
Histiocytóza z Langerhansových buněk u dětí a dospívajících
Langerhans cell histiocytosis in children and adolescents
The histiocytosis represents the rare diseases with large biological behavior and clinical symptoms, as well. The Langerhans cell histiocytosis is the most frequent entities in children in this category. The paper does the summary of the etiology, biology, clinical aspects, diagnostic possibilities, and therapeutic outcomes of LCH. The crucial prognostic value represents disease extension: single-system in compare with multi-system LCH. The role of treatment in some LCH forms is questinable but in other forms the chemotherapy is important. Histiocytosis affects all age groups but more than 50% cases concern children up to 15 years of age. There are not gender predominance but exist age dependent predisposition (e. g. bone disease in children 5 – 15 years of age or multi-system disease in children less than 2 years of age). Multi-system LCH incidence is aproximately 2.6 children/ 1 milion children/ year (9 in chidren less to 1 year of age and 0.7 in age group 10 – 14 years). In the Czech Republic supposed 10 – 20 children per year. Histiocytosis characterized abnormal accumulation mononuclear phagocytic cells (dendritic cells and macrophages) which are derivate from primary CD34+ hematopoietic stem cells. The basic point in the differentiation process has large scope of cytokines. LCH was considered as malignant disease, reactive or aberrant immunological answer of organism in the previous time. The last review showed disease with pathological Langerhans cells in granulomatous lesion together with common inflamantory elements as eosinophils, lymphocytes and macrophages. The cell infiltration and clinical symptoms are explained as outcome of aberant secretion these cells and activated T-lymphocytes. Therefore clinical symptoms showed different type of manifestation from spontaneous regression through recurrent behavior to malignant multi-system disease with fatal outcome. Diagnosis is done by histopathological investigation with proof of intracellular Birbecks granules, imunohistochemical S-100 protein positivity, and CD1a+ as well. But the Langerhans cell histiocytosis etiology and pathogenesis in unknown so far. The treatment strategy depends on the spread of disease.
Key words:
Langerhans cell histiocytosis – children – adolescents
Autoři:
H. Mottl
Působiště autorů:
Klinika dětské hematologie a onkologie 2. lékařské fakulty UK a FN Motol Praha, přednosta prof. MU Dr. Jan Starý, DrSc.
Vyšlo v časopise:
Vnitř Lék 2010; 56(Supplementum 2): 64-73
Kategorie:
Histiocytóza z Langerhansových buněk a některá další vzácná hematologická onemocnění
Souhrn
Histiocytózy jsou vzácná onemocnění s velkou biologickou variabilitou a širokým spektrem klinických projevů. U dětí se nejčastěji vyskytuje histiocytóza z Langerhansových buněk (LCH). V práci je podán přehled současných znalostí o epidemiologii, biologii, klinických projevech, diagnostických postupech a léčebných možnostech LCH. Zásadní prognostický význam má rozsah postižení – monosystémové onemocnění ve srovnání s postižením multisystémovým. U některých forem LCH je úloha léčby diskutabilní, u jiných je třeba agresivní onkologická terapie. Histiocytóza postihuje všechny věkové skupiny, avšak ve více než 50 % se vyskytuje u dětí do 15 let věku. Není patrná predominance pohlaví, ale je zřejmá věková predispozice u některých forem. Příkladem je častá kostní forma u dětí od 5 do 15 let nebo systémové postižení u dětí do 2 let věku. Incidence multisystémové formy LCH je uváděna asi 2,6 nových onemocnění na 1 milion dětí za rok, z toho 9 nových onemocnění u dětí do 1 roku věku a 0,7 ve věkové skupině 10 – 14 let. V České republice předpokládáme 10 – 20 nových onemocnění (všechny formy) za rok. Histiocytóza je onemocnění charakterizované abnormální akumulací buněk mononukleárního fagocytárního systému (dendritické buňky a makrofágy). Jednotlivé komponenty mononukleárního fagocytárního systému jsou derivovány z primární CD34+ hematopoetické kmenové buňky. Zásadní úlohu v diferenciaci má široká škála cytokinů. Onemocnění má mnoho projevů od spontánní regrese přes recidivující podobu až po život ohrožující multisystémové postižení s rychlým až fatálním průběhem. V průběhu let byla LCH považována za onemocnění nádorové, reaktivní nebo za aberantní imunitní odpověď organizmu. V současné době je zřejmé, že Langerhansovy buňky mají patologické rysy, avšak v granulomatózní lézi jsou akumulovány i běžné zánětlivé elementy jako eozinofily, lymfocyty a makrofágy. Buněčná infiltrace a klinické projevy jsou tedy vysvětlitelné jako výsledek aberantní cytokinové sekrece uvedených buněk a aktivovaných T-lymfocytů. Onemocnění má proto širokou škálu projevů od spontánní regrese přes recidivující podobu až po život ohrožující multisystémové postižení s rychlým až fatálním průběhem. Diagnóza je stanovena histopatologickým vyšetřením s průkazem intracelulárních Birbeckových granulí a imunohistochemickým průkazem S-100 proteinu a CD1a+. Přes pokrok znalostí o LCH však etiologie a patogeneze onemocnění zůstává nejasná. Léčebná strategie závisí na rozsahu postižení.
Klíčová slova:
histiocytóza z Langerhansových buněk – děti – dospívající
Úvod
Histiocytózy jsou vzácná onemocnění s velkou biologickou variabilitou a širokým spektrem klinických projevů (tab. 1) [79]. Nejčastější jednotkou je histiocytóza z Langerhansových buněk (LCH) [3,93]. Je to onemocnění dříve známé jako histiocytóza X [60] s podjednotkami eozinofilní granulom, choroba Handova - Schüllerova - Christianova a Lettererova - Siweova. V roce 1868 publikoval Langerhans práci popisující kožní infiltráty způsobené dendritickými buňkami, které nyní nesou jeho jméno [54]. První pozorování v souvislosti s projevy LCH, a to impetiginózní kožní změny a okrouhlé kostní defekty na lebce, popsal Smith v roce 1865 [17]. Avšak již Hippokrates zmiňuje bolestivé rezistence na kalvě ve spojitosti s onemocněním, které má benigní průběh [23]. Jednotlivé projevy onemocnění popsali Hand [42], Schüller [82], Christian [16], Letterer [57] a Siwe [84]. Následně Farber [29] uvažoval o identickém onemocnění s různým biologickým chováním a konečně Lichtenstein [60] použil pro celé spektrum onemocnění název histiocytóza X [4,76,77]. Po rozpoznání charakteru buněk v infiltrátech onemocnění histiocytózou X [76] jako buněk Langerhansových byl doporučen název histiocytóza z Langerhasových buněk [31,45]. Tento název se mezinárodně ujal a je běžně používán, včetně závazné WHO klasifikace nádorů krve a krvetvorných orgánů [46]. V roce 1987 Histiocyte Society [99], za výrazné aktivity G. J. D’Angia, stanovila diagnostická kritéria LCH.
![Klasifikace histiocytóz. Modifikováno podle [31].](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image/9df7609e7e2fd00ef4c6cb7796a003f9.jpg)
Klinická manifestace sahá od monoložiskového postižení se spontánní regresí přes opakovaně recidivující chorobu až po život ohrožující, rychle progredující multisystémové onemocnění s minimální reakcí na intenzivní onkologickou léčbu [1].
Histiocytóza je onemocnění charakterizované abnormální akumulací buněk mononukleárního fagocytárního systému (dendritické buňky a makrofágy) [93]. Jednotlivé komponenty mononukleárního fagocytárního systému jsou derivovány z primární CD34+ hematopoetické kmenové buňky. Zásadní úlohu v diferenciaci má široká škála cytokinů, např. interleukin1 - α, interferon - γ, interleukin-10 nebo růstové faktory [19,28,75]. Vědecké poznatky posledních let ukazují, že většina mononukleárních fagocytů pochází z myeloidní linie, ale ve zralé dendritické buňky a makrofágy se mohou vyvíjet i lymfoidní a mezenchymální progenitorové buňky [102].
V průběhu let byla LCH považována za onemocnění nádorové, reaktivní nebo za aberantní imunitní odpověď organizmu [10]. V současné době je zřejmé, že jde o klonální onemocnění Langerhansových buněk, které mají patologické rysy. Kromě toho jsou v různé míře zastoupeny i reaktivní zánětlivé elementy jako eozinofily, lymfocyty a makrofágy. Buněčná infiltrace a klinické projevy jsou tedy vysvětlitelné jako výsledek aberantní cytokinové sekrece všech uvedených buněk a aktivovaných T-lymfocytů [53].
Epidemiologie, biologie a genetické aspekty
Histiocytóza z Langerhansových buněk postihuje všechny věkové kategorie od novorozeneckého období po dospělý věk, avšak ve více než v 50 % se vyskytuje u dětí do 15 let. Není výrazná predominance pohlaví, mírně však převažují chlapci [69,93]. U některých forem je však zřejmá věková predispozice. Příkladem je častá kostní forma u dětí od 5 do 15 let nebo systémové postižení u dětí do 2 let věku. Předpokládaná prevalence v dětském věku je 1 : 50 000 [15], tedy 2 – 5/ 1 milion dětí do 15 let věku. Jiné studie však uvádějí incidenci 8,9/ 1 milion dětí s nárůstem jak multisystémové formy, tak monosystémové formy. U multisystémového postižení [58] je nízké riziko (LR) – věk nad 2 roky, bez postižení plic, jater, sleziny a hematopoetického systému a vysoké riziko (HR) – věk pod 2 roky a multiorgánové postižení nebo věk nad 2 roky s postižením plic, jater, sleziny nebo hematopoetického systému. Monosystémové formy LCH existují jako jednoložiskové postižení kosti, kůže nebo lymfatické uzliny, či víceložiskové postižení skeletu nebo lymfatických uzlin [49]. V České republice v letech 2004 – 2005 předpokládáme asi 23 nově diagnostikovaných onemocnění u dětí (všechny formy) [71]. Celkově u dětí v 62 – 70 % převažuje monosystémové postižení, a to zejména skeletu s predominancí v oblasti kalvy [67,89].
Histiocytóza z Langerhansových buněk je na základě práce Willmana et al [95] považována za klonální nádorové onemocnění s velmi rozdílnou biologickou aktivitou na základě genetických mutací. Klonální charakter onemocnění má jak mimosystémová, tak multisystémová forma. Výjimkou je plicní postižení u dospělých kuřáků, u kterého klonalita není potvrzena [100,101], a je předpoklad, že na patogenezi plicní formy LCH nemají vliv ani chromozomální aberace [18]. Na základě těchto poznatků vznikla hypotéza o genetické alteraci proliferace, regulaci buněčného cyklu a/ nebo apoptózy u postižených buněk jako možných příčin vzniku LCH [6]. Byla prokázána vysoká exprese proliferačního markeru Ki - 67 v LCH lézích, což znamená, že buněčný cyklus není blokován [5,81]. Není však vysvětlen nízký počet mitóz ve většině LCH lézí při vysoké expresi Ki - 67 [5,53,80]. Nedaří se také dlouhodobá kultivace LCH buněk in vitro nebo u imunodeficitních myší. Vysvětlením může být vliv faktorů „buněčného mikro-environmentu“, které se podílejí na progresi LCH lézí [81].
Současné práce věnují velkou pozornost možným genetických mechanizmům podmiňujícím vznik LCH. Jedním z nich je aberantní imunitní odpověď na infekční agens. Zatím se však nepodařilo definitivně prokázat virový původ LCH [47,50,85], nicméně je u pacientů s LCH pozorován zvýšený výskyt lidského herpes viru (HHV - 6), což podporuje hypotézu o sekundární virové infekci lymfocytů s následným rozvojem onemocnění [36].
U monosystémové formy byla v HLA systému (Human Leukocyte Antigen) prokázána vyšší frekvence fenotypu HLA-DRB1*03 a další znaky HLA-A*01, - B*08, - DRB1*03. Nabízí se úvaha, že HLA-DRB1*03 má protektivní účinek proti rozvoji multisystémové formy LCH [8]. Podobně vysoká incidence HLA-DR4 nebo - CW7 u pacientů, zejména s monoložiskovým postižením skeletu, vede k úvaze o imunitní dysfunkci, která hraje při vzniku LCH důležitou roli [63].
LCH buňky jsou považovány za fenotypicky nezralé Langerhansovy buňky (LC), ale přesné stimulační mechanizmy nejsou detailně známé. Nepochybně se mezi aktivační mechanizmy LCH buněk řadí:
- chromozomální alterace,
- klonální proliferace LCH buněk, která ale ještě nemá definitivní vysvětlení na molekulární úrovni a
- zvýšená cytokinová produkce v LCH lézích [6].
V LCH ložiscích jsou prokázány vysoké hladiny cytokinů IL-2α, IL-1β, IL-3, IL-4, IL-5, IL-7, IL-10, GM-CSF, TGF-α, TGF-β, TNF-α a TNF-γ [24,28]. Cytokiny se podílejí na proliferaci a diferenciaci progenitorů LCH buněk, a brání tak jejich vyzrávání. Následné studie však musí objasnit patologii a mechanizmy celkového cytokinového působení na LCH buňku u různých forem onemocnění.
Zatím nebyla nalezena zásadní predispoziční změna v genetické výbavě, která by souvisela se vznikem histiocytózy z Langerhansových buněk [10]. Nicméně zvýšený výskyt různorodých nádorových onemocnění u pacientů s LCH [27] vede k úvaze o přítomnosti zatím neprokázaných genetických změn.
Na rozdíl od LCH byl u jiné formy histiocytózy učiněn v posledních letech značný pokrok v určení biologického a cytogenetického původu onemocnění. Jedná se o familiární formu hemofagocytární lymfohistiocytózy (FHL), u které jsou detekovány genové změny u 3 ze 4 dosud popisovaných forem. U FHL1 je prokázán defekt genu pro perforin, u FHL2 je to Munc 13-4 a u FHL4 se jedná o syntaxin 11. Zatím nebyla definována genetická změna u FHL3. U histiocytózy z Langerhansových buněk podobné poznatky zatím nejsou k dispozici.
Klinické projevy a diagnostický postup
Klinický obraz histiocytózy z Langerhansových buněk je velmi heterogenní a zahrnuje širokou škálu příznaků, z nichž mnohé v iniciálním období vedou k úvahám o řadě jiných onemocnění. Mezi klinické projevy patří např. obraz chronické otitidy, diabetes insipidus, z celkových příznaků jsou to teploty a neprospívání s váhovým úbytkem. Právě variabilita a malá specifičnost příznaků jsou důvodem často obtížného stanovení správné diagnózy.
Skelet
Postižení skeletu je prokázáno asi u 80 % pacientů a je nejčastější formou LCH [51]. Z klinických projevů dominuje bolestivost, otok, vzácně patologická fraktura. Nejčastějším místem postižení je lebka (asi 50 %). Léze je často náhodným nálezem při RTG vyšetření z jiných důvodů (např. úraz). Na lebce jsou typickými lokalizacemi oblast kalvy a kost skalní s projevy chronické sekrece ze zvukovodu. V RTG obraze dominuje poměrně přesně ohraničené projasnění v postiženém místě (obr. 2, 3). Dalšími lokalizacemi jsou skelet horních končetin (20 %), pánev a lopatka (12 %) a obratle (10 %).
Základní vyšetřovací metodou je prostý RTG snímek. Komplementární vyšetřovací metodou je scintigrafie skeletu [20,44] nebo magnetická rezonance (MRI) [37], případně pozitronová emisní tomografie (PET). Dosud však chybí výsledky studií vyšetření PET u větších souborů pacientů [11]. Definitivní diagnóza je stanovena histopatologickým vyšetřením bioptického vzorku tkáně (obr. 1 – 3).



Kůže
Představuje druhý nejčastěji postižený orgán (30 – 60 %) [10]. V místě léze se kůže šupí, má erytematózní až seborrhoický vzhled, někdy se objeví červené papuly lokalizované zejména retroaurikulárně, v axile, inguině nebo perianálně. Případné petechie jsou typické pro LCH a jen výjimečně se vyskytují u afekcí jiné etiologie. Zvýšené opatrnosti je třeba zejména u kojenců s přetrvávajícím seborrhoickým charakterem ekzému nebo „plenkovou“ dermatitidou nereagující na běžnou léčbu. Diferenciální diagnóza je značně široká a zahrnuje seborrhoickou dermatitidu, toxický erytém, herpetickou infekci, ale také scabies. Při nejasném nálezu je nutné provést bioptické vyšetření [55] (obr. 4a – 4c).


Lymfatické uzliny
Postižení bývá často pouze reaktivní, např. při kožní formě LCH, jindy jsou spádové uzliny sekundárně v různé míře postiženy infiltrací LCH. Mimosystémová infiltrace lymfatických uzlin je vzácná [48] a k stanovení diagnózy je nutná biopsie. Základní vyšetřovací metodou je ultrazvukové vyšetření (UZ), případně další zobrazovací metody podle lokalizace postižení. (obr. 5).

Hematopoetický systém
Postižení hematopoetického systému patří k častým a závažným projevům multisystémové formy LCH. Postihuje 60 – 70 % pacientů s fatálním průběhem onemocnění [32,94]. Nepříznivý prognostický význam má zejména neutropenie a trombocytopenie. Samotná infiltrace kostní dřeně je však vzácná, a to i při výrazné pancytopenii (obr. 1). Příčinou cytopenie je spíše sekundární hemofagocytární syndrom z makrofágové aktivace při základním onemocnění [32]. Při známkách cytopenie je třeba provést punkci a trepanobiopsii kostní dřeně.
Játra a slezina
Společně s hematopoetickým systémem a plícemi patří do skupiny tzv. rizikových (prognosticky nepříznivých) orgánů při LCH. Hepatosplenomegalie může být způsobena jak primárním postižením, tak sekundárně, např. při lymfadenopatii v oblasti porty hepatis. Pro jaterní postižení je typická infiltrace prostorů kolem žlučovodů s následnou cholestázou až progresí do sklerozující cholangoitidy (v laboratorním obraze je patrná elevace GMT, ALP, TAG a cholesterolu), která při progresi může být indikací k transplantaci jater [12]. Hypoalbuminemie, případně provázená ascitem, je projevem jaterní dysfunkce, stejně jako hyperbilirubinemie, elevace jaterních testů nebo koagulopatie s prodlouženým protrombinovým časem. Kritériem pro postižení sleziny je zvětšení +2 cm a jater +3 cm pod oblouk žeberní (ultrazvuk nebo magnetická rezonance) [37] a/ nebo popsané biochemické změny [97]. Při průkazu uvedených kritérií nebývá nutná histologická verifikace postižených orgánů.
Plíce
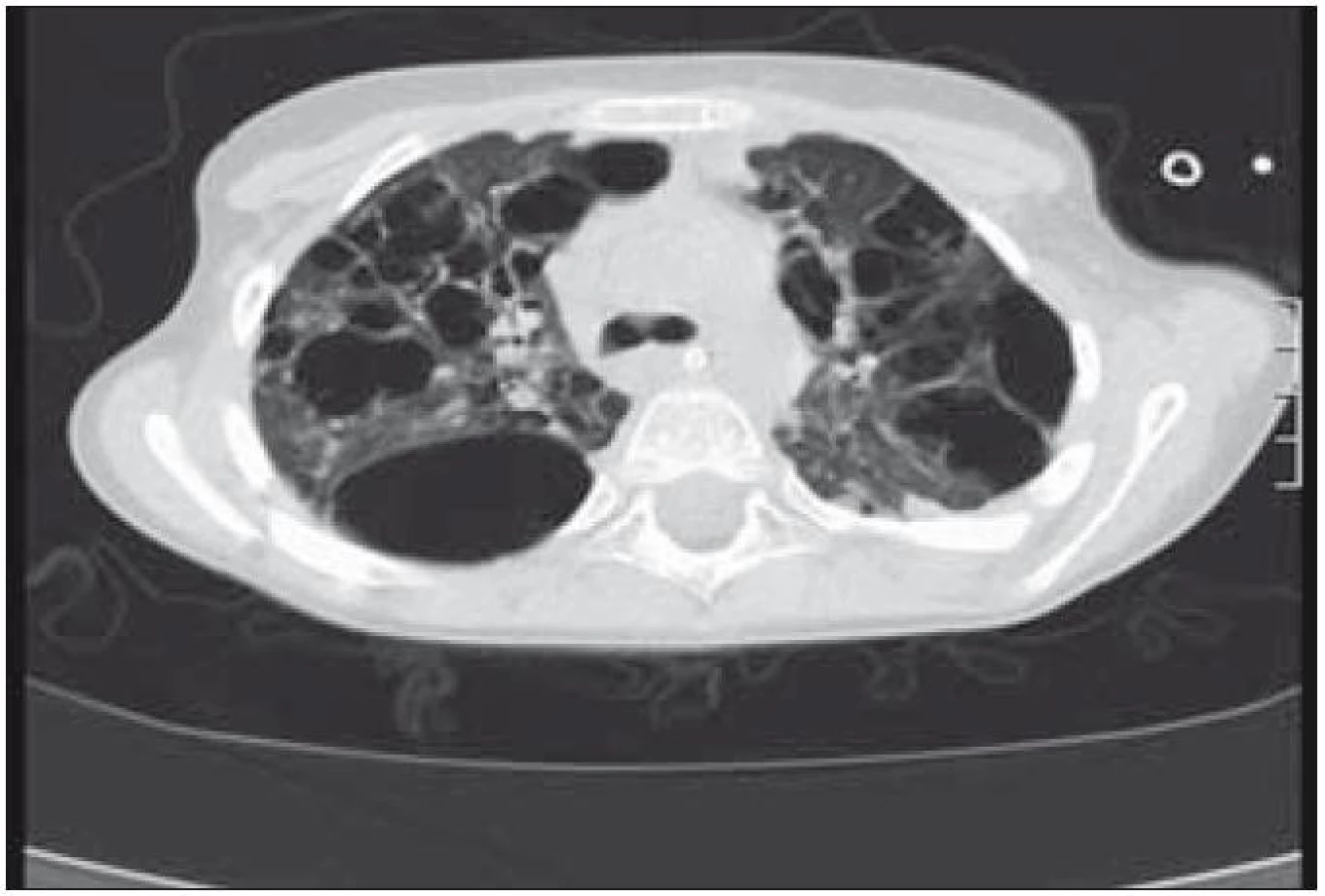
Plicní forma je vzácná a je prokazována asi u 15 % dětí s LCH. Infiltrace postihuje intersticiální i alveolární struktury plicní tkáně. Ložiska nádorové infiltrace regredují, jizví se a vzniká výrazný paracikatrikózní emfyzém až do obrazu tzv. voštinovité plíce (obr. 8). V klinickém obraze dominuje suchý kašel (> 50 %), dále dyspnoe a bolest hrudníku. Asi v 10 % je prvním příznakem spontánní pneumotorax [68]. V RTG obraze dominují v počáteční fázi intersticiální změny. Diagnóza je stanovena biopticky (obr. 7) nebo CT vyšetřením s vysokým rozlišením (HRCT – mikronodulární změny s cystickou progresí) (obr. 6), které je i vhodnou metodou pro sledování dynamiky plicních změn [13].


Gastrointestinální trakt
Základním projevem je neprospívání způsobené malabsorpcí. Dále je to zvracení, průjmovité stolice a exsudativní enteropatie s hypoproteinemií. Správná diagnóza je možná pouze z biopsie střeva [74].
Endokrinní systém
Nejčastější endokrinopatií je diabetes insipidus, který bývá patrný ještě před průkazem intrakraniálních změn, může se ovšem objevit až v průběhu léčby, případně po jejím ukončení. Diabetes insipidus provází zejména postižení v oblasti orofaciální, temporální a baze lební. U části pacientů je dále pozorována růstová retardace, předčasná puberta, hypotalamický syndrom, případně galaktorea při postižení hypotalamo-hypofyzární oblasti [21,22]. Při endokrinní symptomatologii je třeba provést MRI mozku a komplexní endokrinologické vyšetření.
Centrální nervový systém
Známky akutního postižení CNS, jakými jsou intrakraniální hypertenze a křeče, jsou vzácné. K detekci změn v oblasti CNS je optimální zobrazovací metodou MRI (obr. 9) s propracovaným klasifikačním systémem (tab. 3) [40], kterou doplní vyšetření neurologické, oční a endokrinologické. Postižení mozku má dvě formy. Jednak jsou to granulomatózní změny s prokázanou aktivitou LCH buněk, které jsou lokalizované zejména v oblasti hypotalamu, ale mohou se objevit supratentoriálně i infratentoriálně a jsou považovány za aktivní formu onemocnění. Naopak častěji popisované změny zejména v oblasti mozečku jsou charakteru degenerativních změn a mohou se objevit až několik let po stanovení diagnózy LCH. Až 10 % pacientů trpí neurologickými projevy různé intenzity v podobě progresivní ataxie, dysartrie, nystagmu, hyperflexie, dysfagie, neostrého vidění nebo obtíží při postižení hlavových nervů [23,42,78].

![Klasifikace postižení CNS při histiocytóze z Langerhansových buněk podle MRI nálezu [40].](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image/2608fc47e528686d1d32da1e59118a96.jpg)
Vzácně postižené orgány
Jen zcela výjimečně byla při generalizované formě LCH popsána infiltrace ledvin, močového měchýře a pankreatu [23].
Léčebná strategie
Terapie histiocytózy z Langerhansových buněk je určována rozsahem postižení. Obecně má monosystémové onemocnění (skelet nebo lymfatické uzliny nebo kůže) benigní charakter se značnou tendencí k spontánnímu hojení v průběhu měsíců až let [33,66,91]. Zcela zásadní význam má pečlivé vstupní vyšetření (staging) k vyloučení dalších ložisek. Léčba monosystémové formy LCH nemá jednotně stanovený protokol a používají se různé léčebné modality (lokální aplikace kortikoidů – intralezionálně nebo mast, monoterapie – kortikoidy, vinblastin, kyretáž, radioterapie) [25,35]. U izolovaného postižení kůže, zejména u malých dětí, je vysoká pravděpodobnost spontánního zhojení, a proto je možno s intenzivní léčbou vyčkat podle klinického průběhu. Nicméně u části pacientů onemocnění progreduje do multisystémové formy LCH, a proto je třeba pečlivá dispenzarizace na specializovaném pracovišti, které má dlouhodobé zkušenosti s histiocytózou z Langerhansových buněk. Podobně není třeba při izolovaném postižení kosti okamžitě indikovat systémovou léčbu, kterou je možné zahájit až při progresi onemocnění nebo při lokalizaci, kde hrozí fraktura nebo komprese míchy či optického nervu, případně při rozsáhlé extra-oseální expanzi [9,14,26,52,86,91,98]. Uplatnění radioterapie je u ložiskového postižení kosti výrazně limitováno vzhledem k pozdním následkům. Používá se nízká dávka ozáření (6 – 8 Gy), a to zejména na afekce v oblasti optického nervu a míchy [38,39,83]. Zvláštní zmínku zasluhuje plicní forma LCH. Postihuje zejména dospělé pacienty, mezi kterými výrazně převažují kuřáci. Nicméně onemocnění se vzácně vyskytuje i u dětí a má při systémové terapii příznivou prognózu [62,70,72,90,92].
U mnohočetného kostního postižení je také indikována systémová léčba. Po zavedení kombinace kortikoidů a vinblastinu v léčebném protokolu DAL - HX 83 se podařilo snížit reaktivaci onemocnění na 18 %, tedy podobně jako u jednoložiskové kostní formy LCH [91]. Na menších souborech pacientů byl prokázán příznivý léčebný efekt indometacinu, bifosfonátů a kombinace 6-merkaptopurinu a metotrexátu [30,73,96].
Léčba multisystémové formy je v současné době stále ještě stratifikována podle protokolu LCH III [59], který navazuje na předchozí studie LCH I (1991 – 1995) a LCH II (1996 – 2000). Pacienti léčení protokolem LCH III jsou rozděleni do 3 skupin (G 1 – 3) podle lokalizace a rozsahu postižení (tab. 2). Na rozdíl od LCH II není v nové klasifikaci posuzován věk (</ > 2 roky) a v léčebním režimu je vynechán etoposid, u kterého nebyl prokázán signifikantní léčebný účinek, ale představuje vyšší riziko vzniku sekundární leukemie [56,61]. Výsledky předchozích studií prokázaly zásadní prognostický význam časné odezvy po 6 týdnech léčby [34,65], a proto je v LCH III hodnocení odezvy na léčbu po 6 týdnech. Má určující význam pro pokračování protokolu. Při dobré odpovědi („no active disease“ – bez patrné aktivity onemocnění nebo „better“ – regrese onemocnění) pokračuje léčba udržovací chemoterapií, jinak se znovu opakuje iniciální část protokolu. Základní kombinací systémové léčby je kombinace prednisonu (40 mg/ m2) a vinblastinu (6 mg/ m2).

Riziko reaktivace onemocnění je zejména u multisystémové formy a pohybuje se v rozmezí 27 – 62 % [59]. Pro léčbu reaktivovaného onemocnění je v současné době připravován nový protokol s kombinací 2 - chlorodeoxy-adenosinu (2 - CdA) a cytosin-arabinosidu (Ara - C). Výsledky předchozí studie LCH - S - 98, která doporučovala monoterapii 2 - CdA, zatím nebyly publikovány, ale zkušenosti s oběma preparáty na malých souborech jsou povzbuzující [3,7,69,88,93].
Léčebné výsledky u monosystémové formy jsou velmi příznivé s celkovým přežitím 95 – 100 %, zatímco u multisystémového postižení je přežití do 81 % [20,64].
Navzdory pokroku ve znalostech o biologickém chování LCH, a tím zlepšení terapeutického ovlivnění onemocnění zůstává značný prostor pro další zlepšení léčebných výsledků multisystémového postižení a rezistentní formy onemocnění. Zejména u pacientů s reaktivací LCH je nadějnou metodou vysokodávkovaná chemoterapie s následnou alogenní transplantací kostní dřeně [87].
V závěru je třeba zmínit pozdní následky, které jsou častější u multisystémového postižení (71 %) než u monosystémové formy LCH (24 %). Ve výčtu pozdních následků dominuje diabetes insipidus (24 %), ortopedické obtíže (20 %), poškození sluchu (13 %) a neurologické obtíže (11 %), jak dokumentuje studie Histiocytární společnosti [43], případně plicní změny (11 %) [7].
Závěr
Léčebné výsledky u monosystémové formy jsou velmi příznivé s celkovým přežitím 95 – 100 %, zatímco u multisystémového postižení je přežití do 81 %. Výzvou pro další výzkum tedy zůstává léčba multisystémového onemocnění, zejména s postižením tzv. rizikových orgánů (plíce, játra, slezina a hematopoetický systém). Je zapotřebí začlenit molekulárně genetické studie v rámci mezinárodních protokolů, které by mohly umožnit průkaz případných markerů či prognostické faktory a pomohly cíleně stratifikovat léčebný režim. Jen tak je možné včas odlišit formy onemocnění, které jsou primárně rezistentní nebo indolentní k používaným léčebným schématům.
Podporováno výzkumným záměrem VZMZ 64203/ 6715.
doc. MUDr. Hubert Mottl, CSc.
www.fnmotol.cz
e-mail: hubert.mottl@fnmotol.cz
Doručeno do redakce: 9. 9. 2010
Zdroje
1. Adam Z, Kavan P, Koutecký J. et al. Histiocytóza z Langerhansových buněk. Čs Pediatr 1997; 52 : 899 – 905.
2. Arceci RJ, Brenner MK, Pritchard J. Controversies and new approaches to treatment of Langerhans cell histiocytosis. Hematol Oncol Clin North Am 1998; 12 : 339 – 357.
3. Aricò M, Egeler RM. Clinical aspects of Langerhans cell histiocytosis. Hematol Oncol Clin North Am 1998; 12 : 247 – 258.
4. Avery ME, McAfee JG, Guild HG. The course and prognosis of reticuloendotheliosis (eosinophilic granuloma, Schüller - Christian disease and Letterer - Siwe disease): a study of forty cases. Am J Med 1957; 22 : 636 – 652.
5. Bank MI, Rengtved P, Carstensen H et al. Langerhans cell histiocytosis: an evaluation of histopathological parameters, demonstration of proliferation by Ki - 67 and mitotic bodies. APMIS 2003; 111 : 300 – 308.
6. Bechan GI, Egeler RM, Arceci RJ. Biology of Langerhans cells and Langerhans cell histiocytosis. Int Rev Cytol 2006; 254 : 1 – 43.
7. Bernard F, Thomas C, Bertrand Y et al. Multi-centre pilot study of 2-chlorodeoxyadenosine and cytosine arabinoside combined chemotherapy in refractory Langerhans cell histiocytosis with haematological dysfunction. Eur J Cancer 2005; 41 : 2682 – 2689.
8. Bernstrand C, Carstensen H, Jakobsen B et al. Immunogenetic heterogeneity in single-system and multi-system langerhans cell histiocytosis. Pediatr Res 2003; 54 : 30 – 36.
9. Berry DH, Gresik M, Maybee D et al. Histiocytosis X in bone only. Med Pediatr Oncol 1990; 18 : 292 – 294.
10. Bhatia S, Nesbit ME Jr, Egeler RM et al. Epidemiologic study of Langerhans cell histiocytosis in children. J Pediatr 1997; 130 : 774 – 784.
11. Binkovitz LA, Olshefski RS, Adler BH. Coincidence FDG - PET in the evaluation of Langerhans’ cell histiocytosis: preliminary findings. Pediatr Radiol 1997; 33 : 598 – 602.
12. Braier J, Ciocca M, Latella A et al. Cholestasis, sclerosing cholangitis, and liver transplantation in Langerhans cell histiocytosis. Med Pediatr Oncol 2002; 38 : 178 – 182.
13. Brauner MW, Grenier P, Tijani K et al. Pulmonary Langerhans cell histiocytosis: evolution of lesions on CT scans. Radiology 1997; 204 : 497 – 502.
14. Broadbent V, Gadner H. Current therapy for Langerhans cell histiocytosis. Hematol Oncol Clin North Am 1998; 12 : 327 – 338.
15. Carstensen H, Ornvold K. The epidemiology of Langerhans cell histiocytosis in children in Denmark. Med Pediatr Oncol 1993; 21 : 387 – 388.
16. Christian HA. Defects in membranous bones, exophthalmos and diabetes insipidus: an unusual syndrome of dyspituitarism. Med Clin North Am 1920; 3 : 849 – 871.
17. Coppes - Zantinga A, Egeler RM. Historical review the 9. Langerhans cell histiocytosis X fines revealed. Br J Haematol 2002; 16 : 3 – 9.
18. Dacic S, Trusky C, Bakker A et al. Genotypic analysis of pulmonary Langergans cell histiocytosis. Hum Pathol 2007; 34 : 1345 – 1349.
19. de Graaf JH, Tamminga RY, Dam - Meiring A et al. The presence of cytokines in Langerhans’ cell histiocytosis. J Pathol 1996; 180 : 400 – 406.
20. Dogan AS, Conway JJ, Miller JH et al. Detection of bone lesions in Langerhans cell histiocytosis: complementary roles of scintigraphy and conventional radiography. J Pediatr Hematol Oncol 1996; 18 : 51 – 58.
21. Donadieu J, Rolon MA, Pion I et al. Incidence of growth hormone deficiency in pediatric - onset Langerhans cell histiocytosis: efficacy and safety of growth hormone treatment. J Clin Endocrinol Metab 2004; 89 : 604 – 609.
22. Donadieu J, Rolon MA, Thomas C et al. Endocrine involvement in pediatric - onset Langerhans’ cell histiocytosis: a population-based study. J Pediatr 2004; 144 : 344 – 350.
23. Donadieu J, Egeler RM, Pritchard J. Langerhans cell histiocytosis: a clinical update. In: Weitzman S, Egeler RM (ed). Histiocytic disorders of children and adults. Cambridge: Cambridge University Press 2005 : 5 – 129.
24. De Graaf JH, Tamminga RY, Dam - Meiring A et al. The presence of cytokines in Langerhans’ cell histiocytosis. J Pathol 1996; 180 : 400 – 406.
25. Egeler RM, Thompson RC Jr, Voûte PA et al. Intralesional infiltration of corticosteroids in localized Langerhans’ cell histiocytosis. J Pediatr Orthop 1992; 12 : 811 – 814.
26. Egeler RM, D’Angio GJ. Langerhans cell histiocytosis. J Pediatr 1995; 127 : 1 – 11.
27. Egeler RM, Neglia JP, Aricò M et al. The relation of Langergans cell histiocytosis to acute leukemia, lyphomas, and other solid tumors. The LCH - Malignancy Study Group of the Hisitocyte Society. Hematol Oncol Clin North Am 1998; 12 : 369 – 378.
28. Egeler RM, Favara BE, van Meurs M et al. Differential In situ cytokine profiles of Langerhans-like cells and T cells in Langerhans cell histiocytosis: abundant expression of cytokines relevant to disease and treatment. Blood 1999; 94 : 4195 – 4201.
29. Farber S. The nature of solitary or eosinophilic granuloma’ of bone. Am J Pathol 1941; 17 : 625 – 629.
30. Farran RP, Zaretski E, Egeler RM. Treatment of Langerhans cell histiocytosis with pamidronate. J Pediatr Hematol Oncol 2001; 23 : 54 – 56.
31. Favara BE, McCarthy RC, Mierau GW. Histiotosis X. Hum Pathol 1983; 14 : 663 – 676.
32. Favara BE, Jaffe R, Egeler RM. Macrophage activation and hemophagocytic syndrome in Langerhans cell histiocytosis: report of 30 cases. Pediatr Dev Pathol 2002; 5 : 130 – 140.
33. Gadner H, Grois N. The histiocytosis syndromes. In: Fitzpatrick TB, Eisen AZ, Wolff K et al (eds). Dermatology in general medicine. Vol. II. 4th ed. New York: McGraw - Hill, Inc. 1993 : 2003 – 2017.
34. Gadner H, Heitger A, Grois N et al. Treatment strategy for disseminated Langerhans cell histiocytosis. DAL HX - 83 Study Group. Med Pediatr Oncol 1994; 23 : 72 – 80.
35. Gadner H, Ladish S. The treatment of Langerhans cell histiocytosis. In: Weitzman S, Egeler RM (eds). Histiocytic disorders of children and adults. Cambridge: Cambridge University Press 2005 : 229 – 253.
36. Glotzbecker MP, Carpentieri DF, Dormans JP. Langerhans cell histiocytosis: a primary viral infection of bone? Human herpes virus 6 latent protein detected in lymphocytes from tissue of children. J Pediatr Orthop 2004; 24 : 123 – 129.
37. Goo HW, Yang DH, Ra YS et al. Whole - body MRI of Langerhans cell histiocytosis: comparison with radiography and bone scintigraphy. Pediatr Radiol 2006; 36 : 1019 – 1031.
38. Gramatovici R, D’Angio GJ. Radiation therapy in soft - tissue lesions in histiocytosis X (Langerhans’ cell histiocytosis). Med Pediatr Oncol 1988; 16 : 259 – 262.
39. Greenberger JS, Cassady JR, Jaffe N et al. Radiation therapy in patients with histiocytosis: management of diabetes insipidus and bone lesions. Int J Radiat Oncol Biol Phys 1979; 5 : 1749 – 1755.
40. Grois NG, Favara BE, Mostbeck GH et al. Central nervous system disease in Langerhans cell histiocytosis. Hematol Oncol Clin North Am 1998; 12 : 287 – 305.
41. Grois N, Prayer D, Prosch H et al. Neuropathology of CNS disease in Langerhans cell histiocytosis. Brain 2005; 128 : 829 – 838.
42. Hand A. Polyuria and tuberculosis. Arch Pediatr 1893; 10 : 673 – 675.
43. Haupt R, Nanduri V, Calevo MG et al. Permanent consequences in Langerhans cell histiocytosis patients: a pilot study from the Histiocyte Society - Late Effects Study Group. Pediatr Blood Cancer 2004; 42 : 438 – 444.
44. Howarth DM, Mullan BP, Wiseman GA et al. Bone scintigraphy evaluated in diagnosing and staging Langerhans’ cell histiocytosis and related disorders. J Nucl Med 1996; 37 : 1456 – 1460.
45. Jaffe R. Patology of histiocytosis X. Perspect Pediatr Pathol 1987; 9 : 4 – 47.
46. Jaffe ES, Harris NL, Stein H et al. World Health Organization classification of tumours. Pathology and genetics. Tumors of Haematopoietic and lymphoid tissues. Lyon: IARC Press 2001 : 32 – 41.
47. Jenson HB, McClain KL, Leach CT et al. Evaluation of human herpesvirus type 8 infection in childhood Langerhans cell histiocytosis. Am J Hematol 2000; 64 : 237 – 241.
48. Kakkar S, Kapila K, Verma K. Langerhans cell histiocytosis in lymph nodes. Cytomorphologic diagnosis and pitfalls. Acta Cytol 2001; 45 : 327 – 332.
49. Karis J, Bernstrand C, Fadeel B et al. The incidence of Langerhans cell histiocytosis in children in Stockholm County, Sweden 1992 – 2001. Proceedings of the XIX Meeting of Histiocyte Society. Philadephia 2003.
50. Kawakubo Y, Kishimoto H, Sato Y et al. Human cytomegalovirus infection in foci of Langerhans cell histocytosis. Virchows Arch 1999; 434 : 109 – 115.
51. Kilpatrick SE, Wenger DE, Gilchrist GS et al. Langerhans’ cell histiocytosis (histiocytosis X) of bone. A clinicopathologic analysis of 263 pediatric and adult cases. Cancer 1995; 76 : 2471 – 2484.
52. Ladish S, Gadner H. Treatment of Langerhans cell histiocytosis – evolution and current approaches. Br J Cancer Suppl 1994; 23: S41 – S46.
53. Laman JD, Leenen PJ, Annels NE et al. Langerhans - cell histiocytosis ‘insight into DC biology’. Trends Immunol 2003; 24 : 190 – 196.
54. Lampert F. Langerhans cell histiocytosis. Historical perspectives. Hematol Oncol Clin North Am 1998; 12 : 213 – 219.
55. Lau L, Krafchik B, Trebo MM et al. Cutaneous Langerhans cell histiocytosis in children under one year. Pediatr Blood Cancer 2006; 46 : 66 – 71.
56. Le Deley MC, Vassal G, Taïbi A et al. High cumulative rate of secondary leukemia after continuous etoposide treatment for solid tumors in children and young adults. Pediatr Blood Cancer 2005; 45 : 25 – 31.
57. Letterer E. Aleukamische retikulose (ein Beitrag zu den proliferativen Erkrankungen des reticuloendothelialapparates). Frankfurter Zeitschritte Pathol 1924; 30 : 377 – 394.
58. LCH Study Group of the Histiocyte Society. LCH II – Treatment Protocol of the Second International Study for Langerhans cell histiocytosis. Protocol 1996.
59. LCH Study Group of the Histiocyte Society. LCH III – Treatment Protocol of the Second International Study for Langerhans cell histiocytosis. Protocol 2001.
60. Lichtenstein L. Hisitocytosis X: integration of eosinophilic granuloma of bone, Letterer - Siwe disease and Schüller - Christian disease as related manifestations of a single nosologic entity. AMA Arch Pathol 1953; 56 : 84 – 102.
61. Lopes LF, de Camargo B. Secondary acute promyelocytic leukemia after treatment with etoposide for Langerhans cell histiocytosis (LCH). Med Pediatr Oncol 1999; 32 : 315.
62. McClain KL, Gonzales J, Jonkers R et al. Need for a cooperative study: pulmonary Langerhans cell histiocytosis and its management in adults. Med Pediatr Oncol 2002; 39 : 35 – 39.
63. McClain KL, Laud P, Wu WS et al. Langerhans cell histiocytosis patients have HLA Cw7 and DR4 types associated with specific clinical presentations and no increased frequency in polymorphism of the tumor necrosis factor alpha promoter. Med Pediatr Oncol 2003; 41 : 502 – 507.
64. Minkov M, Grois N, Heitger A et al. Treatment of multisystem Langerhans cell histiocytosis. Results of the DAL - HX 83 and DAL - HX 90 studies. DAL - HX Study Group. Klin Pediatr 2000; 212 : 139 – 144.
65. Minkov M, Grois N, Heitger A et al. DAL - HX Study Group. Response to initial treatment of multisystem Langerhans cell histiocytosis: an important prognostic indicator. Med Pediatr Oncol 2002; 39 : 581 – 585.
66. Mottl H, Mráček J, Kabelka Z et al. Hisitiocytóza z Langerhansových buněk u děti. Čs Pediatr 1992; 47 : 530 – 533.
67. Mottl H, Ganevová M, Radvanská J et al. Léčebné výsledky histiocytózy z Langerhansových buněk protokolem LCH II. Čas Lék Čes 2005; 144 : 753 – 755.
68. Mottl H, Nekolná M, Starý J et al. Plicní forma histiocytózy z Langerhansových buněk u dětí. 15. Konference dětských hematologů a onkologů ČR a SR. České Budějovice, Abstrakta 2005 : 14.
69. Mottl H, Starý J, Šmelhaus V et al. Současná strategie léčby histiocytózy z Langerhansových buněk u dětí. XXX. brněnské onkologické dny 2006 : 140.
70. Mottl H, Stary J, Snajdauf J et al. Pulmonary Langerhans cell histiocytosis in children and adolescents. Pediatr Blood Cancer 2006; 47 : 451.
71. Mottl H, Starý J. Histiocytóza z Langerhansových buněk u dětí – klinické projevy, diagnostika a současná léčba. Čs Pediatr 2007; 62 : 220 – 225.
72. Mottl H, Stary J, Snajdauf J et al. Primary pulmonary Langerhans cell histiocytosis: Outcome in children and adolescents. 23rd Annual Meeting of Histiocyte Society, 23. 9. – 25 .9. 2007. Cambridge, UK. Abstract.
73. Munn SE, Olliver L, Broadbent V et al. Use of indomethacin in Langerhans cell histiocytosis. Med Pediatr Oncol 1999; 32 : 247 – 249.
74. Nanduri VR, Kelly K, Malone M et al. Colon involvement in Langerhans’ cell histiocytosis. J Pediatr Gastroenterol Nutr 1999; 29 : 462 – 466.
75. Neumann C, Schaumberg - Lever G, Döpfer R et al. Interferon gamma is a marker for histiocytosis X cells in the skin. J Invest Dermatol 1988; 91 : 280 – 282.
76. Nezelof C. Histiocytosis X: a histological and histogenetic study. Perspect Pediatr Pathol 1979; 5 : 153 – 178.
77. Osband ME, Lipton JM, Lavin P et al. Histiocytosis - X. N Engl J Med 1981; 304 : 146 – 153.
78. Prayer D, Grois N, Prosch H et al. MR imaging presentation of intracranial disease associated with Langerhans cell histiocytosis. AJNR Am J Neuroradiol 2004; 25 : 880 – 891.
79. Pritchard J, Broadbent V. Histiocytosis – an introduction. Br J Cancer Suppl 1994; 23: S1 – S3.
80. Schmitz L, Favara BE. Nosology and pathology of Langerhans cell histiocytosis. Hematol Oncol Clin North Am 1998; 12 : 221 – 246.
81. Schouten B, Egeler RM, Leenen PJ et al. Expression of cell cycle-related gene products in Langerhans cell histiocytosis. J Pediatr Hematol Oncol 2002; 24 : 727 – 732.
82. Schüller A. Uber eigenartige schadeldefekte im jugendalter. Fortschritte Auf der Gebiete Rontgenstrahlen 1915; 23 : 12 – 18.
83. Selch MT, Parker RG. Radiation therapy in management of Langerhans cell histiocytosis. Med Pediatr Oncol 1990; 18 : 97 – 102.
84. Siwe SA. Die Reticuloendotheliose-ein neues krankheitsbild unter den hepatosplenomegalien. Zeitschrift für Kinderheilkunde 1933; 55 : 212 – 247.
85. Slacmeulder M, Geissmann F, Lepelletier Y. No association between Langerhans cell histiocytosis and human herpes virus - 8. Med Pediatr Oncol 2002; 39 : 187 – 189.
86. Slater JM, Swarm OJ. Eosinophilic granuloma of bone. Med Pediatr Oncol 1980; 8 : 151 – 164.
87. Steiner M, Matthes - Martin S, Attarbaschi A et al. Improved outcome of treatment-resistant high-risk Langerhans cell histiocytosis after allogeneic stem cell transplantation with reduced - intensity conditioning. Bone Marrow Transplant 2005; 36 : 215 – 225.
88. Stine KC, Saylors RL, Saccente S et al. Efficacy of continuous infusion 2 - CDA (cladribine) in pediatric patients with Langerhans cell histiocytosis. Pediatr Blood Cancer 2004; 43 : 81 – 84.
89. Stuurman KE, Lau I, Doda W et al. Evaluation of the natural history and long term complications of patients with Langerhans cell histiocytosis of bone. Proc XIX meeting Histiocyte Society. Philadelphia 2003.
90. Tazi A, Soler P, Hance AJ. Adult pulmonary Langerhans’ cell histiocytosis. Thorax 2002; 55 : 405 – 416.
91. Titgemeyer C, Grois N, Minkov M et al. Pattern and course of single-system disease in Langerhans cell histiocytosis data from the DAL - HX 83 - and 90 - study. Med Pediatr Oncol 2001; 37 : 108 – 114.
92. Vassallo R, Ryu JH, Colby TV et al. Pulmonary Langerhans’ - cell histiocytosis. N Engl J Med 2000; 342 : 1969 – 1978.
93. Weitzman S, Egeler RM. Histiocytic disorders of children and adults: introduction to the problem, overview, historical perspective and epidemiology. In: Weitzman S, Egeler RM (eds). Histiocytic disorders of children and adults. Cambridge: Cambridge University Press 2005 : 1 – 13.
94. Willis B, Ablin A, Weinberg V et al. Disease course and late sequelae of Langerhans’ cell histiocytosis: a 25 year experience at the University of California, San Francisco. J Clin Oncol 1996; 14 : 2073 – 2082.
95. Willman CL, Busque L, Griffith BB et al. Langehans’ cell histiocytosis (histiocytosis X) a clonal proliferative disease. N Engl J Med 1994; 331 : 154 – 160.
96. Womer RB, Anunciato KR, Chehrenama M. Oral methotrexate and alternace - day prednisone for low - risk Langerhans cell histiocytosis. Med Pediatr Oncol 1995; 25 : 70 – 73.
97. Wong A, Ortiz - Neira CL, Reslan WA et al. Liver involvement in Langerhans cell histiocytosis. Pediatr Radiol 2006; 36 : 1105 – 1107.
98. Woo KI, Harris GJ. Eosinophilic granuloma of the orbit: understanding the paradox of aggressive destruction responsive to minimal intervention. Ophthal Plast Reconstr Surg 2003; 19 : 429 – 439.
99. Writing Group of the Histiocyte Siociety. Histiocytosis syndromes in children. Lancet 1987; 1 : 208 – 209.
100. Yousem SA, Colby TV, Chen YY et al. Pulmonary Langerhans’ - cell histiocytosis. Molecular analysis of clonality. Am J Surg Pathol 2001; 25 : 630 – 636.
101. Yu RC, Chu C, Buluwela L et al. Clonal proliferation of Langerhans cells in Langerhans cell histiocytosis. Lancet 1994; 343 : 767 – 768.
102. Zouny JW. Cell fate development in the myeloid system. In: Lotze MT, Thomson AW (eds). Dendritic cells. San Diego: Academic Press 1999 : 29 – 49.
Štítky
Diabetologie Endokrinologie Interní lékařstvíČlánek vyšel v časopise
Vnitřní lékařství

2010 Číslo Supplementum 2
- Plicní hypertenze – syndrom mnoha tváří – vyžaduje přesnou diagnostiku a specializovanou léčbu
- Proces hojení ran krok za krokem a co ho může zkomplikovat
- Limity glykovaného hemoglobinu a význam dalších glykovaných proteinů
- Korelace dávky levothyroxinu s titrem autoimunitních protilátek u primární hypotyreózy
- Zeolit-jodový komplex pomáhá v péči o infikované rány
Nejčtenější v tomto čísle
- Hemofagocytující lymfohistiocytóza
- Erdheimova-Chesterova nemoc v obrazech
- Systémová mastocytóza
- Histiocytóza z Langerhansových buněk u dětí a dospívajících
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy