
Syndrom VEXAS: autoinflamatorní onemocnění dospělého věku s kožními projevy. Přehledný článek
VEXAS Syndrome: an Adult-Onset Autoinflammatory Disease with Cutaneous Manifestations. A Review
VEXAS syndrome (vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic mutation) is a chronic progressive disease first described in 2020. It is an X-linked autoinflammatory disease that is caused by a somatic mutation in the UBA1 gene in hematopoietic cells. The disease typically affects older men. VEXAS syndrome is characterized by multi-organ inflammation, presenting with general symptoms such as fever and elevated inflammatory markers, as well as organ-specific symptoms. Almost any organ can be affected, including the eyes, lungs, joints, cartilage, or blood vessels in the form of vasculitis. Skin manifestations are not only the most common but also often the first sign of the disease. Diagnosis requires the detection of a pathogenic variant of the UBA1 gene.
Keywords:
VEXAS syndrome – UBA1 mutation – autoinflammatory neutrophilic dermatosis
Autoři:
T. Sychrovská 1; H. Mann 1
Působiště autorů:
Revmatologický ústav a Klinika revmatologie 1. LF UK, Praha
1
Vyšlo v časopise:
Čes-slov Derm, 100, 2025, No. 4, p. 170-175
Kategorie:
Novinky v medicíně
Souhrn
Syndrom VEXAS (vakuoly, E1 enzym, X-vázané, autoinflamatorní, somatická mutace) je chronické progresivní onemocnění, poprvé popsané v roce 2020. Jedná se o X-vázané autoinflamatorní onemocnění vznikající na podkladě somatické mutace UBA1 genu v hematopoetických buňkách. Onemocnění nejčastěji postihuje muže vyššího věku. Pro syndrom VEXAS je charakteristický multiorgánový zánět, projevující se celkovými příznaky, jako horečky a zvýšení zánětlivých parametrů, stejně jako orgánově specifické příznaky. Postižen může být téměř jakýkoli orgán včetně očí, plic, kloubů, chrupavek nebo cév ve formě vaskulitidy. Kožní postižení je nejčastějším, a zároveň často i prvním klinickým projevem syndromu VEXAS. Pro diagnózu onemocnění je nutný průkaz patogenní varianty UBA1 genu.
Klíčová slova:
syndrom VEXAS – UBA1 mutace – autoinflamatorní neutrofilní dermatóza
ÚVOD
Syndrom VEXAS je chronické, progresivní, autoinflamatorní onemocnění dospělého věku způsobené somatickými mutacemi UBA1 genu v myeloidních progenitorových buňkách kostní dřeně. Název odkazuje na hlavní rysy onemocnění. Mutace UBA1 ovlivňuje funkci E1 enzymu, vedoucí v důsledku k hromadění proteinů, jinak určených k degradaci, v buňce ve formě vakuol. Na hromadění těchto proteinů reaguje organismus zánětlivou odpovědí – autoinflamací. UBA1 gen se nachází na X chromozomu, vzhledem k somatickému charakteru mutace – mutace vznikající během života, onemocnění nacházíme zejména u starších mužů [4]. Ukazuje se, že různé mutace mají vztah k fenotypu a závažnosti onemocnění, což má prognostický význam [17]. Ač je syndrom VEXAS vzácné onemocnění, ve skupině mužů starších 50 let je odhadovaná prevalence 23,4/100 000 [14]. Lze tedy předpokládat, že v ČR žije více než 600 pacientů se syndromem VEXAS, kteří dosud nebyli diagnostikováni.
KLINICKÉ MANIFESTACE
Klinické projevy syndromu VEXAS lze rozdělit na zánětlivé a hematologické. Zánětlivá aktivita onemocnění se projevuje celkovými příznaky (horečky a hubnutí) provázenými zvýšením zánětlivých parametrů, a orgánově specifickými projevy (nejčastěji kožní projevy, plicní infiltráty, oční záněty, chondritida a artritida). V rámci syndromu VEXAS může být postižen téměř jakýkoliv orgán (obr. 1). Nejčastějšími hematologickými projevy jsou makrocytární anemie a trombocytopenie vznikající v důsledku dysfunkce kostní dřeně. Pacienti mají predispozici k hematologickým malignitám, především myelodysplastickému syndromu [4].
![Nejčastější projevy syndromu VEXAS a jejich prevalence dle [4]](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image/52b5c8d3c5eb6a05c191bc47f06e8923.png)
Dermatologické projevy
Kožní projevy jsou nejčastější orgánově specifickou manifestací syndromu VEXAS, vyskytující se u 88 % pacientů. V 63 % případů je kožní postižení prvním příznakem onemocnění [15]. Závažnost a charakter kožních projevů se mění v čase s obdobími relapsů a remisí [18]. Typická je rovněž velmi dobrá reakce kožních projevů na systémové glukokortikoidy [15]. Nejčastější kožní manifestací jsou mnohočetná, erytematózní, edematózní ložiska připomínající Sweetův syndrom (Sweet syndrome-like), s maximem na trupu a končetinách (obr. 2) [3, 18].

Asi u třetiny pacientů mají tyto projevy semilunární tvar (obr. 3). K dalším kožním projevům patří podkožní noduly podobné jako u erythema nodosum anebo hmatná purpura způsobená vaskulitidou. V histopatologickém vyšetření projevů připomínajících akutní neutrofilní dermatózu (Sweetův syndrom) v kožní biopsii typicky nacházíme obraz neutrofilní dermatózy s infiltráty z nevyzrálých myeloidních buněk, různým podílem neutrofilů s menším podílem lymfocytů [18,19]. Tyto většinou perivaskulární neutrofilní infiltráty leukocytoklasií a s intersticiálním edémem typicky postihují celou dermis, vzácněji zasahují až do hypodermis. Diferenciálně diagnosticky může pomoci skutečnost, že léze u syndromu VEXAS obvykle nejsou bolestivé, i když vzácněji se vyskytla jak bolestivost, tak svědivost [18].
Obraz kožního postižení je ale velmi heterogenní a vzácněji se může objevit livedo reticularis, livedo racemosa, vezikuly nebo pustuly, periorbitální otok, a dokonce urtikariální projevy (urticaria-like) (obr. 4–6) [7, 13]. V jistém smyslu typickou kožní manifestací u syndromu VEXAS je závažná lokální reakce na podání anakinry (reakce v místě vpichu injekce), objevující se asi u třetiny pacientů, nejčastěji ve formě zarudlých ložisek, vzácněji až ulcerací a abscesů [4, 9]. Typ klinického postižení, rozsah a závažnost se však mohou interindividuálně velmi lišit [17]. Jednotliví pacienti pak často mají kombinaci těchto kožních projevů [15]. Současný výskyt (koexistence) těchto kožních projevů u jednoho pacienta by měl vzbudit podezření na syndrom VEXAS. U pacientů jsou relativně časté vaskulitidy (dle některých autorů stejně časté jako neutrofilní dermatózy [17]), postiženy mohou být cévy jakéhokoliv kalibru. Mohou klinicky imponovat jako polyarteritis nodosa nebo kožní vaskulitida [17]. Nejčastější je leukocytoklastická vaskulitida, nicméně dle některých prací mají kožní léze v histologickém obraze vysoký stupeň leukocytoklazie, bez přítomnosti fibrinoidní nekrózy, a nesplňují tedy kritéria pro diagnózu leukocytoklastické vaskulitidy [18]. Tato„perivaskulární dermatitida“ se klinicky projevuje purpurou a bývá též častým projevem syndromu VEXAS [17]. Postižení pojivové tkáně zahrnuje artralgie nebo artritidy a chondritidy (obr. 7).




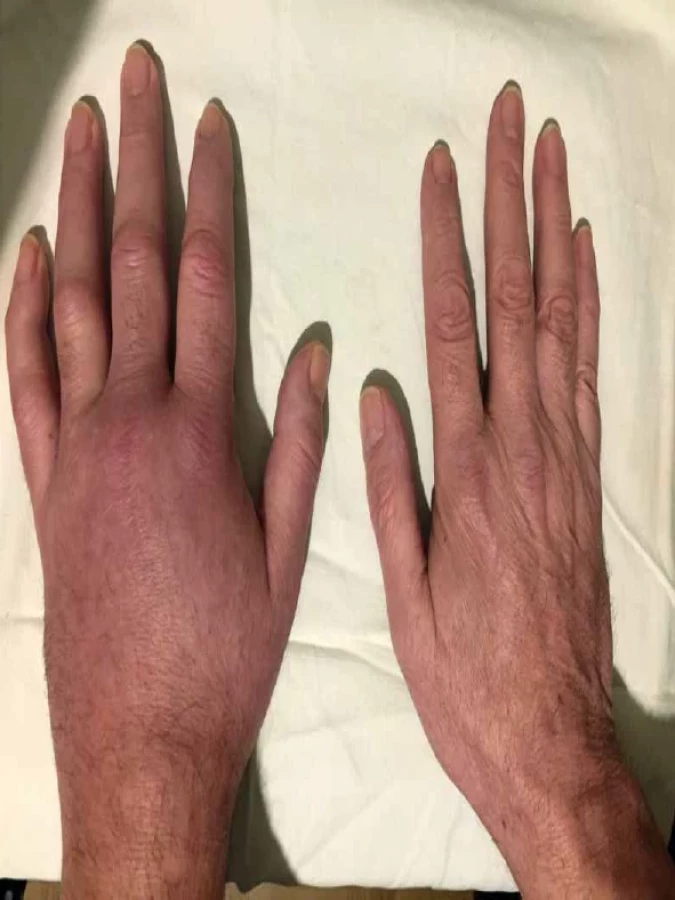
Chondritida v rámci syndromu VEXAS postihuje v podstatě výhradně chrupavky nosu a ušního boltce. Pacienti často naplní klasifikační kritéria pro recidivující polychondritidu [12]. Nicméně postižení chrupavek dýchacích cest a kostochondritida popisované u recidivující polychondritidy byly u syndromu VEXAS pozorovány pouze ojediněle [8]. V porovnání s pacienty s recidivující polychondritidou mají pacienti se syndromem VEXAS častěji kožní projevy, horečky a postižení oka. Postižení oka může být u syndromu VEXAS jak nezánětlivé, ve formě periorbitálního otoku, tak zánětlivé. Zánět může postihnout v podstatě jakoukoliv část oka ve formě unilaterální skleritidy, episkleritidy nebo uveitidy [18]. Klinicky významná je predispozice k tromboembolickým příhodám, často rekurentním [7]. Plicní postižení se může projevit dušností a/nebo kašlem, může být ale i zcela asymptomatické, nicméně změny na HRCT nacházíme téměř u všech pacientů [5].
LÉČBA
Syndrom VEXAS má obvykle progresivní charakter, na léčbu odpovídá jen částečně a je spojený s vysokým rizikem infekčních, tromboembolických a maligních komplikací a rizikem orgánových selhání [11, 17]. Zánětlivé projevy onemocnění obvykle dobře odpovídají na léčbu středními dávkami glukokortikoidů, ale u většiny pacientů onemocnění časem progreduje. Imunosupresivní léčiva jako metotrexát, azathioprin, cyklosporin a mykofenolát mofetil nebývají účinná a jejich použití se nedoporučuje. Z dostupných biologických léčiv se nejslibněji jeví blokáda IL-6 [6]. Údaje o účinnosti léčby blokátory IL-1 jsou nejednoznačné, navíc aplikace anakinry může vést k závažným lokálním reakcím. Řada kazuistických sdělení svědčí o dobré účinnosti inhibitorů JAK, především ruxolitinibu [6, 10]. Zejména u nemocných se závažnými hematologickými projevy nebo s myelodysplastickým syndromem se užívá léčba nukleosidovým analogem azacytidinem [1] nebo alogenní transplantace krvetvorných buněk [2], které mohou vést k eradikaci mutovaného klonu.
KDY POMÝŠLET NA SYNDROM VEXAS?
Pro syndrom VEXAS zatím nejsou k dispozici žádná klasifikační ani diagnostická kritéria. Vzhledem k velmi heterogenním klinickým projevům je diagnostika složitá. Zásadní je průkaz patogenní varianty UBA1, ten je možný ze vzorku periferní krve. Vzhledem k relativně nízké prevalenci, by ale měl být podmínkou pro indikaci genetického vyšetření vysoký stupeň klinického podezření. Lze použít například níže uvedený algoritmus (obr. 8).
![Diagnostický algoritmus při podezření na VEXAS syndrom [12]](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image/bebd50abee38193cce04db00a3d33687.png)
Kožní projevy jsou u nemocných se syndromem VEXAS velmi časté a časné – bývají přítomné již v počátku onemocnění, dermatologové proto hrají důležitou roli při diagnostice tohoto vzácného, ale závažného onemocnění. Podezření na syndrom VEXAS bychom měli mít především u mužů středního a vyššího věku s hematologickými projevy (cytopenie) a zánětlivými příznaky (FW, CRP) a s kožními projevy typu kožní vaskulitidy, neutrofilní dermatózy či chondritidy chrupavky ucha či nosu [17]. Pro diagnózu je nutný průkaz patogenní varianty UBA1 genu, tu je možné testovat na několika pracovištích v ČR. Při klinickém podezření na syndrom VEXAS můžete kontaktovat Revmatologický ústav na e-mailové adrese VEXAS@revma.cz [16].
Pro lepší porozumění chorobě, včasnou diagnostiku, prognózu i účinnější léčbu je zásadní mezioborová spolupráce dermatologů, hematologů, imunologů a revmatologů.
Tab. 1 Klinická charakteristika a četnost kožních lézí ve skupině 37 pacientů se syndromem VEXAS (upraveno dle [16])
|
Popis |
makulopapuly a noduly |
100% |
|
pustuly |
13% |
|
|
vesikuly/buly |
11% |
|
|
livedo reticularis |
8% |
|
|
livedo racemosa |
8% |
|
|
Tvar |
oválné |
97% |
|
semilunární |
32% |
|
|
Barva |
růžové |
76% |
|
červené |
73% |
|
|
nafialovělé/ lividní |
49% |
|
|
Velikost |
≥10 mm |
89% |
|
<10 mm |
11% |
|
|
Lokalizace |
trup |
81% |
|
ruce |
86% |
|
|
nohy |
84% |
|
|
obličej |
30% |
|
|
Symptomy |
bolest |
35% |
|
svědění |
24% |
|
|
Průběh |
relabující (exacerbace/ remise) |
81% |
|
perzistující |
11% |
Zdroje
- AALBERS, A. M., VAN DAELE, P. L. A., DALM, V. A. S. H. et al. Long-term genetic and clinical remissions after cessation of azacitidine treatment in patients with VEXAS syndrome. Hemasphere, 2024, 8(8).
- ALI, S. B., GURNARI, C. Allogenic haematopoietic stem cell transplantation in VEXAS: A review of 33 patients. Clinical Rheumatology, 2024, 43(11).
- ARGOBI, Y. VEXAS syndrome with cutaneous nodules. Dermatology Reports, 2022, 14(2).
- BECK, D. B., FERRADA, M. A., SIKORA, Kelsey A. et al. Somatic mutations in UBA1 and severe adult-onset autoinflammatory disease. The New England Journal of Medicine, 2020, 383(27), p. 2628–2638.
- BORIE, R., DEBRAY, M. P., GUEDON, A. F. et al. Pleuropulmonary manifestations of vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic (VEXAS) syndrome. Chest, 2023, 163(3), p. 575–585.
- BOYADZHIEVA, Z., RUFFER, N., KÖTTER, I., KRUSCHE, M. How to treat VEXAS syndrome: A systematic review on effectiveness and safety of current treatment strategies. Rheumatology (Oxford), 2023, 62(11), p. 3518–3525.
- GEORGIN-LAVIALLE, S., TERRIER, B., GUEDON, A. F. et al. Further characterization of clinical and laboratory features in VEXAS syndrome: Large-scale analysis of a multicentre case series of 116 French patients. British Journal of Dermatology, 2022, 186(3), p. 564–574.
- GUERRERO-BERMÚDEZ, C. A., CARDONA-CARDONA, A. F., ARIZA-PARRA, E. J. et al. Vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic syndrome (VEXAS syndrome) with prominent supraglottic larynx involvement: A case-based review. Clinical Rheumatology, 2022, 41(11), p. 3565–3572.
- HARRISON, I., COHEN, O., YI, L. et al. Neutrophilic eccrine hidradenitis associated with VEXAS syndrome: A case report. International Journal of Dermatology, 2024, 63(6), p. 759–761.
- HEIBLIG, M., FERRADA, M. A., KOSTER, M. T. et al. Ruxolitinib is more effective than other JAK inhibitors to treat VEXAS syndrome: A retrospective multicenter study. Blood, 2022, 140(8), p. 927–931.
- KIRINO, Y., MAEDA, A., ASANO, T. et al. Low remission rates and high incidence of adverse events in a prospective VEXAS syndrome registry. Rheumatology (Oxford), 2024.
- KOSTER, M. J., LASHO, T. L., OLTEANU, H. et al. VEXAS syndrome: Clinical, hematologic features and a practical approach to diagnosis and management. American Journal of Hematology, 2024, 99(2), p. 284–299.
- KOSTER, M. J., WARRINGTON, K. J. VEXAS within the spectrum of rheumatologic disease. Seminars in Hematology, 2021, 58(4), p. 218–225.
- LODA, A., BRANDSMA, J. H., VASSILEV, I., SERVANT, N., LOOS, F., AMIRNASR, A. et al. Genetic and epigenetic features direct differential efficiency of Xistmediated silencing at X-chromosomal and autosomal locations. Nat Commun., 2017, 8(1), p. 1–16.
- SAAD, A. J., PATIL, M. K., CRUZ, N. et al. VEXAS syndrome: A review of cutaneous findings and treatments in an emerging autoinflammatory disease. Experimental Dermatology, 2024, 33(3).
- SYCHROVSKÁ, T., STIBŮRKOVÁ, B., MANN, H. Syndrom VEXAS: vzácné a závažné autoinflamatorní onemocnění. Česká revmatologie, 2024, 4(32), p. 152–161.
- TAN, IJ., FERRADA, M. A., AHMAD, S. et al. Skin Manifestations of VEXAS Syndrome and Associated Genotypes. JAMA Dermatology, 2024, 160(8), p. 822–829.
- ZAKINE, E., SCHELL, B., BATTISTELLA, M. et al. UBA1 variations in neutrophilic dermatosis skin lesions of patients with VEXAS syndrome. JAMA Dermatology, 2021, 157(11), p. 1349–1354.
- ZAKINE, E., PAPAGEORGIOU, L., BOURGUIBA, R. et al. Clinical and pathological features of cutaneous manifestations in VEXAS syndrome: A multicenter retrospective study of 59 cases. Journal of the American Academy of Dermatology, 2023, 88(4), p. 917–920.
Poděkování
Tato práce je podpořena grantem AZV ČR č. NU23-1000160 a projektem podpory pro specifický vysokoškolský výzkum SVV 260 764.
Střet zájmů
Autorka v souvislosti s tématem práce v posledních 12 měsících nespolupracovala s žádnou farmaceutickou firmou.
Do redakce došlo dne 13. 6. 2025.
Adresa pro korespondenci:
MUDr. Tereza Sychrovská
Na Slupi 450/4 128 00 Praha 2
e-mail: sychrovska@revma.cz
Štítky
Dermatologie Dětská dermatologieČlánek vyšel v časopise
Česko-slovenská dermatologie

2025 Číslo 4
- Nástroje k hodnocení závažnosti psoriázy v klinických studiích
- Systémová léčba atopické dermatitidy konečně i u dětí
- Inovace v hojení ran: krytí Zetuvit Plus Silicone Border pro optimální management exsudátu z ran
Nejčtenější v tomto čísle
- Ekzém rukou 1. část: subtypy, epidemiologie, diagnostika, prevence a léčba
- Syndrom VEXAS: autoinflamatorní onemocnění dospělého věku s kožními projevy. Přehledný článek
- Generalizované folikulárně vázané papuly
- Zápisnica zo zasadania výboru a dozornej rady Slovenskej dermatovenerologickej spoločnosti zo dňa 14. 3. 2025 o 10.00 hod. v salóniku hotela Turiec Martin
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy