových nádorov mäkkých tkanív a ktorých neoplastická povaha je spochybňovaná.
Medzi nádorové histiocytózy nepatrí spektrum procesov zaradených do skupiny hemofagocytovej, resp. familiárnej lymfohistiocytózy, ani sekundárna hemofagocytová lymfohistiocytóza, hoci ich často akútny a obyčajne fatálny priebeh simuluje obraz agresívneho nádorového ochorenia. Takisto sem nepatria sekundárne hemofagocytové lymfohistiocytózy. Hoci v prípade hemofagocytovej lymfohistiocytózy sú známe genetické abnormality (mutácia génu pre perforín a génu Munc 13 - 4), tak uvedené „lymfohistiocytózy“ predstavujú nenádorové histiocytové proliferácie [12,45]. Tieto procesy recentne podrobne v tomto časopise opísali Adam et al [3]. Takisto medzi nádorové histiocytózy nepatrí tzv. „sínusová histiocytóza s masívnou lymfadenopatiou“, alebo Rosaiova - Dorfmanova choroba (tiež podrobne diskutovaná v zmienenom článku Adama). Je to polyklónové histiocytové ochorenie nejasnej etiológie, so zmiešaným fenotypom buniek (viď vyššie – črty makrofágov aj DCs), jeho histologický obraz však môže byť pre menej skúseného patológa problematický aj z hľadiska diferenciálnej diagnózy pravých histiocytových nádorov a fagocytujúcich nenádorových histiocytóz [14].
Z uvedených dôvodov tu uvádzame „len“ problematiku histiocytového sarkómu a stručne komentujeme lézie typu retikulohistiocytómu a multicentrickej retikulohistiocytózy.
Histiocytový sarkóm (ICD - O M 9755/ 3)
Histiocytový sarkóm bol v minulosti (na úrovni vtedajších poznatkov) neraz klasifikovaný v rámci dnes už obsolentnej kategórie „malígnej histiocytózy“, pričom tu často išlo o dnešné kategórie veľkobunkových ML T alebo B radu. Skutočný histiocytový sarkóm je definovaný ako zhubný nádor, ktorý vzniká primárnou klonovou proliferáciou buniek monocytového - histiocytového (makrofágového) radu. Podľa klasifikácie SZO z roku 2008 je definovaný ako nádor, ktorý vykazuje morfologické a fenotypové znaky zrelých tkanivových histiocytov.
Histogeneticky má tak síce blízko k iným nádorom monocytového krvotvorného radu, a to akútnej monocytovej (prípadne myelomonocytovej) a monoblastovej leukémii, ale bunky týchto akútnych leukémií sú obyčajne na nižšom diferenciačnom či vývojovom stupni. Morfologická a fenotypová analýza individuálneho nádoru však neumožnia odlíšiť túto leukémiu a histiocytový sarkóm, pretože znaky ako CD68 a CD163 sú pozitívne na rôznych stupňoch diferenciačného vývoja uvedeného radu. Pre odlíšenie je nevyhnutná znalosť klinických a laboratórnych údajov, a preto je žiadúce vždy najprv zvážiť, či nádor nie je extramedulárnou propagáciou akútnej monocytovej leukémie alebo blastickej fázy myeloproliferatívnej neoplázie („myelosarkóm“ s fenotypom monocytov). Tieto extramedulárne manifestácie primárnych nádorov krvotvorby nepatria medzi histiocytové sarkómy [82]. Navyše, keďže histiocyty nie sú schopné recirkulácie, tak pravý histiocytový sarkóm by nemal byť schopný leukemizácie [3,46].
Vo všeobecnosti je histiocytový sarkóm vzácny nádor, aj najväčšie publikované súbory uvádzajú len nepočetné skupiny prípadov (najviac 14 pacientov v súbore Hornicka et al [38]). Môže vzniknúť u detí aj u dospelých, a to primárne buď v nodálne (menej často), alebo častejšie v extranodálnej lokalizácii ako sarkóm kože, mäkkých tkanív, brušných orgánov, najmä črevného traktu (tenkého aj hrubého čreva), sleziny a pod. [9,27,51,54,65,66].
V minulosti sa zrejme časť prípadov histiocytového sarkómu dnešnej definície, ako sme tu uviedli, označovala aj ako „malígna histiocytóza“. Z pohľadu dnešnej úrovne poznatkov je zrejmé, že pravá „malígna histiocytóza“ neexistuje. Avšak aj v recentnej literatúre sa niekedy tento názov objaví, zväčša ako synonymný názov mnohopočetného či disseminovaného histiocytového sarkómu so systémovými príznakmi [78]. V zásade teda klinická manifestácia histiocytového sarkómu môže byť v spektre od „monolokalizácie“ so solitárnou masou až po disseminovaný multifokálny a multiorgánový postih typu „malígnej histiocytózy“. Histiocytový sarkóm má potenciál pre agresívny klinický priebeh, najmä pri postihu viacerých skupín LU alebo pri primárne extranodálnej lokalizácii s dissemináciou. Naopak prípady, ktoré sa prezentujú ako klinicky lokalizované ochorenie, majú pomerne priaznivé dlhodobé prežívanie [38]. Preto je pravdepodobné, že možným prognostickým faktorom je veľkosť nádoru, resp. klinické štádium procesu [38,90]. Niekedy je klinická manifestácia bizarná, čo môže spôsobiť diagnostické rozpaky – napr. Cao et al [15] pozorovali primárny histiocytový sarkóm CNS s relapsom v mediastíne. U časti prípadov vznikajú prejavy pancytopénie.
U niektorých pacientov vzniká histiocytový sarkóm v súvise s predošlým alebo synchrónnym iným zhubným nádorovým ochorením, čo vyvoláva otázky o možnom histogenetickom súvise oboch nádorových procesov. V zásade tu prichádzajú do úvahy 2 rôzne možnosti:
- a) kombinácia s nádorom z germinálnych buniek s teratómovou komponentou, obyčajne tento nádor býva lokalizovaný v mediastíne [78]. Ak histiocytový sarkóm vznikne sekundárne po aplikácii chemoterapie, nutne vzniká otázka možnosti sekundárneho snáď terapiou indukovaného nádoru. Je známe, že teratómové bunky sú schopné pluripotentného vývoja a diferenciácie rôznymi cestami diferenciácie epitelových, mezenchýmových a neuroektodermových vývojových radov a in vitro aj v rámci krvotvorných bunkových radov. Preto tu prichádza do úvahy aj iný pôvod – mohlo by ísť o vznik histiocytového nádoru z nezrelej zárodočnej bunky [82]. Takéto „preskočenie“ diferenciácie nádorovej bunky na iný vývojový rad sa niekedy nazýva aj transdiferenciácia. Genetické testy môžu podporiť úvahy o spoločnom klonálnom pôvode, keď sa v bunkách oboch nádorov identifikuje i12p (izochromozóm 12p – genetický „marker“ nádorov germinálnych buniek). Táto chromozómová zmena však v nádorovej histiocytovej bunke nie je vždy vyznačená [78].
- b) ešte pravdepodobnejšia je transdiferenciácia v prípade histiocytového sarkómu u pacienta s primárnou diagnózou inej malignity krvotvorného alebo lymfatického tkaniva (najčastejšie lymfómy, leukémie, alebo myelodysplastický syndróm), keďže prinajmenej v niektorých prípadoch molekulovo - genetické analýzy dokazujú identický klonový pôvod oboch nádorov. Najčastejšie boli histiocytové sarkómy doteraz verifikované u pacientov s malobunkovými B - NHL, vrátane folikulového NHL, pričom vznik sarkómu sprevádza strata pan - B - znaku PAX5 [9,10,29]. Menej časté sú správy o vzniku histiocytového sarkómu u pacientov s ALL, obzvlášť ALL B radu, a to niekedy aj s viacročným intervalom po pôvodnej diagnóze leukémie [22,30]. Výnimočné sú prípady asociované s ALL T radu [19]. Jeden takýto prípad sme pozorovali aj my, kedy sme diagnostikovali histiocytový sarkóm čreva u detského pacienta s predtým verifikovanou a liečenou ALL T radu [54].
Napokon na tomto mieste je potrebné uviesť hoci ojedinelú recentnú kazuistiku, ktorá zachytáva ďalšiu možnú klinickú situáciu: pacient od ranného detstva s diagnostikovaným autoimúnnym lymfoproliferatívnym syndrómom (ktorý je známy častou asociáciou s malígnymi lymfómami), vznikom klasického Hodgkinovho lymfómu vo veku 6 rokov, ďalej manifestáciou „sínusovej histiocytózy s masívnou lymfadenopatiou“ a následným vznikom histiocytového sarkómu [86]. Išlo teda o vznik dvoch nádorov rôznej bunkovej línie v teréne uvedenej autoimúnnej lymfoproliferácie.
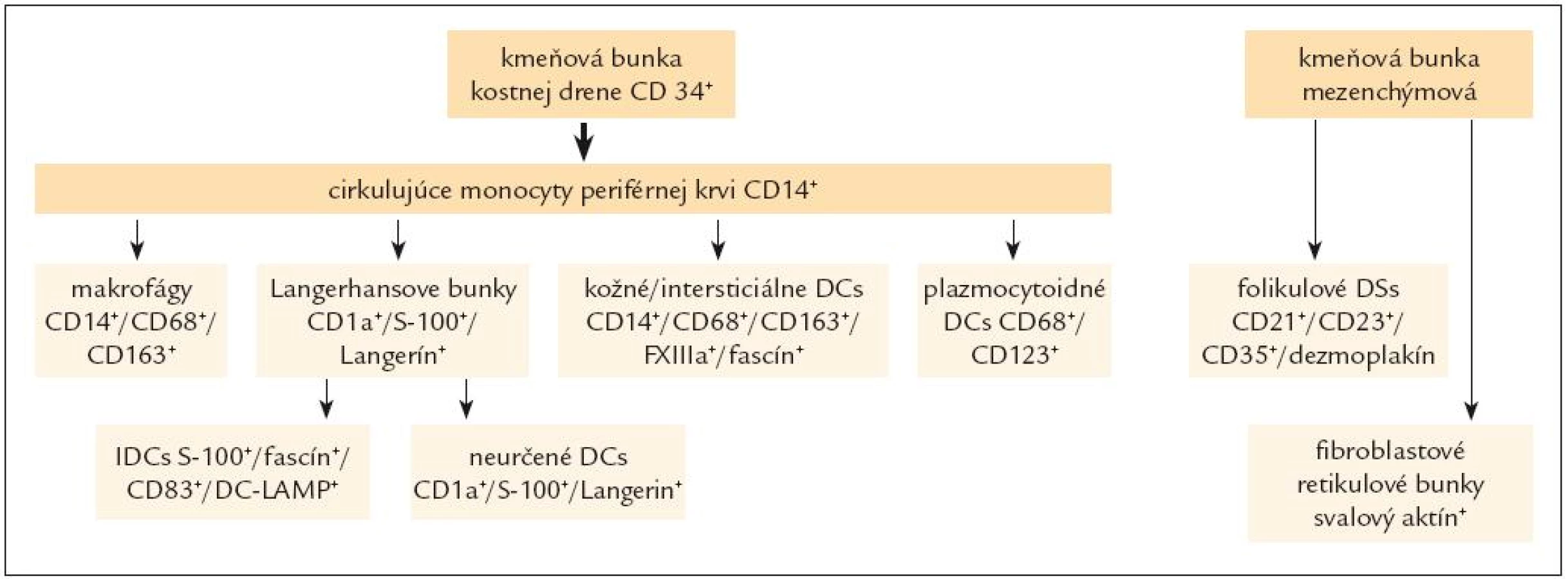
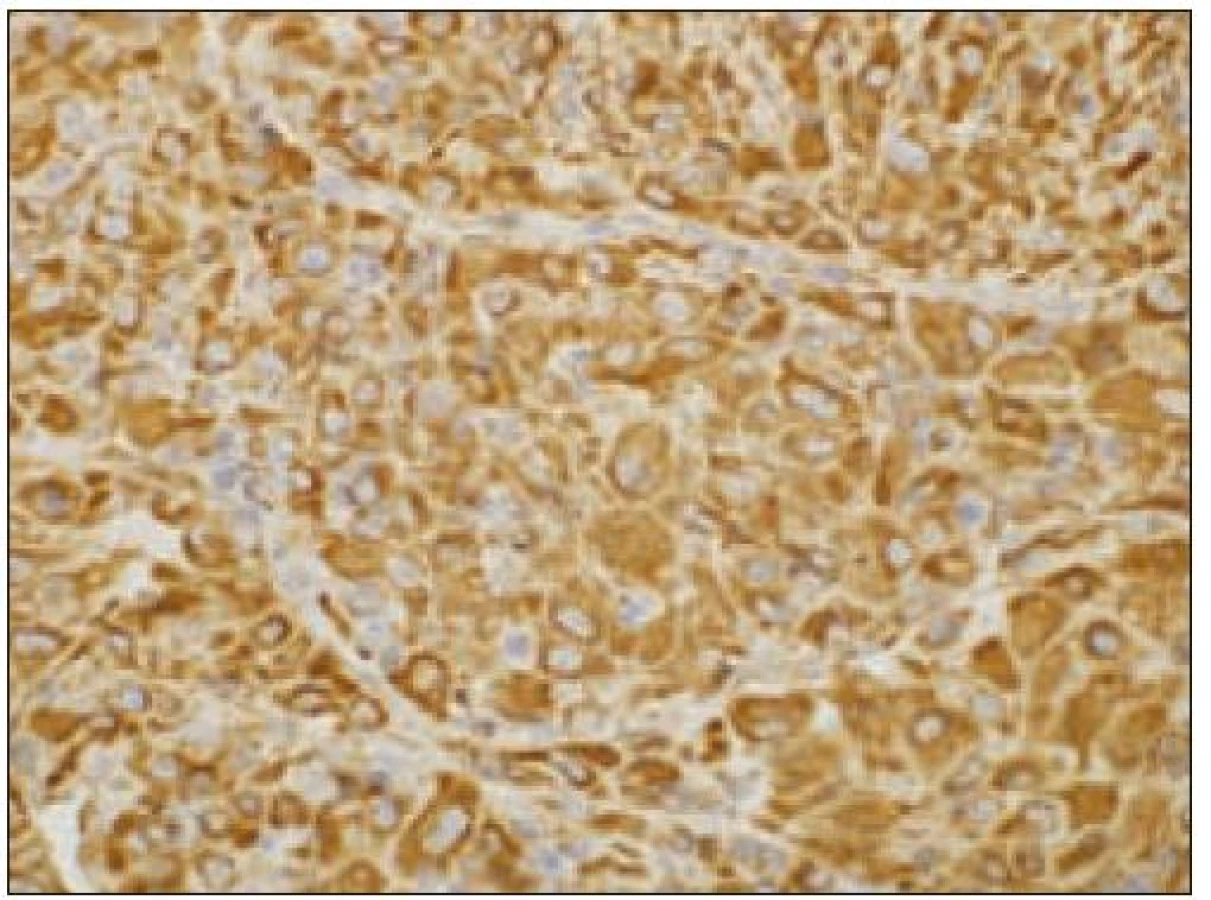
Všetky pre patológa - bioptika potrebné údaje bioptického obrazu histiocytového sarkómu sú podrobne uvedené v klasifikácii SZO z roku 2008. Tu len stručne uvádzame niektoré pre patológa hierarchicky zoradené charakteristiky (obr. 3 – 6):
- a) topografia nádorovej infiltrácie: vo väčšine opísaných prípadov nodálneho alebo splenického výskytu sa objavuje difúzna nádorová infiltrácia so zotretím pôvodnej štruktúry a jej zóny. V niektorých prípadoch sa pozoruje intrasinusoidálna propagácia nádorových buniek v infiltrovaných hematopoetických orgánoch (pečeni, slezine) a v LU.
- b) histocytologický obraz: ide o nádor s pomerne pestrou morfológiou, v ktorom obyčajne dominujú veľké a pleomorfné bunky so širokou eozinofilnou cytoplazmou, nepravidelným až bizarným jadrom a zväčša nápadnými jadierkami. Pre uvedený vzhľad bývajú bunky nádoru označované aj ako „epiteloidné“ [35]. Niekedy je v nádore nápadná hemofagocytóza [65], alebo mnohé z buniek sú viacjadrové. Mitotická aktivita býva vysoká. Pri výskyte v oblasti hlavy a krku sa zdôrazňuje možnosť vretenobunkovej morfológie tohto nádoru [7]. Výsledný obraz tak môže patológovi pripomínať morfológiu iných nádorov, najmä melanómu, veľkobunkového anaplastického lymfómu, prípadne menej diferencovaných karcinómov, či tumorov s „rabdoidnou“ morfológiou, čo zvyšuje nároky na dôkladnú imunohistochemickú analýzu. Ďalším diagnostickým problémom, ktorý je pomerne častým javom všetkých tu diskutovaných non-LCH nádorov, je nález početných reaktívnych buniek, najmä lymfocytov, eozinofilov a pod. Táto situácia sčasti pripomína imunitnú a zápalovú reakciu hostiteľa pri vzniku klasického Hodgkinovho lymfómu. Niekedy je táto reakcia tak pokročilá, že „prekrýva“ obraz nádoru, najčastejšie bol tento jav pozorovaný pri výskyte v CNS [15].
- c) histologický obraz a iné parametre vo vzťahu k odhadu prognózy: nie sú známe histologické parametre, ktoré by umožnili „grading“ nádoru, aj keď všeobecne možno predpokladať, že zvýšená anaplázia a rastová (proliferačná a mitotická) aktivita môžu byť indikátorom prognózy. Prognosticky dôležitá je zrejme už diskutovaná „masa“ nádoru.
-
d) fenotyp nádorových buniek: ako už bolo uvedené, dôležitá je imunohistochemická analýza fenotypových znakov a odlíšenie nádorov inej histogenézy, t.j.:
- pozitívny dôkaz CD45 a znakov histiocytových buniek, ako sú lyzozým CD68 a CD163, pričom pozitivita lyzozýmu a CD68 býva cytoplazmatická a granulárna a pozitivita CD163 membránová alebo cytoplazmatická [90]. Nádorové bunky často vykazujú aj pozitivitu CD43, CD14 a CD4 (35),
- negatívny dôkaz markerov karcinómu, melanómu, veľkobunkových lymfómov a i.,
- ako aj negatívny dôkaz markerov iných dendritických buniek. Niekedy sa v bioptickom obraze zisťuje fokálna a slabá pozitivita S - 100 proteínu časti buniek nádoru [35,45].
- e) genetické analýzy: nie sú známe pozitívne nálezy, resp. výsledky molekulových štúdií ostávajú zatiaľ kontroverzné [90]. T bunkový a Ig receptor sú per definitiam bez prestavby [82]. Napriek tomu sa recentne vyskytol údaj, že v nemalej časti prípadov sporadického histiocytového sarkómu (až do 40 % prípadov) možno identifikovať klonálnu prestavbu Ig receptora pri absencii súčasnej klonovej B bunkovej lymfoproliferácie [42]. Možno tu očakávať ďalšie údaje a hypotézy, či tieto nálezy povedú k zmene nazerania na histogenézu histiocytového sarkómu a na jeho definíciu.
- f) diferenciálna diagnostika z pohľadu patológa: viď vyššie.



Retikulohistiocytóm a multicentrická retikulohistiocytóza
Úvodom stručného prehľadu týchto 2 jednotiek znovu zdôrazňujeme, že nie sú súčasťou klasifikácie SZO z roku 2008. Obe jednotky, retikulohistiocytóm aj multicentrická retikulohistiocytóza sú označením pre zriedkavé a nie vždy celkom presne definované histiocytové proliferácie v koži alebo mäkkotkanivových štruktúrach. Zdá sa, že predstavujú 2 formy toho istého benígneho nádoru, pričom solitárny retikulohistiocytóm je izolovaný kožný nádor obyčajne diagnostikovaný u novorodencov a mladých dospelých, zatiaľ čo multicentrická retikulohistiocytóza, ako naznačuje už názov, je mnohopočetný nádor kožných lokalizácií a kĺbov spojených s deformujúcou polyosteoartropatiou, ktorej príznaky prevládnu v asi 1/ 2 postihnutých pacientov [45]. Vyskytuje sa u starších pacientov ženského pohlavia, v takmer 1/ 2 prípadov v asociácii s hyperlipidémiou, autoimúnitným alebo zhubným onkologickým ochorením.
Tento nádor (resp. oba tieto nádory) morfologicky v zásade zodpovedá fibróznemu histiocytómu klasifikácie fibrohistiocytových nádorov mäkkých tkanív (tab. 4, obr. 7 – 11). Tvoria ho veľké bunky so širokou svetlou až sklovitou eozinofilnou a slabo PAS+ cytoplazmou, bez atypií, medzi nimi sú disperzné zápalové bunky. Pri imunohistochemickej analýze bunky nádoru vykazujú rôzny stupeň pozitivity CD68+ (pozitivita býva paranukleárne akcentovaná), pričom dôkaz S - 100 proteínu, CD1a a fascínu je negatívny [45]. Recentne bol opísaný údajne nový podtyp retikulohistiocytómu, v ktorom prevládajú morfologicky epiteloidné histiocyty, autori [62] nazvali ho „solitárny epiteloidný histiocytóm“ a navrhli jeho zaradenie do skupiny histiocytových chorôb. V jeho histologickom obraze sa typicky okrem dominujúcich epiteloidných buniek nachádzajú aj početnejšie primiešané lymfocyty a neutrofilné leukocyty. Epiteloidné histiocyty vykazovali fenotyp makrofágu (CD163+, CD68+ a vimentín+), ale súčasne aj parciálne fenotyp kožných a intersticiálnych DCs (pozitivita F XIIIa) a S - 100 proteínu. Podľa ich mienky ide o benígnu pravdepodobne reaktívnu nenádorovú proliferáciu histiocytov neznámej etiológie. Je zrejmé, že tu ide o určité spektrum hyperplastických reaktívnych a pravých nádorových benígnych „histiocytových proliferácií“, ktorých fenotyp sa môže líšiť zrejme aj v závislosti od aktivácie pravých histiocytov versus aktivácie kožných DCs. Tým sa charakter procesu v rámci vyššie diskutovanej plasticity môže blížiť zmenám typu xantogranulómu – viď ďalej (obr. 12). V klasických histologických (patologických) deskripciách nádoru sa neuvádza penovitosť cytoplazmy buniek nádoru, ani účasť xantómových buniek, napriek tomu mnohé zdroje [3,39] nádor typu retikulohistiocytómu priamo radia do skupiny či spektra juvenilného xantogranulómu.






Nádory dendritických buniek myeloidného pôvodu
Histogeneticky a v zmysle koncepcie SZO klasifikácie z roku 2008 (tab. 3, obr. 2) sem patria:
- histiocytóza a sarkóm Langerhansových buniek, ktoré sú referované samostatne,
- nádory interdigitujúcich dendritických buniek,
- nádory z neurčených dendritických buniek,
- nádory z dermálnych a intersticiálnych buniek a
- nádory plazmocytoidných dendritických buniek.
Sarkóm interdigitujúcich dendritických buniek (ICD - O M 9757/ 3)
Interdigitujúce dendritické bunky sú, ako sme už uviedli, špecializované DCs T zóny lymfatických uzlín, resp. lymfatického tkaniva. Nádor, ktorý tvorí nádorová proliferácia vretenovitých až ovoidných buniek s im podobným fenotypom, sa podľa klasifikácie SZO z roku 2008 nazýva sarkóm z interdigitujúcich DCs (ďalej IDCS).
Tento sarkóm patrí medzi veľmi zriedkavé nádory, publikované zväčša len ako kazuistiky (v česko - slovenskom písomníctve napr. Adam et al [2]), pričom sa odhaduje, že celkove bolo doteraz publikovaných len niekoľko desiatok prípadov IDCS (Cossu et al v roce 2006 [17] identifikovali len 39 prípadov IDCS) . Tie sa vyskytli u pacientov od detského až stareckého veku, s miernou prevahou pacientov mužského pohlavia. Časť prípadov (7 z 39 podľa uvedených autorov) bola diagnostikovaná v asociácii s inou hematologickou malignitou. Išlo o nádory T radu (T - ALL, mycosis fungoides) alebo B radu (najmä folikulový ML a CLL [33]). Asociácia s EBV infekciou (na rozdiel od sarkómu folikulových DCs) nebýva obyčajne identifikovaná.
IDCS sa diagnostikuje častejšie ako primárne nodálny asymptomatický nádor a zriedkavejšie ako nádor so systémovými prejavmi ochorenia, ako sú strata na váhe, zvýšená teplota a pod. Okrem nodálnych foriem miestom primárneho vzniku IDCS môžu byť aj extranodálne lokalizácie viscerálne, ako sú tonzily, pľúca, brušné orgány, najmä pankreas, duodenum, pečeň, slezina, obličky a iné [33,47,50,82]. Ďalej môže ísť o sarkóm kosti a mäkkých tkanív s metastatickým postihom lokoregionálnej uzliny, ako tomu bolo aj v prípade Adama et al [2]. Aj keď podľa limitovaných dostupných údajov prognóza IDCS všeobecne je zlá, tak vnútrobrušná lokalizácia sa zdá byť negatívnym prognostickým faktorom.
Histomorfológia IDCS, dôležitá prepatológa a ním vedenú bioptickú dia-gnostiku, je podrobne opísaná v klasifikácii SZO z roku 2008. Na tomto mieste len stručne pripomíname niektoré základné histologické črty procesu:
- a) topografia nádorovej infiltrácie: v prípade nodálneho výskytu nádor vzniká v kôre, a to v T zóne uzliny, spočiatku môže v nej byť ešte zachované reziduá reaktívnych lymfatických folikulov. Čoskoro sú však tieto, podobne ako ostatné súčasti, nádorom infiltrované. Výsledný obraz nodálneho IDCS tak závisí od včasnosti, resp. pokročilosti procesu (a teda odberu LU na biopsiu). V prípade extranodálnych foriem ide o čiastočný alebo difúzny postih celého orgánu, napr. sleziny [50].
- b) histocytologický obraz: individuálne nádorové bunky môžu vykazovať morfologické spektrum od štíhlych vretenovitých buniek až po viac „napučané“ histiocytoidné až epiteloidné bunky s nápadnejším jadrom a širokou eozinofilnou cytoplazmou. Mitotická aktivita v IDCS býva nízka, obyčajne nižšia než 5 – 10 mitotických figúr/ 10 HPF (údaj 10 mitóz na 10 veľkých zorných polí je považovaný za hranicu medzi G1 a G2 sarkómov mäkkých tkanív). V nádore sa môžu vyskytnúť aj obrovské viacjadrové bunky, nekróza nebýva častá. Môžu vykazovať usporiadanie do storiformných štruktúr alebo pruhov (najmä v prípade prevahy vretenobunkovej morfológie), alebo viac rast na spôsob „solídneho“ nádoru (najmä v prípade prevahy skôr ovoidnej až epiteloidne bunkovej morfológie), zriedkavo intrasinusoidálny rast. Na pozadí nádoru sa môže vyskytnúť rôzne rozsiahla fibrotizácia až hyalinizácia. Za typickú známku nádoru sa považuje prítomnosť rozptýlených lymfoidných buniek po nádore (najmä lymfocyty a plazmocyty), v nádore sa môže vyskytnúť aj hemofagocytóza [50].
- c) histologický obraz vo vzťahu k odhadu prognózy: podľa klasifikácie SZO z roku 2008 histologické parametre IDCS (na rozdiel od FDCS) nie sú prognosticky významné, zatiaľ čo niektoré údaje zdôrazňujú, že zvýšenú mitotickú aktivitu nad uvedenú hranicu možno považovať za negatívny prognostický faktor.
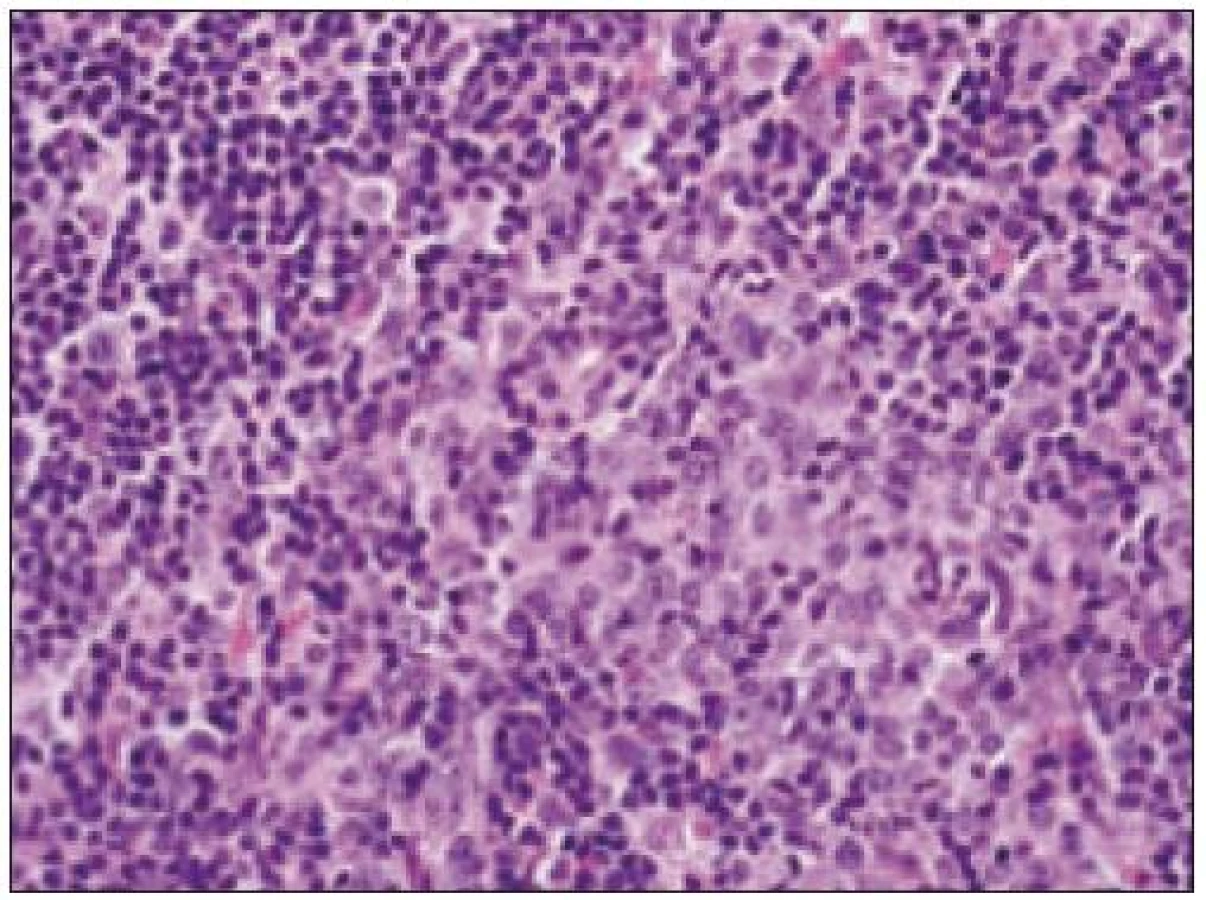
- d) fenotyp nádorových buniek: bunky IDCS bývajú pozitívne pri dôkaze vimentínu a S - 100 proteínu (obr. 1). V časti prípadov sa identifikuje pozitivita dezmoplakínu, CD68, lyzozýmu, zatiaľ čo faktory, ako sú antigén antigén LCH (langerín), antigény FDCs (klusterín, CD21, CD23, CD35), ako aj antigény iných morfologicky v diferenciálno - diagnostickú úvahu prichádzajúcich nádorov sú negatívne (napr. negativita dôkazu MPO, epitelových, melanómových, lymfocytových a endotelových markerov). V ultraštrukturálnom obraze sú zrejmé už uvedené interdigitujúce výbežky cytoplazmy a rôzny počet lyzozómov [33]. Sprievodné lymfocyty sú obyčajne T pôvodu.
- e) genetické analýzy: nie sú známe pozitívne nálezy, T bunkový a Ig receptor sú bez prestavby.
- f) diferenciálna diagnostika z pohľadu patológa: bioptická analýza IDCS si jednak pre ich zriedkavosť, ale aj pre prelínanie niektorých čŕt bioptického obrazu vždy vyžaduje dôkladnú a neľahkú diferenciálnu diagnostiku, najmä odlíšenie od FDCs, v brušnej oblasti aj od nádorov zo spektra GIST.
Nádor z indeterminovaných dendritických buniek (ICD - O M 9757/ 3)
Podľa klasifikácie SZO z roku 2008 ide o nádory nazývané aj histiocytózy z indeterminovaných („záhadných“) buniek, ktoré vznikajú neoplastickou proliferáciou buniek vretenovitého až ovoidného tvaru s fenotypovými črtami, ktoré zodpovedajú črtám normálnych indeterminovaných buniek. Tie sa považujú za údajné prekurzory Langerhansových buniek (obr. 1 a 2).
Ak v podstate pre každý z nádorov DCs platí, že je zriedkavý, tak snáď najzriedkavejšia zo všetkých nádorových histiocytóz je práve táto kategória nádoru. Vyskytuje sa obyčajne v koži, skôr ako disseminovaný než lokalizovaný proces vzhľadu početných lézií s morfológiou papúl až plošných rozsiahlejších lézií, pričom infiltráty prestupujú celú dermis. Podľa niektorých údajov aj tento nádor sa môže vyskytnúť v asociácii s malobunkovými B - NHL, najmä typu folikulového lymfómu. Aj tu možno uvažovať o fenoméne „transdiferenciácie“, kedže translokácia t(14;18) bola identifikovaná aj v bunkách tohto nádoru [75].
Nádor môže mať aj vretenobunkovú morfológiu a pripomínať skôr sarkóm folikulových DCs [76]. Keďže nádorové bunky majú zodpovedať prekurzorom buniek LCH, tak neprekvapuje, že podľa väčšiny dostupných údajov tento nádor tak morfologickým vzhľadom (vrátane cytologických detailov jadra, širokej eozinofilnej cytoplazmy, sklonom k tvorbe viacjadrových buniek a pod.), lokalizáciou v koži a prípadne v LU a aj fenotypove (pozitivita S - 100 proteínu a CD1a) viac než napodobňuje nádor typu LCH. Líši sa „len“ chýbaním Birbeckových granúl a podľa niektorých aj chýbaním expresie langerínu [75]. O to ťažšia je jeho bioptická diferenciálna diagnostika.
Prognóza nádoru je rôzna od prípadu k prípadu, pohybuje sa v spektre od možnej spontánnej regresie cez možnosť protrahovaného priebehu s početnými relapsami až po agresívne formy [43]. Problémom pre určenie prognózy je nielen chýbanie spoľahlivých kritérií, vrátane histologických, ale aj skutočná vzácnosť tohto nádoru a do istej miery aj otáznosť správnej definície niektorých v literatúre opisovaných prípadov.
Nádory intersticiálnych/ dermálnych dendritických buniek
Podľa klasifikácie SZO z roku 2008 ide síce o samostatné, ale vzájomne súvisiace nádory v spektre non-LCH histiocytóz: disseminovaný juvenilný xantogranulóm (ďalej disseminovaný JXG) je nádor pacientov detského veku a Erdheimova - Chesterova choroba (ďalej ECC) je považovaná za adultnú formu tohto nádoru.
Postihnuté tkanivá a orgány pri disseminovanom JXG a rovnako aj pri ECC vykazujú podobný morfologický obraz. Tvoria ho lézie nazývané všeobecne „xantogranulómy“, v ktorých sú miestne nahromadené histiocyty, ďalej pomerne veľké makrofágy so širokou penovitou cytoplazmou – tzv. penovité histiocyty alebo xantómové bunky, obrovské viacjadrové bunky Toutonovho typu, primiešané sú zápalové bunky, najmä lymfocyty a prípadne aj fibroblasty s potenciálom fibroprodukcie. Nahromadenie opísaných buniek často pripomína granulomatózny zápal, z toho vznikol názov xantogranulóm. Dominujúce histiocyty, aj tie s vakuolizovanou cytoplazmou, vykazujú fenotyp buniek monocytového - histiocytového radu (pozitivita znakov CD68 a CD163) a chýbajú im znaky typické pre fenotyp buniek LCH – vykazujú negativitu dôkazu CD1a S - 100 proteínu (ten môže byť slabo pozitívny v časti buniek) a pri elektrónovomikroskopickom vyšetrení v ich cytoplazme obyčajne chýbajú Birbeckove granulá [82].
Napriek uvedeným faktom sa stále diskutuje o histogenéze, aj o klonálnej povahe tak JXG ako aj ECC. Hoci podľa fenotypu možno predpokladať, že predstavujú nádory histiocytového - makrofágového pôvodu, tak niektorí autori sa na základe nie-ktorých fenotypových odlišností buniek JXG (napr. pozitivita faktoru XIIIa a fascínu – Swerdlow et al 2008) prikláňajú skôr k pôvodu v kožných/ intersticiálnych DCs. Okrem toho niektorí diskutujú o možnosti, že bunky disseminovaného JXG vznikajú z CD4+ plazmocytoidných dendritických buniek [55]. Nevyriešenou ostáva aj klonalita procesu. Ak totiž za definujúci znak nádorového procesu akceptujeme dôkaz jeho klonovej povahy, tak povaha JXG a ECC nie je jednoznačná. Nahromadené histiocyty ECC často vykazujú prítomnosť t(12;15;20)(q11;q24;p13.3), alebo ďalšie numerické chromozómové abnormality [86], čo by podporilo úvahy o klonovej (t.j. neoplastickej) povahe procesu. Iní autori v proliferujúcich bunkách izolovaných tkanivovou mikrodisekciou klonalitu nedokázali a prikláňajú sa skôr k nenádorovej povahe ECC a JXG [34]. Je zrejmé, že príčinou uvedených rozporov môžu byť tak rôzne aplikované metódy, ich rôzna senzitivita a špecificita, ako aj malý počet prípadov na testovanie klonality.
Podstatou histomorfologických zmien pri oboch týchto nádoroch sú uvedené xantómové bunky a xantogranulómy, resp. „xantogranulomatózne lézie“ rôzneho rozsahu s potenciálom infiltrovať postihnuté štruktúry.
Orientáciu čitateľa v tejto oblasti negatívne ovplyvňuje nielen zriedkavosť uvedených 2 foriem nádoru, ale a rovnako aj to, že existujú niektoré terminologicky blízke, ale patogenézou aj histogeneticky odlišné chorobné jednotky (niektoré z nich sme už uviedli v úvode k histiocytovým nádorom a v tab. 4). Viaceré z nich môžu navyše spôsobiť aj diferenciálno - diagnostické problémy, pretože klinickou manifestáciou imitujú nádory tejto skupiny a po chirurgickom odstránení spôsobujú diagnostické problémy pri ich histopatologickej analýze. V snahe napomôcť čitateľovi v terminologickej orientácii tu najprv zaraďujeme krátky „slovník“ so zjednodušeným vysvetlením jednotlivých pojmov, ako sú xantóm, xantelasma, xantogranulóm, xantofibróm a pod. Táto krátka stať nemá a nemôže mať ambíciu nahradiť extenzívnejšie špecializované texty o tejto problematike.
Xantoma (xantóm): xantóm je ohra-ničený, lokalizovaný až tumoriformný zhluk buniek tkanivových histiocytov, ktoré v cytoplazme obsahujú tuk. Bunky xantómu – tzv. xantómové bunky – vznikajú procesom intracelulárnej depozície cholesterolu a jeho esterov do cytoplazmy makrofágov, ktoré sa tak menia na bunky s penovitou cytoplazmou. Xantóm nie je pravým nádorom, v tab. 4 je síce v spektre benígnych fibrohistiocytových lézií mäkkých tkanív klasifikovaný ako nádor, v skutočnosti je to pseudotumor [93]. Je to reaktívna histiocytová proliferácia, ktorá vzniká miestne ako odpoveď na alterácie sérových lipidov [dôsledok väčšiny primárnych hyperlipoproteinémií (typ I – V so známymi alebo neurčenými genetickými defektami) a niektorých sekundárnych hyperlipopoteinémií – napr. pri diabetes mellitus, pri cholestatických ochoreniach a pod.]. Podstatná je teda systémová porucha metabolizmu tukov (ktoré vždy pochádzajú z krvi), pričom xantómy vznikajú ako lokalizované zmeny zrejme v súvise s miestnym zvýšením cievnej permeability (napr. trauma, alebo iné miestne poškodenia vedúce k uvoľneniu histamínu). V tejto súvislosti stojí za zmienku, že aj pri ECC býva hypercholesterolémia a dochádza k nadmernému prívodu tuku do histiocytov.
V protiklade ku xantómom asociovaným s hyperlipidémiou existujú aj ďalšie podobné a z hľadiska incidencie podstatne vzácnejšie lézie u jedincov s normálnou hladinou lipidov v sére. Sem patrí napr. tzv. xantomatóza (mnohopočetné xantómy kože) u niektorých pacientov s monoklónovou gamapatiou nejasného významu alebo inými lymfoproliferatívnymi chorobami [6,93]. Jej patogenéza je neobjasnená, diskutuje sa tu napr. o možnej asociácii monoklónového Ig a lipidov, o možnej infekčnej genéze procesu a pod. [6]. Je pravdepodobné, že tento typ ochorenia by mohol alebo mal byť zaradený do spektra nižšie uvedeného disseminovaného JXG.
Súčasťou histologického obraz xantómu môže byť aj prímes elementov chronického zápalu, prípadne fibrotizácia, pričom sa predpokladá sa, že fibróza môže byť dôsledkom fibrogénnych vlastností extracelulárne uvoľnených degradovaných lipidických substancií a cholesterolu. Xantómy vznikajú ako solitárne alebo mnohopočetné útvary v:
- a) povrchových častiach tela, napr. v periokulárnej oblasti, najmä na viečkach (tzv. xantelasma), v koži (napr. gluteálnej oblasti ako tzv. eruptívne xantómy), v podkožnej oblasti okolo kĺbov (tuberózne xantómy v oblasti lakťa, kolena, prstov),
- b) v hlbších častiach mäkkotkanivových štruktúr (blízko šliach a synovie) a zriedka aj v kosti ako tzv. periartikulárne alebo šľachové xantómy.
Xantómové bunky sa môžu vyskytovať aj v asociácii s inými procesmi a zmenami. Sú napr. súčasťou lézie známej ako nodulárna (teno-)synovitída (seu obrovskobunkový nádor šľachových pošiev), ktorá je na šlachy prstov lokalizovaný proces (jeho difúznym proťajškom je pigmentovaná vilonodulárna synovitída postihujúca kolenné a i. kĺby). Pri nej vzniká na šlache uzlovitý útvar, tvorený zmesou vretenovitých buniek (často s bohatými depozitami hemosiderínu), penovitých xantómových buniek a obrovských viacjadrových buniek, preto sa niekedy tento proces označoval aj ako xantofibróm alebo fibroxantóm.
Xantogranuloma (xantogranulóm), resp. kožný juvenilný xantogranulóm (JXG), je v dermatológii a dermatopatológii dobre a dlho známy (solitárny, nie mnohopočetný) proces s typickým histologickým „xantogranulomatóznym“ obrazom (obr. 13 a 14), vrátane výskytu toutonoidných obrovských viacjadrových buniek. Vzhľadom k tendencii spontánneho zániku a „benígnemu“ charakteru sa dlho diskutovalo, či ide o pravý nádor. V rámci nádorov mäkkých tkanív je zaraďovaný medzi benígne fibrohistiocytové nádory mäkkých tkanív (viď vyššie a tab. 4), pričom biologicky ide o proces na rozhraní hyperplastického a benígneho nádorového procesu.


V časti prípadov xantogranulómu sa vyskytujú nekrobiotické zmeny, resp. rôzne rozsiahla koagulačná nekróza – tieto relatívne vzácne až raritné prípady sa označujú ako tzv. nekrotizujúci (nekrobiotický) xantogranulóm. Vyskytuje sa v kožnej alebo mimokožnej forme, kedy môže postihnúť rôzne orgány (najčastejšie mäkkotkanivové štruktúry a zriedkavejšie vnútorné orgány), a spôsobiť tak aj rôznu klinickú symptomatológiu [83]. Veľmi často sa vyskytuje v asociácii s hematologickými malignitami, najmä s lymfoproliferatívnymi ochoreniami s klonálnou proliferáciou plazmocytov a produkciou monotypického Ig [45]. Jeho prejavy sa neraz blížia zmenám pri disseminovanom JXG [6], asociáciou s klonálnou plazmocytovou proliferáciou sa (prinajmenej z hľadiska patogenézy ukladania lipidov v bunkách) približuje problematike xantomatózy. Pôvodne bol opísaný ako ochorenie s progredujúcimi periorbitálnymi xantómami a diskutovalo sa, či príčinou nekrózy sú imúnne komplexy (z cirkulujúcich Ig), ktoré po tkanivovej depozícii spúšťajú zápalovú reakciu [83], alebo spirochétové infekcie (v časti prípadov bola v léziách identifikovaná Borrelia spirochetes [95]). Na tomto mieste je vhodné z hľadiska diferenciálnej diagnostiky aspoň naznačiť, že existuje nesmierne široké spektrum procesov, ktoré možno zaradiť medzi „nekrotizujúce“ alebo „nekrobiotické granulómy“, ktoré však zrejme vznikajú rôznymi inými patogenetickými cestami (pri systémových imunopatologických ochoreniach, kožné granulomatózne ochorenia, kožný nekrotizujúci granulóm u diabetikov a pod. [32]). V rámci sústredenia sa na problematiku prehľadu non-LCH histiocytóz tu nie je priestor pre ďalšiu podrobnú diskusiu v tomto smere, preto tu čitateľa tu odkazujeme na špecializovanú najmä dermatologickú a dermatopatologickú literatúru. Túto diskusiu možno tu uzavrieť s tým, že oblasť opísaných (nielen) kožných xantomatóznych zmien sa značne prelína s problematikou disseminovaného JXG a ECC.
Atypický fibroxantóm (tab. 4) je vretenobunkový nádor, ktorý vzniká najčastejšie v aktinicky poškodenej koži starších ľudí, najmä v oblasti hlavy a krku. Aj keď podľa viacerých koncepcií sa považuje za povrchovú formu spektra malígneho fibrózneho histiocytómu a je radený medzi malígne fibrohistiocytové nádory mäkkých tkanív, tak jeho prognóza je pomerne priaznivá [93].
Xantogranulomatózna zápalová reakcia vzniká niekedy ako sekundárna miestna zápalová reakcia pri procesoch spojených s nekrotickým rozpadom buniek a tkanív a uvoľňovaním lipidov do interstícia [45]. Príčinou rozpadu môžu byť aj infekčné, obzvlášť bakteriálne agens (patogenéza podobné tej, o ktorej sa diskutuje pri xantomatóze). Uvoľnené tukové látky sú fagocytované makrofágmi, ktoré tak získavajú penovitý vzhľad „xantómových buniek“. Tieto bunky spolu s ostatnými elementmi chronickej zápalovej reakcie (lymfocytami, plazmocytami, fibroblastami a pod.) vytvárajú útvary podobné granulómu, z čoho vznikol názov xantogranulomatózna zápalová reakcia. Tak napr. xantogranulomatózna pyelonefritída je zriedkavým variantom chronickej tubulointersticiálnej nefritídy, pri ktorej v interstíciu obličky vznikajú ložiská xantogranulomatóznej zápalovej reakcie. Podobné zmeny sa vyskytujú pri iných chronických zápalových reakciách (napr. xantogranulomatózna cholecystitis, xantogranulomatózna oophoritis, cystiis, orchitis a i.). Ďalej ich môže patológ pozorovať aj v rôznych tkanivách v susedstve primárneho nádoru, alebo jeho sekundárneho (metastatického) nádorového ložiska [84], pri ktorých dochádza v dôsledku rýchleho rastu nádoru alebo iných príčin k nekróze a rozpadu nádorového tkaniva. Ide len o sprievodnú zápalovú reakciu, ktorá nemá vplyv na prognózu základného ochorenia, aj keď v poslednej dobe sa množia údaje o viacerých skupinách nádorov, podľa ktorých histiocytová „stromálna“ reakcia hostiteľa na nádor býva negatívnym prognostickým znakom. Výsledný histologický obraz však môže tak byť problematický, keďže sprievodná reakcia môže prekryť vlastný nádorový proces. V praxi asi najväčšie problémy a diagnostické rozpaky môže spôsobiť xantogranulomatózna (zápalová) reakcia v asociácii s v retroperitoneu lokalizovaným malígnym fibrohistiocytovým nádorom (malígnym fibróznym histiocytómom, a to najmä jeho tzv. inflamatórnym typom [93]). Xantogranulomatózna zápalová reakcia vzniká aj pri Erdheimovej - Chesterovej chorobe – viď ďalej.
V klasifikácii SZO z roku 2008 sa medzi histiocytovými nádormi čiže procesmi klonálneho pôvodu vyskytuje len disseminovaný JXG, ktorý sa považuje za „klonálny proces charakterizovaný proliferáciou histiocytov podobných dermálnemu JXG“ (obr. 13 a 14). Hoci doteraz nebola opísaná progresia solitárneho kožného JXG do disseminovanej formy [21], tak teoreticky je veľmi pravdepodobné, že obe formy JXG, kožný aj disseminovaný, môžu predstavovať rôzne manifestácie, resp. kontinuum toho istého procesu. V oboch formách je v zásade prítomný identický už vyššie opísaný histomorfologický obraz. V minulosti sa pre léziu typu disseminovaného JXG používali rôzne iné alebo synonymne používané názvy, ako napr. xanthoma disseminatum (kože a slizníc, ale aj CNS [61]), benígna cefalická histiocytóza, progresívna nodulárna histiocytóza [48].
Disseminovaný JXG sa vyskytuje najčastejšie u detí do 10 rokov, pričom 1/ 2 všetkých prípadov sa objavuje už v priebehu 1. roka života a časť prípadov reprezentuje kongenitálny typ nádoru. V časti prípadov sa objavuje asociácia s neurofibromatózou typu I, alebo niektorými inými nádormi monocytového histiocytového pôvodu (LCH, juvenilná myelomonocytová leukémia a i.).
Klinicky aj morfologicky podobné ak nie identické xantogranulómy, aj disseminované, sa vyskytujú aj v dospelom veku, a to v 2 formách:
- v asociácii so vznikom bronchiálnej astmy v dospelosti – tzv. adult - onset xanthogranuloma [26],
- alebo v asociácii s hematologickými malignitami [26,79].
Asociácia disseminovaného JXG s nádormi krvotvorby a lymfatického systému pri súčasnej podobnosti viacerých čŕt klinickej manifestácie a morfologického obrazu vyššie diskutovaných jednotiek typu „xantomatózy“ a „nekrotizujúceho“ či „nekrobiotického“ xan-togranulómu vzbudzujú otázku, či všetky diskutované lézie predsa len nepredstavujú súčasť spektra podobných ak nie identických procesov. Napokon klasifikácia histiocytárnych chorôb podľa Histiocytovej spoločnosti [28,39] a rovnako aj opakovane zmienená práca Adama et al [3] hovoria o JXG a nekrobiotickom xantogranulóme ako o „skupine histiocytových chorôb z fagocytujúcich buněk, pre ktoré je typická fagocytóza lipidov“.
Klinické prejavy disseminovaného JXG detí často závisia od veľkosti nádorových ložísk a od miestnych pomerov, ktoré určujú event. vznik dôsledku z narastajúcej masy nádoru a jeho tlaku na okolité štruktúry. Disseminovaný JXG sa vyskytuje buď len vo forme početných kožných nádorov, alebo aj v inej než kožnej lokalizácii, prípadne v ich kombináciách. Kožná forma býva lokalizovaná najmä na koži hlavy, a to okolo očných viečok, na čele alebo inde na tvári. Mnohopočetné kožné JXG sú časté najmä u pacientov vo veku menej ako 6 mesiacov. Kožné lézie majú vzhľad oranžovej alebo červenohnedej papuly, nespôsobujú žiadnu závažnejšiu symptomatológiu a môže aj spontánne regredovať [45,82]. Nenápadnosť prejavov a možný spontánny zánik spôsobujú, že skutočná incidencia tejto lézie je neznáma. Súčasne sú (napr. recentne opäť pod znovuoživeným názvom progresívna nodulárna histiocytóza) známe aj menej časté prípady rozsiahlych kožných nádorových lézií deformujúcich tvár, prípadne sa vyskytujúcich aj v iných vnútorných orgánoch, napr. laryngu, a to aj u dospelých pacientov [48].
Pri vzniku disseminovaného JXG v iných lokalizáciách môžu vzniknúť miestne prejavy z útlaku okolitých štruktúr. Tie môžu a nemusia byť zanedbateľné pri podkožnej mäkkotkanivovej lokalizácii alebo v oblasti kostrového svalstva. Naproti tomu xantogranulómy v očnej oblasti (intraokulárne alebo intraorbitálne) sa spájajú s evolúciou možnej poruchy zraku, ako je glaukóm a slepota [59]. Nádory v pečeni, slezine pľúcach či kosti môžu viesť k výraznej miestnej symptomatológii. Pri infiltrácii CNS (mozgu alebo miechy) to môžu byť diabetes insipidus a iné pituitárne a hypotalamické syndrómy, hydrocefalus, duševné poruchy a pod. [60]. Klinicky najzávažnejšie sú stavy s mnohopočetným alebo systémovým výskytom (v rámci RES). Priame systémové postihnutie kostnej drene, ako aj nepriamy dôsledok všeobecnej aktivácie makrofágového systému môže viesť k redukcii krvotvorby s príslušnými hematologickými komplikáciami a dopadom na obranyschopnosť organizmu. Mnohopočetný a systémový výskyt, napr. v pečeni, slezine a kostnej dreni pri nepriaznivej kombinácii výskytu napriek benígnej povahe procesu môžu, hoci vzácne, spôsobiť aj smrteľné komplikácie a vyžadovať onkologickú liečbu [41].
Z hľadiska koncepčného triedenia faktov viscerálne formy disseminovaného JXG tvoria vlastne akýsi „prechod“ k Erdheimovej - Chesterovej chorobe [3], resp. na oba procesy možno nazerať ako na isté kontinuum či spektrum podobných, ak nie rovnakých procesov.
Erdheimova - Chesterova choroba (ECC) sa považuje, ako sme už uviedli, za adultnú formu disseminovaného JXG, keďže sa vyskytuje u dospelých obyčajne po 40. roku života. Pedia-trické prípady sú extrémne vzácne. Per definitiam je to zriedkavá xantogranulomatózna multisystémová (non-Langerhansova) histiocytóza dospelých pacientov. Etiológia a patogenéza ECC (podobne ako pri JXG) ostáva neznáma, prinajmenej u časti pacientov bývajú v sére biochemické abnormality lipidového metabolizmu, vrátane hypercholesterolémie.
Histologicky sa postihnuté miesta vyznačujú podobnými zmenami ako pri JXG: miestnym nahromadením veľkých makrofágov s penovitou cytoplazmou („xantómových buniek“), obrovských viacjadrových toutoidných buniek, s prímesou lymfocytov, fibroblastov a pod. V postihnutých miestach tak vznikajú xantogranulomatózne štruktúry (s fenotypom CD68+, CD163+, CD1a– ). Preto sa často v literatúre objavujú deskriptívne názvy ako napr. xantogranulóm tej - ktorej oblasti pacienta s ECC [25]. V ložiskách xantogranulómov môže aktiváciou fibroblastov dochádzať k fibrotizácii so sekundárnym ďalším postihnutím tejto oblasti jazvením. Len zriedkavo sa v xantogranulomatóznej lézii objavuje koagulačná nekróza, napr. nekróza v kostnodreňových léziách [52]. Aj tu potom, podobne ako sme to už uviedli pri JXG, možno uvažovať o podobnosti či zhode s léziou typu nekrotického xantogranulómu (viď vyššie). Súčasťou diagnostického algoritmu môže byť bioptické vyšetrenie danej lokality, správna diagnóza vyžaduje dobrú erudíciu patológa, znalosť tejto vzácnej jednotky a neraz nie ľahkú diferenciálnu diagnózu.
Klinické prejavy ECC sú značne heterogénne a závisia:
- od morfologickej fázy vývoja xantogranulomatóznych lézií,
- od tlakového lokálneho efektu nahromadenej masy xantogranulómov,
- od infiltratívneho charakteru procesu a rozsahu postihnutia daných štruktúr,
- od sekundárne indukovaných najmä fibrotických a osteosklerotických zmien postihnutých miest (patogenéza tejto indukcie nie je objasnená),
- od možného systémového efektu z aktivácie monocytového - fagocytového systému (podobne ako pri JXG),
- ako aj od miesta postihu tohto multisystémového ochorenia.
V zásade možno pri ECC pozorovať:
- a) oseálny postih je prítomný takmer u všetkých pacientov s ECC, ale je asymptomatický u približne 60 % z nich [23]. Prejavuje sa ako symetrická bilaterálna osteoskleróza najmä dlhých kostí končatín, s prevahou postihu oblasti metafýzy a diafýzy kostí dolných končatín ako najčastejšia rádiologicky detekovateľná abnormalita pri ECC. Postih menších kostí a chrbtice môže viesť k osteporóze. Podstatou kostných zmien je xantogranulomatózna infiltrácia drene so zvýšenou denzitou zhrubnutých lamiel spongióznej kosti a so sklerotickým zhrubnutím corticalis. Preto aj v postihnutých kostiach dochádza (podobne ako pri medulárnej propagácii iných hematologických a nehematologických malignít) k redukcii pôvodnej krvotvorby so vznikom sprievodných cytopénií.
-
b) extraoseálny postih je prítomný u približne 50 % pacientov, preferenčne sú postihnuté extraoseálne mäkkotkanivové štruktúry, preto niektorí hovoria o tropizme ECC buniek pre spojivové, tukové a perivaskulárne tkanivo [39]. Najviac bývajú postihnuté:
- mäkké tkanivá hlavy a krku z nich najviac orbitálne, retroorbitálne a okulárne štruktúry, v ktorých vznikajú xantogranulomatózne lézie a spôsobujú lokálne tlakovú symptomatológiu (exoftalmus, poruchy zraku),
- mäkkotkanivové štruktúry hrudníka a brucha, infiltráty sa môžu rozšíriť pozdĺž celej aorty, prípadne až do srdca (vrátane možnej infiltrácie perikardu aj do myokardu), ďalej do mediastína (vrátane rozšírenia po a. pulmonalis alebo pozdĺž v. cava inferior) a jej hlavných vetvách a retroperitonea [23,89]. Tu prítomné infiltráty indukujú fibrotizáciu postihnutých lokalít, zrejme prostredníctvom rôznych mediátorov a cytokínov. Tak môže vzniknúť fibrotizácia periaortálnej vrstvy aorty v jej celom rozsahu, alebo prevažne oblasti hrudnej a brušnej aorty (tzv. „coated aorta“ v CT obraze) alebo fibróza perirenálnych oblastí alebo celého retroperitonea [77]. Výsledkom fibrotizácie sú možné vážne, až život ohrozujúce komplikácie ako napr. infiltrácia nadobličiek, zlyhanie srdca, tamponáda srdca, renovaskulárna hypertenzia alebo obštrukcia močového systému s možnou hydronefrózou a renálnou insuficienciou.
Xantogranulomatózne lézie vedúce k fibrotizácii retroperitonea môžu byť klinicky aj histomorfologicky ťažko odlíšiteľné od obrazu Ormondovej choroby [81], ešte zložitejšia je diferenciálna diagnostika v prípade pacienta s Ormondovou chorobou a sekundárnym nádorom s fenotypom malígneho fibrózneho histiocytómu, resp. histiocytového sarkómu [69]. To isté platí pre iné intraabdominálne lokalizované alebo difúzne fibroblastické proliferácie zo skupiny predtým nazývanej aj „chronické periaortitídy“, kam patria aj procesy typu mezenterickej fibromatózy [93]. Navyše u časti týchto pacientov býva prítomná hypergamaglobulinémia.
Do extraoseálnych lézií ECC patrí aj možný postih kostrového svalu, menej často sú postihnuté iné orgánové štruktúry, a to najmä pľúca (pľúcne interstícium s následnou možno intersticiálnou fibrózou), oblička, koža, sliznice a i. Samostatnú klinickú problematiku predstavuje postih CNS, ako na to upozorňuje aj klasifikácia nádorov CNS podľa SZO [60]. Intrakraniálne lézie môžu byť intraparenchymatózne (najmä infiltrácia mozočka a oblasti uhla mozočka a pons Varoli, miechy a pod.) alebo extraparenchymatózne (infiltrácie kavernóznych sínusov, plexus chorioideus a obalových meningových štruktúr), infiltrácie zasahujú neraz až do orbity, hypofýzy a pod. Postihnutie hypofýzy s vývojom príznakov diabetes insipidus (podobne ako pri LCH) a vývoj ďalších príznakov zo strany CNS bol recentne opísaný v československom písomníctve Adamom et al [4,5].
Multiorgánové postihnutie pri systémovej ECC je spôsobené so závažnejšou symptomatológiou a horšou prognózou, pričom za indikátor zlej prognózy sa považuje najmä postih pľúc vo forme intersticiálneho pľúcneho procesu [74].
Nádory plazmocytoidných dendritických buniek
Ako sme už v úvode uviedli, ide o skupinu pomerne tajomných buniek, ktoré v rámci nenádorovej proliferácie možno zastihnúť v rámci niektorých reaktívnych lymfoproliferatívnych stavov. Okrem toho je známe, že klonálne proliferované plazmocytoidné DCs sa vyskytujú v rámci dvoch druhov nádorových procesov:
- a) v prípade niektorých myeloidných nádorov (leukémie myeloidného radu, myeloproliferatívne neoplázie, najmä v blastickej fáze) a blasticky transformovaných myelodysplastických stavov sa vyskytujú v obraze biopticky vyšetrovaných tkanív kostnej drene, uzlín a extranodálnych štruktúr (napr. kože), buď samostatne, alebo v súvise s leukemickou infiltráciou [31]. Vtedy vykazujú fenotypový profil CD123+ a granzým B+, ale expresia CD4+/ CD56+ nebýva vôbec alebo dokonale vyznačená, taktiež je otázné, či nezodpovedajú plazmocytoidným DCs1 bunkám. V časti prípadov bunkách týchto DCs bolo aj dokázané, že majú ten istý klonový pôvod ako dominantná myeloidná nádorová populácia [87]. Tieto nádory však formálne nepatria do tu diskutovanej skupiny non-Langerhansových histiocytóz a uviedli sme ich tu len pre úplnosť,
-
b) v prípade nádoru plazmocytoidných DCs (DCs2) buniek, ktorý spĺňa kritériá pravého nádoru DCs. Ten na rozdiel od všetkých ostatných vyššie aj nižšie diskutovaných nádorov DCs podľa klasifikácie SZO z roku 2008 patrí medzi nádory z prekurzorových buniek plazmocytoidných DCs. V rámci klasifikácie SZO sa preto nádory plazmocytoidných DCs nezaraďujú do klasifikačnej schémy „histiocytových nádorov“, ale do skupiny akútnych leukémií nejasného pôvodu (tab. 5), a to z dvoch dôvodov:
- klinicky ochorenie väčšinou prebieha pod obrazom akútnej leukémie,
- koncepčne všetky akútne leukémie sú nádory z prekurzorových buniek (lymfocytového, myeloidného a ďalších bunkových krvotvorných radov).

Vo svetle citovanej klasifikácie SZO z roku 2008 by sme pre tento nádor zrejme mali používať názov „leukémia plazmocytoidných DCs“, ktorá tvorí časť prípadov kategórie „leukémia/ lymfóm NK bunková lymfoblastová“. Je viac než zrejmé, že v tejto oblasti možno očakávať do budúcna ďalšie upresnenia. Celá táto problematika je pre nešpecializovaného čitateľa viac než problematická až konfúzna, lebo terminológia týchto nádorov (podobne ako terminológia samotných buniek) prešla v posledných rokoch až krkolomným vývojom. Tieto nádory boli napr. v spoločnej klasifikácii kožných lymfómov podľa SZO a EORTC [94] a následne v ďalších publikáciách označované nasledovnými názvami: „blastický NK - bunkový lymfóm“, „CD4+/ CD56+ hematodermická neoplázia“, „agranulárna hematodermická neoplasma“, „včasná plazmocytoidná leukémia“, „lymfóm z dendritických buniek“ a pod.
Formálne teda ide o akútnu leukémiu, ktorá sa často manifestuje aj extramedulárnymi, najmä kožnými infiltrátmi [64,72,82]. Tie dokonca bývajú neraz prvou klinickou manifestáciou, pričom v tejto fáze ochorenia, ako sme na to upozornili aj v jednom z prvých opisov tohto nádoru v česko - slovenskom písomníctve [64], nemusí byť prítomná infiltrácia kostnej drene ani leukemický krvný obraz. Ide o situáciu analogickú k iným akútnym leukémiám – známa je možná klinická manifestácii myelosarkómu s myeloidným alebo monocytovým fenotypom či lymfoblastómu a buď súčasná, alebo následná evolúcia AML, resp. ALL. Ochorenie je charakteristické agresivitou priebehu, v nami pozorovanom prípade časový interval od prvej solitárnej kožnej manifestácie cez disseminovanú kožnú manifestáciu po prietokovo - cytometricky a biopticky verifikovanú infiltráciu kostnej drene, resp. periférnej krvi bol len niekoľko týždňov. V priebehu ochorenia dochádza k lokálne neraz značne deštruktívnej progresii procesu (v našom prípade infiltrácia nazálnych, paranazálnych dutín a infiltrácia do CNS), ako aj k leukemickej disseminácii procesu do parenchýmových orgánov. Masívna infiltrácia kostnej drene spôsobuje typické hematologické prejavy, najmä anémiu a neutropéniu, či pancytopéniu s tomu zodpovedajúcimi komplikáciami. Prognóza ochorenia je vo väčšine publikovaných prípadov zlá.
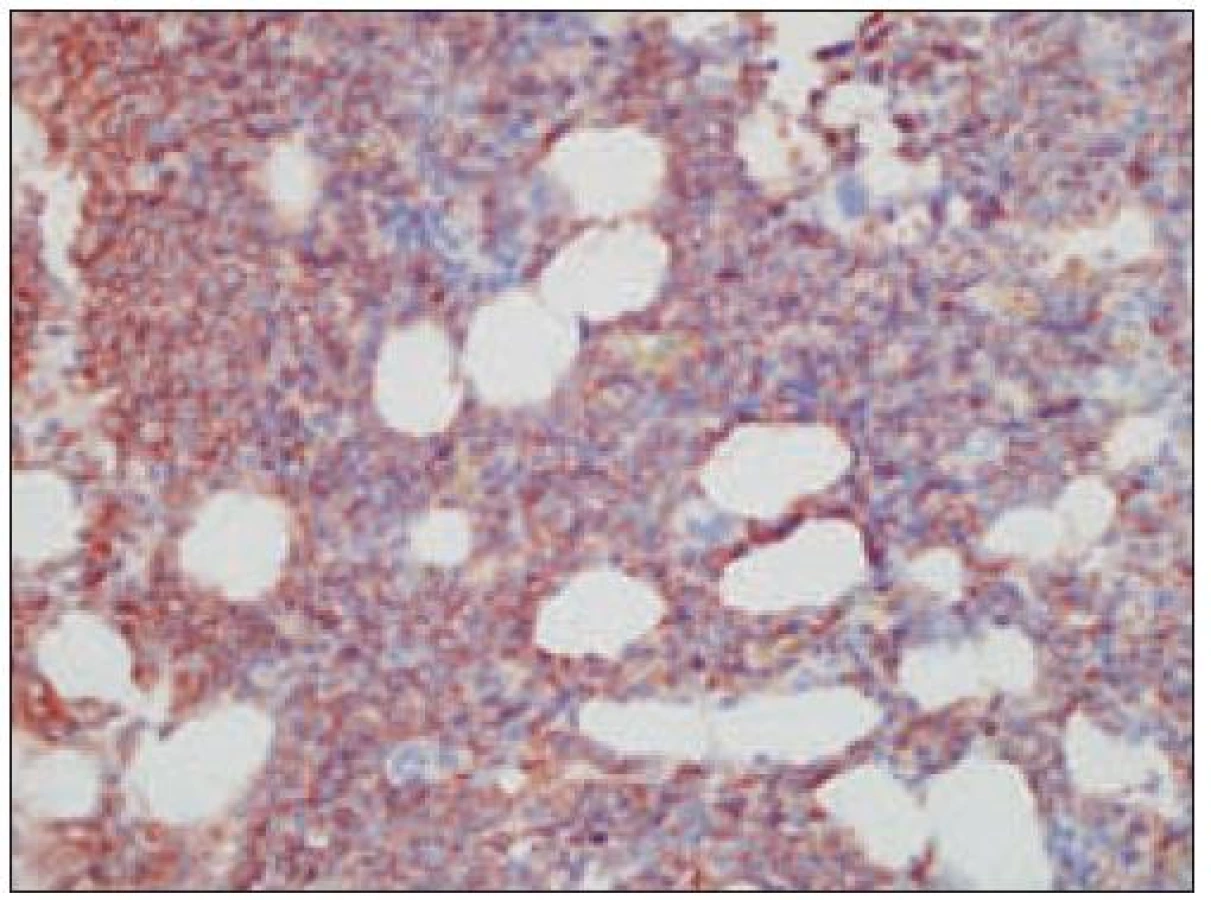
Opätovne aj tu zainteresovaného patológa pre podrobný opis histologického obrazu a imunohistochemického v biopsii identifikovateľného fenotypu odkazujeme na špecializovanú literatúru, resp. publikované kazuistiky [8,35,64,67,85] (obr. 15 – 22). Tu len stručne uvádzame, že nádorové elementy morfologicky zodpovedajú spektru malých až stredne veľkých „blastov“ t.j. morfologicky nezrelých buniek s pleomorfným jadrom a s fenotypom CD4+, CD56+, CD43+. Väčšinou sa zisťuje aj slabá pozitivita LCA (CD45), často pozitivita TdT a negativita znakov myeloidných nádorov a B, resp. T buniek s výnimkou možnej koexpresie CD2 a CD7. Uvedené spektrum imunofenotypovej analýzy väčšinou postačuje pre diagnózu, lebo dôkaz „špecializovaných“ antigénov, ako sú interleukínový 3α - receptor (CD123), antigén DC2 buniek (CD303) a iné, nie je bežne dostupný ani na špecializovaných pracoviskách. Cota et al [18] však recentne upozornili na veľkú možnú variabilitu nielen morfologického obrazu, ale aj fenotypu tohto nádoru. Aj preto sa diskutuje aj o význame detekcie niektorých chromozómových abnormalít pre diagnostiku ochorenia. Výsledky nie-ktorých analýz s použitím FISH a najmä komparatívnej genómovej hybridizácie sa ukazujú byť diagnosticky a diferenciálne - diagnosticky v zmysle odlíšenia iných leukémií (najmä AMMoL) prínosné [35].






Nádory dendritických buniek stromálneho typu
Histogeneticky a v zmysle koncepcie SZO klasifikácie z roku 2008 (tab. 3, obr. 1 a 2) sem patria nádory folikulových dendritických buniek a nádory fibroblastových retikulových buniek.
Sarkóm folikulových dendritických buniek (ICD - O M9785/ 3)
Folikulové dendritické bunky sú, ako sme už uviedli, špecializované DCs B zóny, resp. lymfatických folikulov, lokalizované vnútri ich zárodočných centier. Nádor, ktorý tvorí nádorová proliferácia vretenovitých až ovoidných buniek s im podobným fenotypom sa podľa klasifikácie SZO z roku 2008 nazýva sarkóm z folikulových DCs (ďalej FDCS).
Aj FDCS, podobne ako iné sarkómy dendritických buniek, patrí medzi zriedkavé nádory, ktoré sa môžu vyskytnúť tak u detských, ako aj dospelých pacientov. Niekedy sa objavuje v asociácii s angiolymfoidnou hyperpláziou (seu morbus Castleman), pri ktorej dochádza k nenádorovej proliferácii folikulových DCs. Vtedy toto ochorenie možno dokonca považovať za „prekancerózu“ FDCS, resp. za „presarkomatózny stav“ [40,82]. Iným typom je asociácie EBV infekcie a FDCS, pretože existuje podtyp FDCS, tzv. IPT - podobný FDCS [nazývaný podľa podobnosti so zápalovým pseudotomorom – IPT („inflammatory pseudotumor“)], ktorý je typickým práve pre výskyt u pacientov s EBV infekciou [z pohľadu patológa najľahšie dokázateľné in situ hybridizačnou metódou s použitím sondy EBER („EBV encoded RNA“)]. Nevykazuje žiadne známe rozdiely výskytu podľa pohlavia pacientov, s výnimkou podtypu podobného IPT, ktorý je častejší u pacientov ženského pohlavia.
FDCS patrí medzi indolentné a nízkomalígne sarkómy, agresívne vysokomalígne formy sú zriedkavejšie. Celkove sa chovanie nádoru prirovnáva ku G1 (nízkomalígnym) sarkómom mäkkých tkanív [35]. Lokálne recidívy, niekedy aj oneskorené po rokoch, sa vyskytujú asi u 1/ 2, metastastické šírenie u približne u 1/ 4 pacientov. FDCS sa vyskytuje častejšie (asi 50 – 65 % prípadov) v primárne nodálnej forme. Extranodálne formy nevykazujú predilekčný výskyt – sú známe primárne FDCS oblasti hlavy a krku (najviac ústna dutina, tonzily, resp. nosohltan), prsníka, mediastína, mäkkých tkanív, kože, pečene, sleziny a i. Intraabdominálne formy mávajú horšiu prognózu. Podtyp podobný IPT býva takmer výlučne lokalizovaný vo vnútrobrušnej lokalizácii, najmä v pečeni a slezine a manifestuje sa aj systémovými príznakmi. Tento podtyp však napriek výskytu v brušnej dutine máva indolentný priebeh [91].
Vo vzťahu k histomorfológii FDCS, konštatujeme (rovnako ako v prípade IDCS), že je podrobne opísaná v klasifikácii SZO z roku 2008 a tu stručne sumarizujeme hlavné údaje (obr. 23 – 27):
- a) topografia nádorovej infiltrácie: teoreticky by bolo možné očakávať (analogicky k IDCS), že v nodálnom FDCS je najprv postihnutá B zóna kortikálnej zóny. Ide však o indolentný nádor, zväčša diagnostikovaný v štádiu pokročilej infiltrácie LU bez zachovania pôvodných zónálnych štruktúr. V prípadoch s predchádzajúcou Castlemanovou chorobou, častejšie v plazmocytovom type [8] sú prítomné črty typickej angiofolikulovej hyperplázie, LU je postihnutá parciálne, neskôr dochádza k proliferácii vretenovitých buniek (ktoré nemusia byť vždy identifikované ako proliferácia folikulových DCs a hrozí riziko podobnosti s pravým IPT uzliny) a napokon k infiltrácii celej uzliny [17]. Podobná sekvencia zmien od hyalínovo - vaskulárneho typu Castlemanovej choroby s reaktívnou hyperpláziou folikulových DCs až do rozvinutého FDCS bola pozorovaná v sekvenčných biopsiách aj extranodálne, napr. v nosohltane [40]. Pri malígnej transformácii sa zrejme uplatňuje pôsobenie genetických faktorov, najmä TP53. Na druhej strane by si patológ mal byť vedomý toho, že non-neoplastická proliferácia folikulových DCs môže nastať v rôznych tkanivových prostrediach aj v dôsledku stimulácie nádorom produkovanými cytokínami aj pri iných nádorových ochoreniach, napr. pri periférnom T bunkovom lymfóme typu angioimunoblastového T lymfómu.
- b) histocytologický obraz: FDCS sa obyčajne opisuje ako vretenobunkový sarkóm s rôznymi črtami rastových vzorov – môže byť vírovitý, storiformný, fascikulárny, nodulárny [35]. Jeho nádorové bunky však môžu byť buď štíhle a vretenovité, v snopcoch či zväzkoch, alebo „napučané“ histiocytoidného svetlého vzhľadu so širokou eozinofilnou cytoplazmou, s náznakmi zhlukovatenia až solidizácie. V časti prípadov je morfológia značne odlišná (svetlobunkové formy, onkocytové formy, formy s primiešanými osteoklastom podobnými bunkami a pod. – viď klasifikácia SZO z roku 2008). Častá je heterogenita morfológie aj vnútri jednoho nádoru. V podtypoch podobných zápalovému pseudotumoru (IPT) dominuje obyčajne vretenobunková morfológia [91]. Atypie sú zriedkavé, bunky mávajú skôr blandný cytologický vzhľad a vtedy býva mitotická aktivita (na 10 HPF) nízka. V pleomorfnejších variantoch táto aktivita stúpa a objavujú sa aj atypické formy mitóz [82]. Nekrózy sú častejšie v podtype podobnom IPT. Aj tu sa v obraze nádoru často pozorujú disperzné malé lymfocyty.
- c) histologický obraz a iné parametre vo vzťahu k odhadu prognózy: za negatívne histologicky verifikovateľné prognostické parametre sa považuje vysoký proliferačný a/ alebo mitotický index, výraznejšie cytologické atypie, rozsiahla koagulačná nekróza. To isté platí aj o veľkosti nádoru nad 6 cm a o vnútrobrušnej lokalizácii FDCS. Ak sú tieto negatívne faktory prítomné, tak ochorenie máva prudký priebeh so smrteľným koncom.
- d) fenotyp nádorových buniek: podľa väčšiny známych údajov bunky FDCS imunohistochemicky (obr. 1):
- sú pravidelne pozitívne pri dôkaze markerov folikulových DCs, ako sú klusterín, CD21, CD23 a CD35. Zatiaľ čo pozitivita CD21 a CD23 je spravidla vyznačená, expresia CD35 môže byť rozdielna, a aj preto sa odporúča vyšetriť ich panelovo [35]. Za najspoľahlivejší pri nerovnakej pozitivite iných sa považuje expresia klusterínu [37],
- bývajú obyčajne pozitívne pri dôkaze vimentínu, fascínu a dezmoplakínu a
- sú nejednotne individuálne variabilne pozitívne pri dôkaze CD68, S - 100 proteínu a EMA,
- a sú výnimočne pozitívne pri dôkaze cytokeratínov, CD45 alebo CD20,
- zatiaľ čo sú negatívne pri dôkaze iných markerov iných nádorov, ktorých diferenciálna diagnostika je neraz na základe morfologickej analýzy nevyhnutná (CD1a, MPO, lyzozým, CD34, CD30, HMB45 a pod.).
Sprievodné lymfocyty môžu byť tak T, ako aj B pôvodu, v prípade podtypu podobného IPT sa opisuje nápadná lymfoplazmacytová infiltrácia [91].
- e) genetické analýzy: nie sú známe pozitívne nálezy, T bunkový a Ig receptor sú bez prestavby. Ojedinelé sú správy o asociácii FDCS s proliferáciou fenotypove aj morfologicky nezrelých T buniek, ktoré okrem fenoménov indukovanej autoimunity per se boli biologicky agresívne. Na rozdiel od prípadov transdiferenciácie diskutovanej pri histiocytovom sarkóme však nebol dokázaný klonálny vzťah oboch procesov [53].
- f) diferenciálna diagnostika z pohľadu patológa: aj tu platí, že pozitívna bioptická diagnostika FDCS nie je jednoduchá ani pre patológa pôsobiaceho v špecializovanom centre, vyžaduje si dôkladnú diferenciálnu diagnostiku. Diagnózu FDCS sa patológovi odporúča zvážiť [45,82] v prípade nádoru s nezvyčajnou morfológiou a fenotypom, nádoru pripomínajúceho týmus v mimomediastinálnej, resp. meningeóm v extradurálnej lokalizácii, ako aj v prípade nádoru pripomínajúcom GIST bohatého na lymfocyty, alebo karcinóm štítnej žľazy či oblasti hlavy a krku s vretenobunkovými formáciami (tzv. „CASTLE - tumor“).

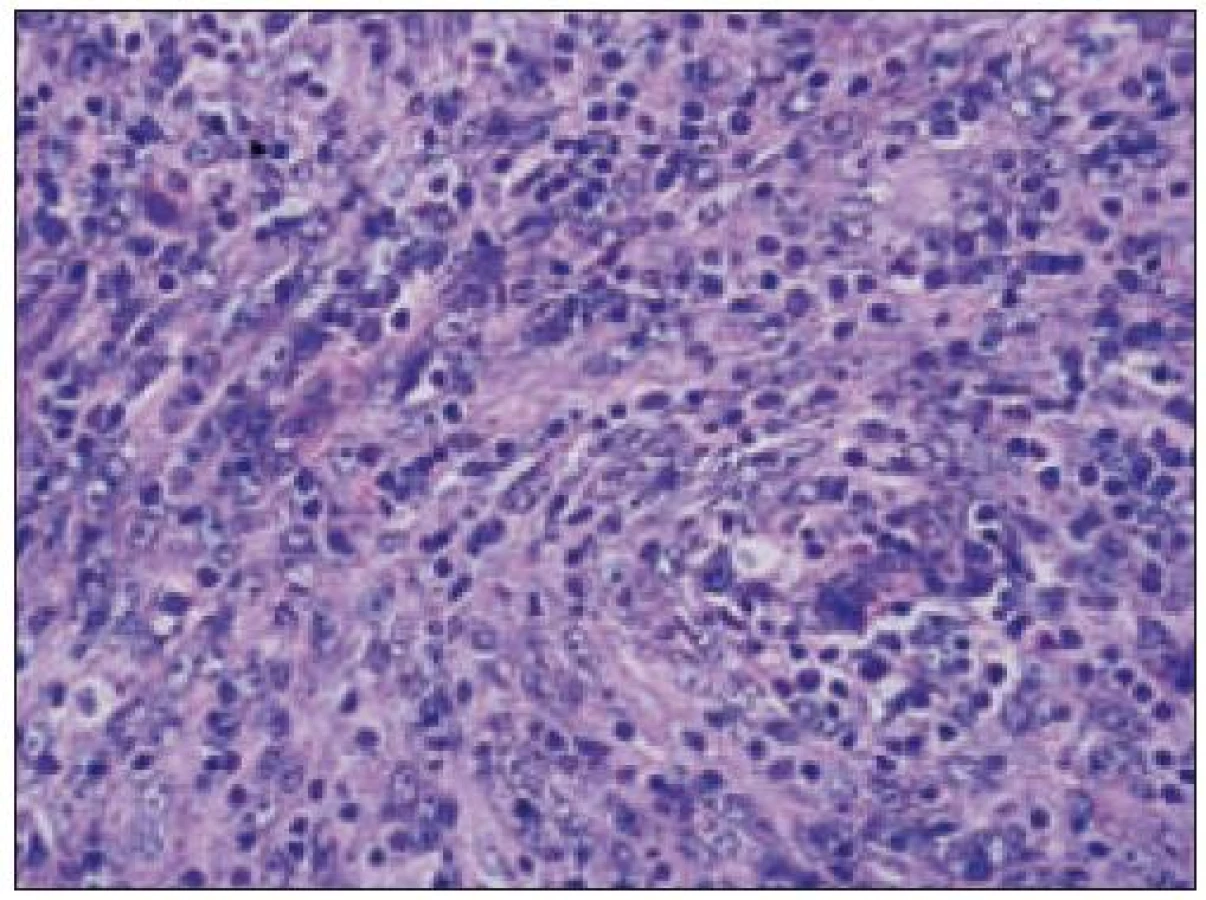

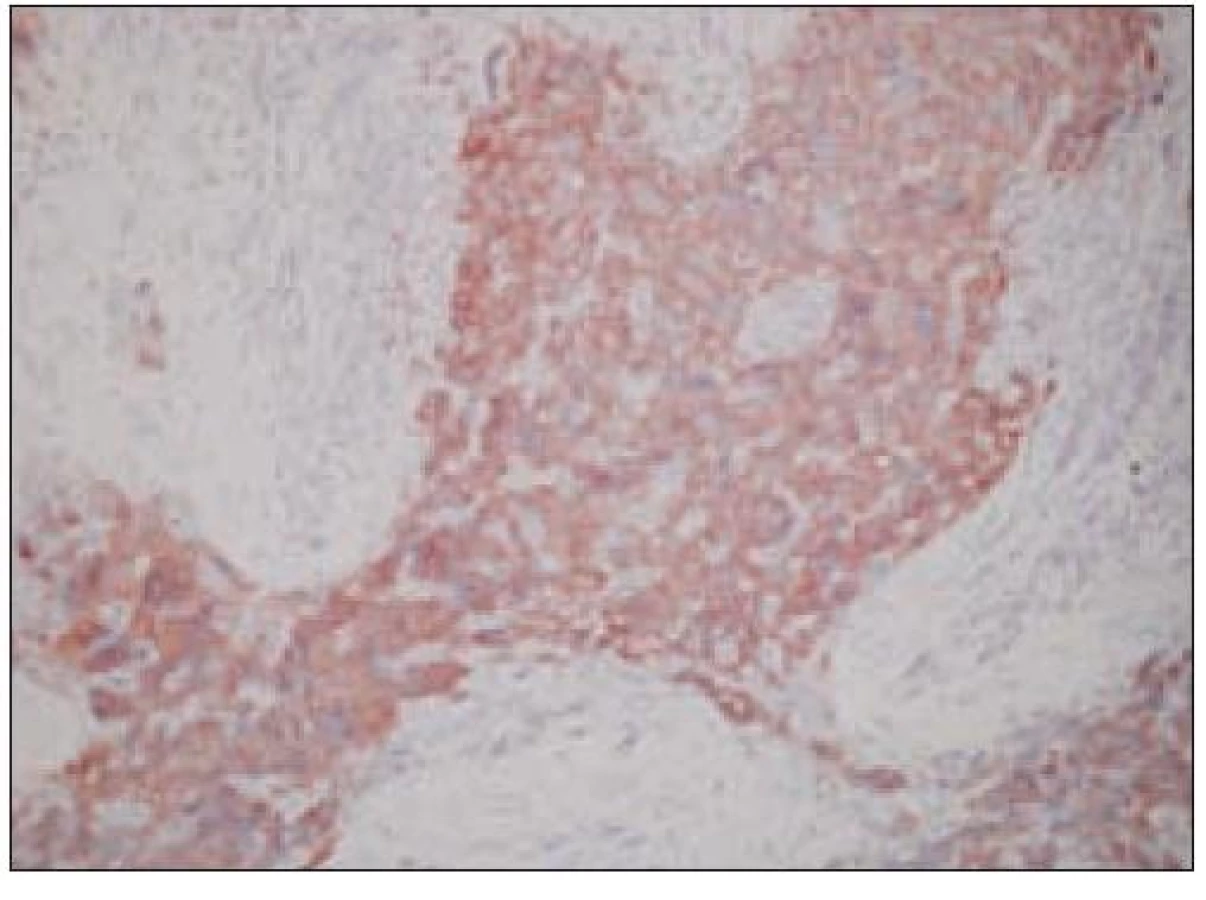

Nádor fibroblastových retikulárnych buniek (ICD - O M 9759/ 3)
Fibroblastové retikulárne bunky predstavujú málo známu populáciu DCs (viď úvodná časť) a jediným, aj to tiež veľmi málo známym nádorom z týchto buniek, je nádor z fibroblastových retikulárnych buniek.
Ide o vzácny typ nádoru, keď podľa Donga et al [24] bolo do roku 2008 známych 14 prípadov v anglicky písanej odbornej tlači. Podľa dostupných údajov môže vzniknúť, podobne ako sarkómy iných DCs, primárne v nodálnej alebo extranodálnej lokalizácii (známe sú primárne nádory v slezine a mäkkotkanivových štruktúrach, najmä retroperitoneu). Prinajmenej teoreticky je potrebné pripustiť, že existuje rozpor medzi ubikvitaritou fibroblastových retikulárnych buniek a vzácnosťou nádorov z nich odvodených a uvedomiť si riziko mylného diagnostického zaradenia tohto „nádoru“ medzi nádory inej proveniencie či histogenézy [24]. O jeho biologickej povahe je k dispozícii málo údajov, je zrejme značne heterogénna: viaceré kazuistiky uvádzajú pomerne indolentný priebeh ochorenia, iné aj veľmi agresívny priebeh [8,24]. Primárne splenická forma vykazuje potenciál pozdných metastáz do pečene [62]. Histologický obraz najmä nodálnych foriem pripomína myofibroblastové nádory uzliny, resp. IPT, alebo je histologicky takmer neodlíšiteľný od FDCS, resp. IDCS s morfológiou oválnych až vretenovitých buniek usporiadaných fascikulárne alebo storiformne [8]. Imunohistochemicky však nevykazuje pre ne typické fenotypové znaky, takže pozitívna diagnóza je veľmi obtiažna a je obyčajne diagnózou per exclusionem. Zahŕňa aj pozitívny dôkaz vimentínu a markerov hladkosvalových buniek (najmä SMA a dezmínu), často sa vyskytuje pozitivita cytokeratínov a CD68 [24,80]. Analogicky k rozdeleniu ich fyziologických proťajškov v LU možno aj nádory týchto buniek rozdeliť na CK negatívne a CK pozitívne, čím sa celý diagnostický a diferenciálno - diagnostický algoritmus ešte viac komplikuje. Navyše, tento nádor zdieľa viaceré spoločné črty so zápalovým myofibroblastovým tumorom, najmä jeho ALK1 negatívnou formou, a je preto možné, že oba patria do spektra toho istého procesu, keď zápalový myofibroblastový tumor by predstavoval jeho nízkomalígny a fibroblastový nádor vysokomalígny variant [24].
Iné histiocytózy dendritických buniek
Klasifikácia SZO z roku 2008 sem zaraďuje kategóriu „neoplázia retikulových buniek NOS“ (angl. not otherwise specified) s neistou či nejednoznačnou expresiou pozitivity len vimentínu a CD68 [8]. Pri tomto údaji možno zvažovať, či nejde o údaj už prekonaný.
Napokon je vhodné uviesť, že podľa niektorých aj keď skutočne ojedinelých údajov bol publikovaný ďalší možný typ nádorov DCs, a to histiocytóza/ sarkóm z folikulových dendritických buniek s črtami buniek vystielajúcich sínusy [imunohistochemická pozitivita tzv. LYVE - 1 (lymphatic vessel endothelium hyaluronan receptor - 1 ako marker lymfatických endotélií)], v rámci ktorej autori zvažujú možnosť nového typu „retikulosarkómu“ nazvaného „sarkóm z buniek vystielajúcich sínusy“ [56].
Podporené projektom OPVaV - 2008/ 2.1/ 01 - SORO – Centrum excelentnosti CEPV I. (IMTS kód 26220120016) a CEPV II. (IMTS kód 26220120036) na Jesseniovej lekárskej fakulte Univerzity Komenského v Martine, ktorý je spolufinancovaný z prostriedkov EÚ.
prof. MUDr. Lukáš Plank, CSc.
www.jfmed.uniba.sk
e-mail: plank@jfmed.uniba.sk
Doručeno do redakce: 9. 9. 2010
Zdroje
1. Adam Z, Adamová Z. Histiocytární choroby a mastocytózy. Diagnostické a léčebné postupy u maligních chorob. Praha: Grada Publishing 2004 : 547 – 551.
2. Adam Z, Pour L, Veselý K et al. Interdigitating dendritic cell sarcoma of the leg. Onkologie 2009; 32 : 364 – 365.
3. Adam Z, Krejčí M, Pour L. Histiocytární choroby. Vnitř Lék 2009; 55 (Suppl 1): S109 – S124.
4. Adam Z, Balšíková K, Pour L et al. Dia-betes insipidus, následovaný po 4 letech dysartrií a lehkou pravostrannou hemiparézou – první klinické příznaky Erdheimovy - Chesterovy nemoci. Popis a zobrazení případu s přehledem informací o této nemoci. Vnitř Lék 2009; 55 : 1173 – 1188.
5. Adam Z, Balšíková K, Krejčí M et al. Centrální diabetes insipidus u dospělých osob – první příznak histiocytózy z Langerhansových buněk a Erdheimovy - Chesterovy choroby. Popis tří případů a přehled literatury. Vnitř Lék 2010; 56 : 138 – 148.
6. Adam Z, Zahradová L, Krejčí M et al. Difuzní plošná normolipemická xantomatóza a nekrobiotický xantogranulom, asociované s monoklonální gamapatií – přínos PET - CT pro stanovení rozsahu nemoci a zkušenosti s léčbou. Popis dvou případ a přehled literatury. Vnitř Lék 2010; 56 : 1158 – 1168.
7. Alexiev BA, Sailey CJ, McClure SA et al. Primary histiocytic sarcoma arising in the head and neck with predominant spindle cell component. Diagn Pahtol 2007; 2 : 7. Available from: www.diagnosticpathology.org/content/2/I/7.
8. Andriko JA, Kaldjian EP, Tsokos M et al. Reticulum cell neoplasms of lymph nodes: a clinicopathologic study of 11 cases with recognition of a new subtype derived from fibroblastic reticular cells. Am J Surg Pathol 1998; 22 : 1048 – 1058.
9. Arai E, Yamamoto T, Shimada S et al. Primary cutaneous true histiocytic sarcoma: A case with an indolent course. J Clin Exp Hematopathol 2003; 43 : 35 – 41.
10. Bassarova A, Trøen G, Fosså A et al. Transformation of B - cell lymphoma to histiocytic sarcoma: somatic mutations of PAX - 5 gene with loss of expression can not explain transdifferentiation. J Hematop 2009; 2 : 135 – 141.
11. Béné MC, Feuillard J, Jacob MC et al. Plasmacytoid dendritic cells: from the plasmacytoid T - cell to type 2 dendritic cells CD4+CD56+ malignancies. Semin Hematol 2003; 40 : 257 – 266.
12. Brunning RD, McKenna RW. Histiocytic proliferations of the bone marrow. In: Brunning RD, McKenna RW (eds). Tumors of the bone marrow. Atlas of tumor pathology. 3rd ed. Washington: AFIP 1993 : 439 – 455.
13. Brychtová S, Bezděková M, Brychta T. Fibroblasty – buňky známé či neznámé? Čes Slov Patol 2010; 46 : 29 – 32.
14. Bubanská E, Plank L, Szépe P et al. Juvenilná myelomonocytová leukémia asociovaná s neurofibromatózou a komplikovaná hemofagocytovým syndrómom. Čes Slov Pediat 2008; 63 : 24 – 32.
15. Cao M, Eshoa C, Schultz C et al. Primary central nervous system histiocytic sarcoma with relapse to mediastinum: a case report and review of the literature. Arch Pathol Lab Med 2007; 131 : 301 – 305.
16. Cossu A, Lissia A, Dedola MF et al. Classic follicular dendritic reticulum cell tumor of the lymph node developing in a patient with a previous inflammatory pseudotumor-like proliferation. Hum Pathol 2005; 36 : 207 – 211.
17. Cossu A, Deiana A, Lissia A et al. Synchronous interdigitating dendritic cell sarcoma and B - cell small lymphocytic lymphoma in a lymph node. Arch Pathol Lab Med 2006; 130 : 544 – 547.
18. Cota C, Vale E, Viana I et al. Cutaneous manifestation of blastic plasmacytoid denderitic cell neoplasm – morphologic and phenotypic variability in a series of 33 patients. Am J Surg Pathol 2010; 34 : 75 – 87.
19. Dalle JH, Leblond P, Decouvelaere A et al. Effect of thalidomide in a child with histiocytic sarcoma following allogeneic bone marrow transplanstation for T - ALL. Leukemia 2003; 17 : 2056 – 2057.
20. Danihel Ľ, Plank L. Nádory krvi v krvotvorného tkaniva. In: Siman J (ed). Princípy chirurgie. Bratislava: SAP 2007 : 742 – 756
21. Dehner LP. Juvenile xanthogranulomas in the first two decades of life: a clinicopathologic study of 174 cases with cutaneous and extracutaneous manifestations. Am J Surg Pathol 2003; 27 : 579 – 593.
22. Dictor M, Warenholt J, György C et al. Clonal evolution to histiocytic sarcoma with the BCR/ ABL rearrangement 14 years after acute lymphoblastic leukemia. Leuk Lymphoma 2009; 50 : 1892 – 1895.
23. Dion E, Graef C, Haroche J et al. Imaging of thoracoabdominal involvement in Erdheim - Chester disease. AJR Am J Roentgenol 2004; 183 : 1253 – 1260.
24. Dong Y, Wu B, Sheng Z et al. Cytokeratin-positive interstitial reticulum cell tumors of lymph nodes: a case report and review of literature. Chin Med J 2008; 121 : 658 – 663.
25. Eble JN, Rosenberg AE, Young RH. Retroperitoneal xanthogranuloma in a patient with Erdheim - Chester disease. Am J Surg Pathol 1994; 18 : 843 – 848.
26. Elner VM, Mintz R, Demirci H et al. Local corticosteroid treatment of eyelid and orbital xanthogranuloma. Trans Am Ophthalmol Soc 2005; 103 : 69 – 74.
27. El - Tatawy R, Mansour L, Gawish K et al. Histiocytic sarcoma, histologic and immuno - phenotypic analysis: a case study. Egyptian Dermatol Online Journals 2008. Available from: www.edoj.org.eg.
28. Favara BE, Feller AC, Pauli M et. al. Contemporary classification of histiocytic disorders. The WHO Committee On Histiocytic/ Reticulum Cell Proliferations, Reclassification Working Group of the Histiocytic Society. Med Pediatr Oncol 1997; 29 : 157 – 166.
29. Feldman AL, Minniti C, Santi M et al. Histiocytic sarcoma after acute lymphoblastic leukaemia: a common clonal origin. Lancet Oncol 2004; 5 : 248 – 250.
30. Feldman AL, Arber DA, Pittaluga S et al. Clonally related follicular lymphoma and histiocytic/ dendritic cell sarcomas: evidence for transdifferentiation of the follicular lymphoma clone. Blood 2008; 111 : 5433 – 5439.
31. Ferry JA, Harris NL. Atlas of lymphoid hyperplasia and lymphoma. Philadelphia: W.B. Saunders 1997.
32. Filo V, Buchvald J, Rasochová E et al. Perforating lipoidic necrosis: successful treatment with cyclosporin A. J Dermatol Treatment 1998; 9 : 41 – 43.
33. Gaertner EM, Tsokos M, Derringer GA et al. Interdigitating dendritic cell sarcoma. A report of four cases and review of the literature. Am J Clin Pathol 2001; 115 : 589 – 597.
34. Gong L, He XL, Li YH et al. Clonal status and clinicopathologic feature of Erdheim - Chester disease. Pathol Res Pract 2009; 205 : 601 – 607.
35. Gorczyca W, Emmons F. Other tumors. In: Gorczyca W, Emmons F (eds). Atlas of differential diagnosis in neoplastic hematopathology. 2nd ed. London: Informa 2008 : 464 – 474.
36. Gould VE, Bloom KJ, Franke WW et al. Increased numbers of cytokeratin-positive interstitial reticulum cells (CIRC) in reactive, inflammatory and neoplastic lymphadenopathies: hyperplasia or induced expression? Virchows Arch 1995; 425 : 617 – 629.
37. Grogg KL, Macon WR, Kurtin PJ et al. A survey of clusterin and fascin expression in sarcomas and spindle cell neoplasms: strong clusterin immunostaining is highly specific for follicular dendritic cell tumor. Mod Pathol 2005; 18 : 260 – 266.
38. Hornick JL, Jaffe ES, Fletcher CD. Extranodal histiocytic sarcoma: clinicopathologic analysis of 14 cases of a rare epithelioid malignancy. Am J Surg Pathol 2004; 28 : 1133 – 1144.
39. Histiocyte society. Available from: www.histiocytesociety.org/site/.
40. Chan AC, Chan KW, Chan JK et al. Development of follicular dendritic cell sarcoma in hyaline - vascular Castleman’s disease of the nasopharynx: tracing its evolution by sequential biopsies. Histopathology 2001; 38 : 510 – 518.
41. Chantranuwat C. Systemic form of juvenile xanthogranuloma: report of a case with liver and bone marrow involvement. Pediatr Dev Pathol 2004; 7 : 646 – 648.
42. Chen W, Lau SK, Fong D et al. High frequency of clonal immunoglobulin receptor gene rearrangements in sporadic histiocytic/ dendritic cell sarcomas. Am J Surg Pathol 2009; 33 : 863 – 873.
43. Cheuk W, Cheung FY, Lee KC et al. Cutaneous indeterminate dendritic cell tumor with a protracted relapsing clinical course. Am J Surg Pathol 2009; 33 : 1261 – 1263.
44. Jaffe ES, Harris NL, Stein H et al. World Health Organization Classification of Tumours. Pathology and Genetics. Tumours of Haematopoietic and Lymphoid Tissue. Lyon: IARC Press 2001.
45. Jaffe R. Disorders of histiocytes. In: Hsi ED (ed). Hematopathology. Philadelphia: Elsevier Inc. 2007 : 513 – 550.
46. Kačírková P, Campr V. Hematoonkologický atlas krve a kostní dřeně. Praha: Grada Publishing 2007.
47. Kanaan H, Al-Maghrabi J, Linjawi A et al. Interdigitating dendritic cell sarcoma of the duodenum with rapidly fatal course: a case report and review of the literature. Arch Pathol Lab Med 2006; 130 : 205–208.
48. Karimi K, Jadidian A, Glavin FL et al. Laryngeal involvement in progressive nodular histiocytosis: a case report. Arch Otolaryngol Head Neck Surg 2010; 136 : 513 – 516.
49. Katakai T, Hara T, Sugai M et al. Lymph node fibroblastic reticular cells construct the stromal reticulum via contact with lymphocytes. J Exp Med 2004; 200 : 783 – 795.
50. Kawachi K, Nakatani Y, Inayama Y et al. Interdigitating dendritic cell sarcoma of the spleen: report of a case with a review of the literature. Am J Surg Pathol 2002; 26 : 530 – 537.
51. Khan AA, Agarwahl A, Chaddha SK et al. Histiocytic sarcoma of the terminal ileum presenting as a large ulcerating lesion: CT diagnosis. Radiol Case Reports Online 2009; 4 : 262 – 266.
52. Kim NR, Ko YH, Choe YH et al. Erdheim - Chester disease with extensive marrow necrosis: a case report and literature review. Int J Surg Pathol 2001; 9 : 73 – 79.
53. Kim WY, Kim H, Jeon YK et al. Follicular dendritic cell sarcoma with immature T - cell proliferation. Hum Pathol 2010; 41 : 129 – 133.
54. Kolenova A, Plank L, Subova Z et al. Histiocytic sarcoma as a second malignancy following T - ALL. Pediat Blood Cancer 2010. V tisku.
55. Kraus MD, Haley RC, Ruiz R et al. „Juvenile“ xanthogranuloma: an immunophenotypic study with reappraisal of histogenesis. Am J Dermatopathol 2001; 23 : 104 – 111.
56. Krokowski M, Merz H, Thorns C et al. Sarcoma of follicular dendritic cells with features of sinus lining cells – a new subtype of reticulum cell sarcoma? Virchows Arch 2008; 452 : 565 – 570.
57. Leenen PJ, Bechan GI, Melis M et al. Heterogeneity in a mouse model of histiocytosis: transformation of Langerin+ dendritic cells, macrophages, and precursors. J Leukoc Biol 2010; 87 : 949 – 958.
58. Lennert K, Remmele W. Karyometrische Untersuchungen an Lymphknotenzellen des Menschen: I. Mitt. Germinoblasten, Lymphoblasten und Lymphozyten. Acta Haematol 1958; 19 : 99 – 113.
59. Lennert K, Mohri N, Stein H et al. Malignant Lymphomas other than Hodgkin’s Disease. Berlin, Heidelberg, New York: Springer Verlag 1978.
60. Liang S, Liu YH, Fang K. Juvenile xanthogranuloma with ocular involvement. Pediatr Dermatol 2009; 26 : 232 – 234.
61. Louis DN, Ohgaki H, Wiestler OD et al. WHO classification of Tumours of the Central nervous System. 4th ed. Lyon: IARC 2007.
62. Martel M, Sarli D, Colecchia M et al. Fibroblastic reticular cell tumor of the spleen: report of a case and review of the entity. Hum Pathol 2003; 34 : 954 – 957.
63. Miettinen M, Fetsch JF. Reticulohistiocytoma (solitary epithelioid histiocytoma): a clinicopathologic and immunohistochemical study of 44 cases. Am J Surg Pathol 2006; 30 : 521 – 528.
64. Minariková E, Plank L, Péč J. Kazuistický prípad pacienta s blastickým NK - bunkovým lymfómom, syn. CD4+/ CD56+ hematodermická neoplázia, syn. včasná plazmocytoidná leukémia/ lymfóm z dendritických buniek. Čes Slov Derm 2008; 83 : 204 – 208.
65. Oka K, Nakamine H, Maeda K et al. Primary histiocytic sarcoma of the spleen associated with haemophagocytosis. Int J Haematol 2008; 87 : 405 – 409.
66. Park MI, Song KS, Kang DY. Histiocytic sarcoma of the rectum: a case report. Korean J Pathol 2006; 40 : 156 – 159.
67. Petrella T, Comeau MR, Maynadié M et al. Agranular CD4+ CD56+ hematodermic neoplasm (blastic NK - cell lymphoma) originates from a population of CD56+ precursor cells related to plasmacytoid monocytes. Am J Surg Pathol 2002; 26 : 852 – 862.
68. Pileri SA, Grogan TM, Harris NL et al. Tumours of histiocytes and accessory dendritic cells: an immunohistochemical approach to classification from the International Lymphoma Study Group based on 61 cases. Histopathology 2002; 41 : 1 – 29.
69. Plank L, Huťka Z, Beseda A et al. Morbus Ormond a sekundárny nádor. Zborník súhrnov XXXIV. celoštátneho zjazdu československých patológov s medzinárodnou účasťou. Banská Bystrica 1983 : 75 – 75.
70. Plank L. Atlas biopsie nelymfómových lymfadenopatií. Martin: Osveta 1990.
71. Plank L, Kodet R, Boudová L. Novinky v klasifikácii lymfoidných neoplázií podľa SZO 2008 (4. vydanie). Transf Hematol 2008; 14 (Suppl 1): 69 – 71.
72. Plank L, Minariková E, Péč J et al. Klasifikácia kožných malígnych lymfómov podľa novej klasifikácie SZO (2008). Onkológia (Bratislava) 2009; 4 : 160 – 163.
73. Podberezin M, Angeles R, Guzman G et al. Primary pancreatic sinus histiocytosis with massive lymphadenopathy (Rosai - Dorfman disease): an unusual extranodal manifestation clinically simulating malignancy. Arch Pathol Lab Med 2010; 134 : 276 – 278.
74. Protopopadakis C, Antoniou KM, Nicholson AG et al. Erdheim - Chester disease: pulmonary presentation in a case with advanced systemic involvement. Respiration 2009; 77 : 337 – 340.
75. Rezk SA, Spagnolo DV, Brynes RK et al. Indeterminate cell tumor: a rare dendritic neoplasm. Am J Surg Pathol 2008; 32 : 1868 – 1873.
76. Rosenberg AS, Morgen MB. Cutaneous indeterminate cell histiocytosis: a new spindle cell variant resembling dendritic cell sarcoma. J Cutan Pathol 2001; 28 : 531 – 537.
77. Serratrice J, Granel B, De Roux B et al. „Coated aorta“: a new sign of Erdheim - Chester disease. J Rheumatol 2000; 27 : 1550 – 1553.
78. Shinoda H, Yoshida A, Teruya - Feldstein J. Malignant histiocytoses/ disseminated histiocytic sarcoma with hemophagocytic syndrome in a patient with mediastinal germ cell tumor. Appl Immunohistochem Mol Morphol 2009; 17 : 338 – 344.
79. Shoo BA, Shinkai K, McCalmont TH et al. Xanthogranulomas associated with hematological malignancy in adulthood. J Am Acad Dermatol 2008; 59 : 488 – 493.
80. Schuerfeld K, Lazzi S, De Santi MM et al. Cytokeratin-positive interstitial cell neoplasm: a case report and classification issues. Histopathology 2003; 43 : 491 – 494.
81. Stašková K, Ježíková A, Michalicová A et al. Morbus Ormond. Rheumatologia 2009; 23 : 99 – 99.
82. Swerdlow SH, Campo E, Harris NL et al. WHO classification of tumours of heamatopoietic and lymphoid tissues. 4th ed. Lyon: IARC 2008.
83. Ugurlu S, Bartley GB, Gibson LE. Necrobiotic xanthogranuloma: long term outcome of ocular and systemic involvement. Am J Ophthalmol 2000; 129 : 651 – 657.
84. Ueda T, Kawano H. Xanthogranulomatous transformation of metastatic testicular tumor. Urology 1983; 22 : 646 – 648.
85. Urosevic M, Conrad C, Kamarashev J et al. CD4+ CD56+ hematodermic neoplasms bear a plasmacytoid dendritic cell phenotype. Hum Pathol 2005; 36 : 1020 – 1024.
86. Vencio EF, Jenkins RB, Schiller JH et al. Clonal cytogenetic abnormalities in Erdheim - Chester disease. Am J Surg Pathol 2007; 31 : 319 – 321.
87. Venkataraman G, McClain KL, Pittaluga S et al. Development of disseminated histiocytic sarcoma in a patient with autoimmune lymphoproliferative syndrome and associated Rosai - Dorfman disease. Am J Surg Pathol 2010; 34 : 589 – 594.
88. Vermi W, Facchetti F, Rosati S et al. Nodal and extranodal tumor - forming accumulation of plasmacytoid monocytes/ interferon producing cells associated with myeloid disorders. Am J Surg Pathol 2004; 28 : 585 – 595.
89. Veyssier - Belot C, Cacoub P, Caparros - Lefebvre D et al. Erdheim - Chester disease: clinical and radiologic characteristics of 59 cases. Medicine (Baltimore) 1996; 75 : 157 – 169.
90. Vos JA, Abbondanzo SL, Barekman CL et al. Histiocytic sarcoma: a study of five cases including the histiocyte marker CD163. Mod Pathol 2005; 18 : 693 – 704.
91. Cheuk W, Chan JK, Shek TW et al. Inflammatory pseudotumor-like follicular dendritic cell tumor: a distinctive low - grade malignant intra - abdominal neoplasm with consistent Epstein-Barr virus association. Am J Surg Pathol 2001; 25 : 721 – 731.
92. Wang E, Hutchinson CB, Huang Q et al. Histiocytic sarcoma arising in indolent small B - cell lymphoma: report of two cases with molecular/ genetic evidence suggestive of a „transdifferentiation“ during the clonal evolution. Leuk Lymphoma 2010; 51 : 802 – 812.
93. Weiss SW, Goldblum JR. Chapter 12 – 14: Benign fibrohistiocytic tumors. Fibrohistiocytic tumors of intermediate malignancy. Malignanf fibrous histiocytoma. In: Enzinger and Weiss’s Soft tissue tumors. 5th ed. Philadelphia: Mosby Elesevier 2008 : 331 – 428.
94. Willemze R, Jaffe ES, Burg G et al. WHO - EORTC classification for cutaneous lymphomas. Blood 2005; 105 : 3768 – 3785.
95. Zelger B, Eisendle K, Mensing C et al. Detection of spirochetal micro-organisms by focus - floating microscopy in necrobiotic xanthogranuloma. J Am Acad Dermatol 2007; 57 1026 – 1030.
Štítky
Diabetologie Endokrinologie Interní lékařstvíČlánek vyšel v časopise
Vnitřní lékařství

2010 Číslo Supplementum 2
- Inovace v hojení ran: krytí Zetuvit Plus Silicone Border pro optimální management exsudátu z ran
- Ivabradin zlepšuje kvalitu života starších pacientů se srdečním selháním
- Clopidogrel je v prevenci kardiovaskulárních příhod přínosnější než kyselina acetylsalicylová
- Atorvastatin rychle snižuje riziko kardiovaskulárních příhod u diabetiků
Nejčtenější v tomto čísle
- Hemofagocytující lymfohistiocytóza
- Erdheimova-Chesterova nemoc v obrazech
- Systémová mastocytóza
- Histiocytóza z Langerhansových buněk u dětí a dospívajících
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy