
Autoprotilátky u systémových onemocnění pojiva a ANCA asociovaných vaskulitid, jejich vztah k intersticiálním plicním procesům a prognóze
Autoantibodies in systemic connective tissue disease and ANCA-associated vasculitis, their relationship to interstitial lung diseases and prognosis
Lung disease, including interstitial lung disease (ILD), is a frequent complication of systemic connective tissue disorders (CTD) and ANCA (anti-neutrophil cytoplasmic antibody) associated vasculitis (AAV). Pulmonary manifestations are prognostic factor of CTDs and vasculitis. Autoantibodies assessment is a part of differential diagnosis algorithm of lung diseases. Autoantibodies importance is mainly clinical-diagnostic. Using detection of some autoantibodies it is possible to determine prognosis of lung involvement, especially in CTDs.
Key words:
autoantibodies – connective tissue disease – interstitial lung diseases – prognosis – vasculitis
Autoři:
Martina Doubková 1; Jana Pokorná 2
Působiště autorů:
Klinika nemocí plicních a tuberkulózy LF MU a FN Brno, pracoviště Bohunice
1; Laboratoř autoimunitní diagnostiky Dermatovenerologického oddělení LF MU a FN Brno, pracoviště Bohunice
2
Vyšlo v časopise:
Vnitř Lék 2017; 63(2): 98-106
Kategorie:
Přehledné referáty
Souhrn
Plicní postižení včetně intersticiálního plicního postižení (IPP) jsou častou komplikací systémových onemocnění pojiva (SOP) a ANCA asociovaných vaskulitid (AAV – antineutrophil cytoplasmic antibody-associated vasculitis). Plicní komplikace jsou prognostickým faktorem SOP a vaskulitid. Vyšetření autoprotilátek je součástí diferenciálně diagnostického algoritmu plicních onemocnění. Jejich význam je převážně klinicko-diagnostický. Pomocí některých autoprotilátek, zejména u SOP, je možné se vyjádřit k prognóze plicní nemoci.
Klíčová slova:
autoprotilátky – intersticiální plicní procesy – prognóza – systémové onemocnění pojiva – vaskulitidy
Úvod
Plicní postižení včetně intersticiálního plicního postižení (IPP) jsou častou komplikací systémových onemocnění pojiva (SOP) a ANCA asociovaných vaskulitid (AAV – antineutrophil cytoplasmic antibody-associated vasculitis). Obsahem práce je klinický a prognostický význam autoprotilátek u SOP a ANCA vaskulitid ve vztahu k plicnímu nálezu, zejména intersticiálnímu plicnímu postižení.
I. Autoprotilátky u systémových onemocnění pojiva
Systémová onemocnění pojiva (SOP), starším názvem kolagenózy, jsou heterogenní skupina imunologicky podmíněných zánětlivých onemocnění projevujících se multiorgánovým postižením [1]. U pacientů se SOP bývá postižena jakákoliv část respiračního ústrojí: dýchací cesty (bronchiektazie, bronchiolitidy), pleura, plicní parenchym (intersticiální plicní postižení – IPP, difuzní alveolární krvácení, aspirační pneumonie, pneumonie/oportunní infekce, malignity), plicní cévní řečiště, dýchací svaly (bránice). IPP jsou heterogenní skupina převážně chronických nenádorových a neinfekčních onemocnění alveolů a intersticia. Fenotypy postižení plicního intersticia jsou pro dané SOP nespecifické a obvykle kopírují fenotypy idiopatických intersticiálních pneumonií (IIP). IIP jsou skupinou akutních, subakutních a chronických intersticiálních plicních nemocí charakterizovaných různým stupněm zánětu a různým stupněm plicní fibrózy [2,3]. Postižení plicního intersticia v rámci SOP je nejčastěji přítomno u systémové sklerodermie (SSc), revmatoidní artritidy (RA), polymyozitidy/dermatomyozitidy (PM/DM), Sjögrenova syndromu (SS), systémového lupus erythematodes (SLE), smíšených chorob pojiva (mixed connective tissue disease – MCTD), nediferencovaných SOP [1]. Frekvence výskytu IPP u SOP, klinický obraz, prognóza a odpověď na léčbu závisí na histopatologickém obrazu (akutní intersticiální pneumonie – AIP, běžná intersticiální pneumonie – UIP, nespecifická intersticiální pneumonie – NSIP, lymfocytární intersticiální pneumonie – LIP, obliterující bronchiolitida s organizující se pneumonií – BOOP/OP, deskvamativní intersticiální pneumonie – DIP, difuzní alveolární hemoragie – DAH, granulomatózní plicní postižení, fibrobulózní postižení a syndrom kombinované fibrózy a emfyzému – CPFE – combined pulmonary fibrosis and emphysema) a na typu SOP [4].
Autoprotilátky u SOP vznikají proti vlastním orgánovým strukturám. Proces jejich vzniku může být vyvolán modifikací vlastních struktur např. zánětem, který vede ke ztrátě imunologické tolerance. Přítomnost anti-topoizomerázy I (anti-Scl-70) u SSc je spojována s vývojem IPP a je častějším nálezem u pacientů s difuzním kožním postižením, nekoreluje však s tíží fibrózy [5–7]. Anticentromerové protilátky jsou častěji spojeny s limitovanou kožní sklerodermií a plicním vaskulárním onemocněním (plicní arteriální hypertenze) a nebývají většinou spojovány s těžkou intersticiální plicní fibrózou [6]. Výskyt IPP – PM/DM bývá přítomen u 73 % nemocných s pozitivitou anti-histidyl-tRNA syntetázy (anti-Jo-1) [8]. Přítomnost protilátek anti-Jo-1 u nemocných s PM/DM znamená vyšší riziko IPP v rámci antisyntetázového syndromu [8].
Patogenetický význam autoprotilátek u SOP
Jejich etiopatogenetický význam není plně objasněn. Základním mechanizmem vzniku plicního postižení u SOP je pravděpodobně autoimunitní reakce, která vede k tvorbě autoprotilátek zaměřených proti vlastním strukturám tělesných tkání se vznikem imunokomplexů. Není to ale jen mechanizmus imunokomplexů podílejících se na vzniku plicního postižení u SOP, protilátkami navozená cytotoxicita a buněčný typ přecitlivělosti se rovněž účastní na rozvoji plicního postižení u SOP [8]. Pod vlivem cytokinů, prozánětlivých a fibroproliferativních, může docházet i k nakupení a proliferaci fibroblastů a k rozvoji plicní fibrózy [9,10].
Laboratorní diagnostika – detekce autoprotilátek a jejich prognostický význam u SOP
Imunologická vyšetření zahrnují průkaz antinukleárních protilátek (ANA) s různými typy fluorescence (homogenní, granulární, nukleolární, centromery aj) a další typizaci antinukleárních protilátek (proti dvouvláknové jaderné deoxyribonukleové kyselině ds-DNA, proti nukleozomům a histonům) a proti extrahovatelným jaderným antigenům ENA [anti-SS-A (Ro), anti-SS-B (La), anti-Sm, ribonukleoproteinu U1-RNP, anti-DNA-topoizomeráze Scl-70, anti-histidyl-tRNA syntetázy]. Dále vyšetřujeme přítomnost revmatoidního faktoru (RF), protilátky proti cyklickému citrulinovému peptidu anti-CCP, anti-Mi-2, „lupus antikoagulans“, antifosfolipidové protilátky. Některé autoprotilátky jsou vysoce specifické a jsou součástí diagnostických kritérií. Základní panel vyšetřovaných autoprotilátek u SOP a detekovatelné protilátky u jednotlivých SOP uvádí tab. 1 a tab. 2.
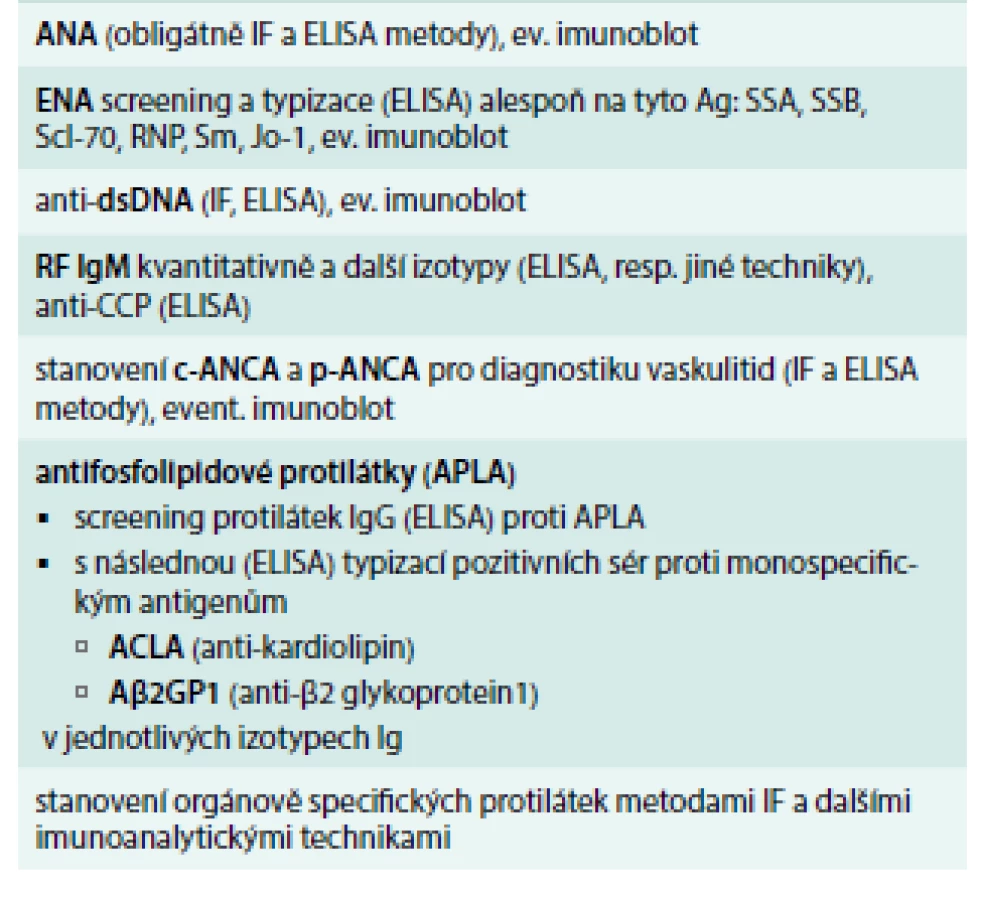
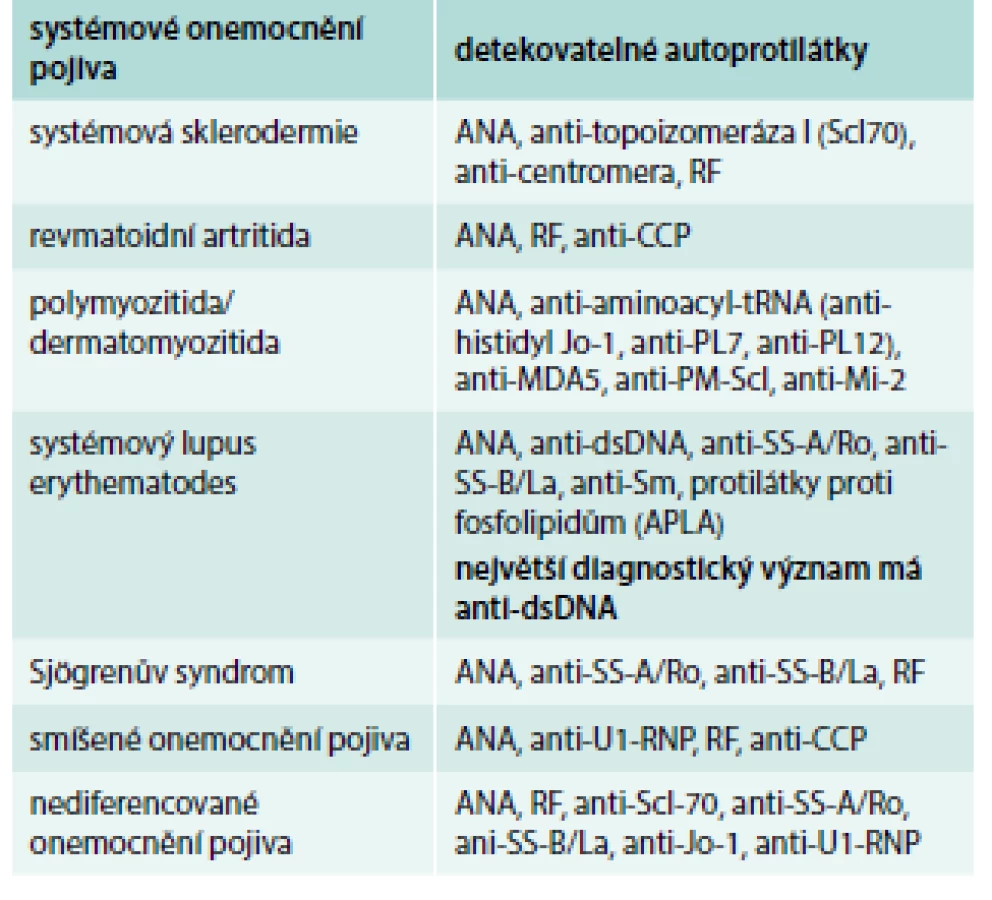
Systémový lupus erythematodes (SLE)
Více než 100 autoprotilátek může být nalezeno u SLE [11]. Protilátky ds-DNA a anti-Smith antigen jsou specifické pro SLE [11,12]. Korelace plicního postižení se žádnou z autoprotilátek nebyla prokázána, i když v jedné studii byl popsán vztah anti-SS-A a chronické intersticiální pneumonitidy a U1-RNP a IPP [11,13,14].
Revmatoidní artritida (RA)
Nalézáme pozitivitu RF, ACPA – protilátky proti citrulinovým peptidům (anticyclic citrullinated peptide anti-CCP, antimutated citrullinated vimentin anti-MCV, fibrinogen), anti-hnRNP (heterogeneous nuclear ribonucleoproteins antibodies). RF a anti-CCP byly prediktorem plicního postižení u RA, včetně IPP [11,15].
Sjögrenův syndrom (SS)
Nacházíme pozitivitu anti-Ro (anti-SS-A) (antiSjögren’s syndrome related antigen A) a anti-La (anti-SS-B). Obě autoprotilátky jsou prediktory plicního postižení [11,16,17].
Systémová sklerodermie (SSc)
Anti-topoizomeráza I (anti-Scl-70) je asociována s plicní fibrózou. Přítomnost Scl-70 značí horší prognózu a ve 20–65 % se vyvine difuzní forma onemocnění. Protilátky proti centromerám se vyskytují u CREST syndromu (C – kalcinóza, R – Raynaudův fenomén, E – porucha motility ezofagu, S – sklerodaktylie, T – teleangiektazie) u 60–80 % a jsou spojeny s plicní hypertenzí a jen vzácně s IPP [7,11]. Anti-RNP (ribonucleoproteins antibodies): anti-U11/U12 RNP mají vztah k IPP [11,18]. AECA (antiendothelial cell antibodies) se mohou vázat na fibroblasty a podílet se na vzniku fibrózy [11,19].
Dermatomyozitida/polymyozitida (DM/PM)
Anti-aminoacyl-tRNA syntetázy (anti-ARS) jsou detekovány u PM/DM a korelují s přítomností IPP [11,20,21]. Mezi anti-aminoacyl-tRNA syntetázy patří: anti-Jo-1 (histidyl-tRNA syntetáza), anti-threonyl (anti-PL7), anti-PL7, anti-PL12, anti-EJ, anti-OJ, anti-KS a jiné t-RNA syntetázy. Přítomnost anti-SRP (anti-signal recognition particle): anti-7SL RNA znamená obecně špatnou prognózu a tito pacienti mají nejkratší přežití [20]. Pacienti s anti-Mi-2 jsou z hlediska mortality na stejné úrovni, jako ti bez přítomnosti této autoprotilátky [21]. Antisyntetázový syndrom je kombinací myozitidy, artritidy, intersticiálního plicního procesu, Raynaudova fenoménu, „prstů mechanika“, horečky a pozitivity některé z antisyntetázových autoprotilátek [22]. IPP je častěji přítomno u pacientů s pozitivitou anti-Jo-1, anti-PL12, anti-KS. U nemocných s pozitivitou anti-PL7 nebyla popsána asociace s rozvojem rychle zhoršujícího se IPP [23]. Anti-MDA5 (melanoma differentiation-associated protein 5) mají ze všech autoprotilátek nejintimnější asociaci s rapidně progredujícím plicním postižením intersticia u dermatomyozitidy, respektive především u amyopatické dermatomyozitidy [24,25]. V 10–15 % případů může být DM/PM projevem paraneoplastického syndromu (karcinom ovarií, plic, kolorektálního karcinomu). Přítomnost anti-TIF-1 (transcriptional intermediary factor 1) je asociována v 50 % s tumorem [26].
Smíšené onemocnění pojiva
MCTD (Mixed Connective Tissue Disease) je kombinací SLE/SSC/PM. Pozitivita anti-U1-RNP a anti-SS-A (anti-Ro52) má vztah k IPP [27].
Diferenciální diagnostika IPP a SOP
Udává se, že 15–20 % pacientů s chronickou IPP může mít frustní (plně nevyjádřenou) formu SOP nebo se může SOP následně plně vyvinout [8]. Homma et al sledovali 68 nemocných s IIP po dobu 1–11 let. U 13 nemocných (19 %) v průběhu sledování došlo k vývoji SOP [28]. Nediferencovaná onemocnění pojiva mají rysy několika SOP, ale nesplňují kritéria jednotlivých SLE, SSc, PM/DM, SS (tab. 3). Překryvné syndromy splňují kritéria 2 nebo více SOP [11,29,30]. Intersticiální pneumonie s autoimunitními rysy (interstitial pneumonitis with autoimmune features – IPAF) zahrnují jak pacienty s IPP při nediferencovaném onemocnění pojiva, tak skupinu nemocných s IPP, kteří nesplňují diagnostická kritéria systémové choroby pojiva, a přesto vykazují známky autoimunitního onemocnění [31].

II. Protilátky proti cytoplazmě neutrofilních granulocytů u systémových vaskulitid
Vaskulitidy jsou heterogenní skupinou onemocnění charakterizovaných zánětem cévní stěny. Na základě převažující velikosti a typu postižených cév se dělí na vaskulitidy malých, středních a velkých cév. Podle revidované nomenklatury vaskulitid z roku 2012 jsou vaskulitidy malých cév dále děleny na ANCA-asociované vaskulitidy (antineutrophil cytoplasmic antibody-associated vasculitis – AAV) a imunokomplexové vaskulitidy [32,33]. ANCA-asociované vaskulitidy jsou vzácná, ale velmi závažná až život ohrožující onemocnění. Mezi AAV řadíme 3 klinické entity: granulomatózu s polyangiitidou (GPA, dříve nazývaná Wegenerova granulomatóza), mikroskopickou polyangiitidu (MPA) a eozinofilní granulomatózu s polyangiitidou (EGPA, dříve zvaná Churgův-Straussové syndrom) [32,33]. Jedná se o primární systémové nekrotizující vaskulitidy postihující cévy malého až středního průsvitu, které jsou asociovány ve většině případů s pozitivitou ANCA. GPA je charakterizována nekrotizující vaskulitidou s extravaskulárním granulomatózním zánětem, který není přítomen u MPA. Ačkoliv plicní a ledvinné postižení je běžné u obou GPA i MPA, granulomatózní postižení horních a dolních cest dýchacích se jeví být přítomno zejména u GPA. Plicní postižení včetně intersticiálního je častou komplikací ANCA vaskulitid a ovlivňuje jejich prognózu.
Protilátky proti cytoplazmě neutrofilů (ANCA) jsou sérologické markery přítomné u některých idiopatických systémových vaskulitid, cév malého průsvitu, jako je GPA nebo MPA, a méně u EGPA. Termín ANCA-asociované vaskulitidy je používán pro primární vaskulitidy, u kterých jsou přítomny cirkulující ANCA protilátky. První popis ANCA je z roku 1982 u nemocných s nekrotizující glomerulonefritidou a symptomy systémové vaskulitidy [34]. V roce 1985 byla ANCA popsána u Wegenerovy granulomatózy [35].
Patogeneze ANCA u AAV
ANCA jsou přítomny u AAV, u jiných vaskulitid (Kawasakiho nemoc nebo obrovskobuněčná temporální arteriitida) nejsou detekovány nebo jen v malých titrech [36]. Tyto autoprotilátky pravděpodobně nebudou hlavní příčinou AAV, nicméně mohou zesilovat zánětlivé procesy u některých pacientů a přispívat k orgánovému postižení. Současné studie ukazují na úlohu ANCA a buněčné imunity v patogenezi AAV. ANCA, T buňky, makrofágy a jiné složky imunitního systému jsou zapojeny do patogeneze onemocnění. V patogenezi onemocnění se mohou rovněž uplatňovat vlivy genetické a zevního prostředí [37–41]. Je popsána genetická vazba mezi MPO-ANCA a HLA-DQ a PR3-ANCA a HLA-DP [37,42].
Na patogenezi ANCA-asociovaných vaskulitid se pravděpodobně podílí více faktorů. Jedním z možných vysvětlení je předpoklad především vlivu virové nebo bakteriální (stafylokokové) infekce, která aktivuje antigen prezentující buňky (antigen presenting cells – APC) k produkci IL23 (interleukin 23). Z naivních T-lymfocytů pod vlivem IL23 vznikají Th17 (T helper 17). Th17 způsobuje uvolnění prozánětlivých cytokinů TNFα (tumor necrotizing factor α) a IL1β z makrofágů, které stimulují neutrofilní granulocyty k expresi PR3 nebo MPO na svém povrchu. To vede k tvorbě ANCA, které reagují s těmito antigeny a způsobují aktivaci, adhezi, degranulaci neutrofilů, uvolnění kyslíkových radikálů a lyzosomálních enzymů vedoucích k poškození cévní stěny. Zhoršená funkce Treg a zvýšené uvolnění BAFF (B-cell activating factor) z aktivovaných neutrofilních granulocytů vede k diferenciaci B buněk v plazmatické buňky produkující ANCA [38].
Laboratorní diagnostika – detekce ANCA a jejich význam u ANCA vaskulitid
ANCA byly poprvé in vitro detekovány technikou nepřímé imunofluorescence (indirect immunofluorescence – IIF), a to na etanolem fixovaných lidských granulocytech. Jednotlivé složky cytoplazmatických granulí se zde redistribuují podle svého elektrického náboje. IF (immunofluorescence) – reakce se jeví jako difuzní, granulární, tj. cytoplazmatická (cytoplasmic pattern, c-ANCA) a perinukleární (perinuclear pattern, p-ANCA), obr. 1 a obr. 2. Klinické indikace testování ANCA uvádíme v přehledu [46,47].


Klinické indikace testování ANCA protilátek:
- glomerulonefritidy
- plicní hemoragie, pulmorenální syndrom
- kožní vaskulitida se systémovými příznaky
- mnohočetné plicní noduly
- destruktivní onemocnění horních cest dýchacích
- dlouhodobá sinusitida a otitida
- subglotická tracheální stenóza
- mononeuritis multiplex
- retroorbitální masa
- plicní fibróza se systémovými příznaky
- episkleritida, uveitida, retinální vaskulitida
Komplikované je zejména přiřazování a hodnocení p-ANCA obrazu nalézaného na etanolem fixovaných granulocytech. Tento typ fluorescence mohou vykazovat také některé protilátky proti jaderným antigenům – ANA. Pozitivita p-ANCA IF pozorovaná na etanolem fixovaných neutrofilních granulocytech je pak výsledkem pouhého „překryvu“ se souběžnou přítomností protilátek ANA. ANA protilátky specifické pro buňky myeloidní řady jsou označovány jako NANA (neutrophil associated nuclear antibodies) nebo GS-ANA (granulocyte-specific anti-nuclear antibodies) a vyskytují se velmi často u zánětlivých onemocnění gastrointestinální soustavy. Před vyslovením závěru, že se jedná o p-ANCA, je důležité verifikovat (respektive vyloučit) je na substrátu granulocytů fixovaných formalinem: p-ANCA IF – obraz/ETOH (etanol) se zde mění na typickou c-ANCA – fluorescenci, zatímco protilátky ANA žádný IF – vzor v cytoplazmě v prostředí s formalinem netvoří. Jednotlivé ANCA lze detekovat a kvantifikovat (stanovením jejich koncentrace) metodou ELISA (enzyme-linked immunosorbent assay), do současné doby ale nebyly rozpoznány všechny antigenní cíle ANCA [43–45]. Cílovým antigenem pro c-ANCA bývá ve většině případů autoprotilátka zaměřená proti serinové proteináze-3 (PR3-ANCA). Nicméně neplatí, že pozitivita c-ANCA rovná se pozitivita anti-PR3-ANCA. PR3 je protein patřící mezi serinové proteázy a je obsažen nejvíce v azurofilních granulech neutrofilních granulocytů. Nejčastějším cílovým antigenem při pozitivitě p-ANCA je protilátka proti myeloperoxidáze (MPO–ANCA). Také v tomto případě platí, že p-ANCA nerovná se pozitivita anti-MPO-ANCA. MPO je protein, enzym charakterizovaný baktericidní aktivitou. Antigeny PR3 a MPO jsou lokalizovány v primárních azurofilních granulech polymorfonukleárů a monocytů (obr 3). Pod obrazem p-ANCA se mohou objevit protilátky proti laktoferinu, katepsinu, lyzozymu, elastáze a BPI (bacterial permeability increasing protein). V běžné praxi tak ANCA zjišťujeme IF s antigen-specifickou konfirmací ELISA, tato kombinace metod dovoluje až 99% specifitu [46–48]. Další metody průkazu ANCA zahrnují chemiluminiscenci, nové metody ELISA [49,50].

Méně častá je přítomnost cytoplazmatických atypických (atypické c-ANCA), a atypických ANCA (a-ANCA, atypické ANCA nálezy perinukleární a cytoplazmické IF). Cílové antigeny jsou obvykle rozdílné od PR3 a MPO a jsou namířeny proti komponentám jádra, cytosolu nebo granulím. Patří sem laktoferin, BPI, katepsin G, elastáza, lyzozym, kataláza, α enoláza, aktin, histon. Jejich přítomnost není všeobecně vztahována k AAV [49]. Souhrnně se jedná o IgG-ANCA protilátky izotypu IgG (třída imunoglobulin IgG) [49]. Senzitivita IF-ANCA pro AAV je vysoká (≥ 80 %), zatímco specifita je nízká (< 80%). To je způsobeno přítomností p-ANCA (nejsou pozitivní protilátky proti MPO, atypická p-ANCA) např. u idiopatických střevních onemocnění, systémových onemocnění pojiva a také interferencí s ANA. P-ANCA pozitivitu/MPO–ANCA negativitu můžeme vidět u zánětlivých střevních onemocnění, systémových onemocnění pojiva, primární sklerotizující cholangiitidy [47]. Pozitivitu PR3-ANCA vidíme i u idiopatického střevního onemocnění (ulcerózní kolitida) [49], v takových případech je většinou pozitivita PR3-ANCA spojena s atypickou p-ANCA (dle IF) [49]; c-ANCA byly zaznamenány také u infekčních onemocnění [46,47]; c-ANCA/PR3-ANCA jsou zejména u pacientů s GPA, zatímco p-ANCA/MPO-ANCA jsou více u MPA a CSS [49]. U 10–20 % pacientů s klasickou WG můžeme prokázat p-ANCA (MPO–ANCA), a dokonce pacienti s MPA nebo EGPA mohou být c-ANCA (PR3-ANCA) pozitivní [46]. ANCA jsou detekovány u 70–90 % aktivních a generalizovaných GPA, ale pouze u 40–50 % lokoregionálních forem [49]. U EGPA jsou ANCA pozitivní jen u 40 % případů [49]. Ve studii Sinico et al EGPA ANCA pozitivita korelovala s jistým orgánovým postižením (glomerulonefritida, mononeuritis multiplex, purpura a plicní hemoragie). ANCA negativita u EGPA byla spojena s vyšší prevalencí srdečního onemocnění a plicního postižení [51].
Souvislost mezi IPP a ANCA, zejména pozitivita MPO, byla popsána v řadě publikací [52,53]. Kromě toho byly také zaznamenány případy pacientů s IPP a ANCA pozitivitou, ale bez jiných projevů systémové vaskulitidy. IPP (až typu plicní fibrózy) může předcházet klinické manifestaci AAV. Ve většině studií pacienti AAV s IPP mají horší prognózu než pacienti bez IPP [54].
Atypická ANCA-IF s cílovým antigenem neutrofilní elastázy je zjistitelná při kokainem indukovaném orgánovém postižení, které může napodobovat klinicky GPA a u 30–40 % je popisována pozitivita anti-PR3 [55]. Další ANCA, jiné než PR3 a MPO, které by mohly být přítomny u vaskulitid, jsou předmětem dalších výzkumů [56,57].
Detekce ANCA má diagnostický a prognostický význam, nicméně vyšetření ANCA pozitivity v čase má nejistý prognostický význam v určení aktivity onemocnění nebo jeho vzplanutí [57–63]. V práci Tomasson et al zjistili, že opakované měření ANCA v měsíčních intervalech během remise má omezený význam pro předpověď relapsu AAV [61]. Verstockt et al ve své práci popisují nízkou předpovědní hodnotu rizika relapsu jen na základě rutinního sledování ANCA v čase bez klinického korelátu. Pravděpodobnost relapsu se zvyšuje při současném klinickém podezření a nárůstu nebo znovuobjevení ANCA. U pacientů s pozitivitou ANCA v době diagnózy je návrat nemoci méně pravděpodobný při negativitě ANCA po dobu sledování [63]. PR3-ANCA ve srovnání s MPO-ANCA jsou spojeny s vyšší mortalitou a jsou nezávislým prediktorem relapsu [63–66].
Diferenciálně diagnostický význam ANCA a jejich uplatnění v klinické praxi shrnuje tab. 4.

Význam ANCA (anti-neutrophil cytoplasm antibodies) k průkazu AAV v klinické praxi – shrnutí
- detekce ANCA má diagnostický a prognostický význam pro AAV v době diagnózy, nicméně pozitivita ANCA bez klinického korelátu a histopatologického potvrzení není diagnostická pro AAV
- ANCA negativita u pacientů s klinickými a histopatologickými příznaky nevylučuje diagnózu AAV
- vyšetření ANCA v čase jsou využívána pro monitoraci aktivity onemocnění, ale mají omezený význam pro predikci relapsů v době remise, proto by jejich zvýšení nebo přetrvávání bez klinického korelátu nemělo být důvodem k rozhodnutí o zahájení léčby
- pozitivita ANCA protilátek: 75–95 % pacientů s GPA nebo MPA, 40–70 % u EGPA
- PR3-ANCA oproti MPO-ANCA jsou nezávislým prediktorem relapsu onemocnění a mortality
- ANCA pozitivita se vyskytuje u infekčních, zánětlivých střevních onemocnění, maligních hematologických onemocnění, cystické fibrózy a při užívání kokainu
- přítomnost ANCA u intersticiálních plicních postižení, bez klinického korelátu AAV, je rizikem vývoje AAV v čase
- ANCA specifita má pravděpodobně vliv na fenotyp klinického onemocnění
- ANCA pozitivita koreluje u EGPA s jistým orgánovým postižením (glomerulonefritida, mononeuritis multiplex, purpura a plicní hemoragie)
- ANCA negativita u EGPA bývá spojena s vyšší prevalencí srdečního onemocnění a plicního postižení
Závěr
Plicní postižení je běžnou komplikací u nemocných se SOP a vaskulitidami. Plicní komplikace jsou prognostickým faktorem SOP a vaskulitid. Vyšetření autoprotilátek je součástí diferenciálně diagnostického algoritmu plicních onemocnění. Pomocí některých autoprotilátek, zejména u SOP, je možné se na základě jejich detekce vyjádřit k prognóze nemoci.
MUDr. Martina Doubková, Ph.D.
doubkovamartina@seznam.cz
Klinika nemocí plicních a tuberkulózy LF MU a FN Brno,
pracoviště Bohunice
www.fnbrno.cz
Doručeno do redakce 11. 8. 2016
Přijato po recenzi 9. 1. 2017
Zdroje
1. Doubková M, Skřičková J. Intersticiální plicní postižení a systémová onemocnění pojiva. Stud Pneumol Phtiseol 2013; 73(2): 76–83.
2. Antoniou KM, Margaritopoulos GA, Tomassetti S et al. Interstitial lung disease. Eur Respir Rev 2014; 23(131): 40–54. Dostupné z DOI: <http://dx.doi.org/10.1183/09059180.00009113>.
3. Antoniou KM, Margaritopoulos G, Economidou F et al. Pivotal clinical dilemmas in collagen vascular diseases associated with interstitial lung involvement. Eur Respir J 2009; 33(4): 882–896. Dostupné z DOI: <http://dx.doi.org/10.1183/09031936.00152607>.
4. [American Thoracic Society; European Respiratory Society]. American Thoracic Society, European Respiratory Society. American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias. This joint statement of the American Thoracic Society (ATS), and the European Respiratory Society (ERS) was adopted by the ATS board of directors, June 2001 and by the ERS Executive Committee, June 2001. Am J Respir Crit Care Med 2002; 165(2): 277–304. Erratum in Am J Respir Crit Care Med 2002; 166(3): 426.
5. Vencovský J. Jak pomáhá vyšetřování autoprotilátek v diagnostice a hodnocení autoimunitních revmatických chorob? Vnitř Lék 2006; 52(7–8): 697–701.
6. Tzelepis GE, Toya SP, Moutsopoulos HM. Occult connective tissue diseases mimicking idiopathic interstitial pneumonias. Eur Respir J 2008; 31(1): 11–20. Dostupné z DOI: <http://dx.doi.org/10.1183/09031936.00060107>.
7. Self SE. Autoantibody testing for autoimmune disease. Clin Chest Med 2010; 31(3): 415–422. Dostupné z DOI: <http://dx.doi.org/10.1016/j.ccm.2010.04.001>.
8. Bradley B, Branley HM, Egan JJ et al. [British Thoracic Society Interstitial Lung Disease Guideline Group, British Thoracic Society Standards of Care Committee; Thoracic Society of Australia; New Zealand Thoracic Society; Irish Thoracic Society]. Interstitial lung disease guideline: the British thoracic in collaboration with the thoracic society of Australia and New Zealand and the Irish thoracic society. Thorax 2008; 63(Suppl 5): v1-v58. Dostupné z DOI: <http://dx.doi.org/10.1136/thx.2008.101691>. Erratum in Thorax 2008; 63(11): 1029.
9. Vašáková M, Polák J, Matěj R. Postižení plic u systémových onemocnění. In: Vašáková M, Polák J, Matěj R. Intersticiální plicní procesy. Od etiopatogeneze přes radiologický obraz k histopatologické diagnóze. Maxdorf: Praha 2011 : 285–312. ISBN 978–80–7345–251–3.
10. Lamblin C, Bergoin C, Saelens T et al. Interstitial lung diseases in collagen vascular disease. Eur Respir J Suppl 2001; 32 : 69s-80s.
11. Bonela F, Costabel U. Biomarkers in connective tissue disease-associated interstitial lung disease. Semin Respir Crit Care Med 2014; 35(2): 181–200. Dostupné z DOI: <http://dx.doi.org/10.1055/s-0034–1371527>.
12. Papiris SA, Kagouridis K, Bouros D. Serologic evaluation in idiopathic interstitial pneumonias. Curr Opin Med 2012; 18(5): 433–440. Dostupné z DOI: <http://dx.doi.org/10.1097/MCP.0b013e3283560840>.
13. Groen H, ter Borg EJ, Postma DS et al. Pulmonary function in systemic lupus erythematosus is related to distinct clinical, serologic, and nailfold capillary patterns. Am J Med 1992; 93(6): 619–627.
14. Boulware DW, Hedgpeth MT. Lupus pneumonitis and anti-SSA(Ro) antibodies. J Rheumatol 1989; 16(4): 479–481.
15. Aubart F, Crestani B, Nicaise-Roland P et al. High levels of anti-cyclic citrullinated peptide autoantibodies are associated with co-occurrence of pulmonary diseases with rheumatoid arthritis. J Rheumatol 2011; 38(6): 979–982. Dostupné z DOI: <http://dx.doi.org/10.3899/jrheum.101261>.
16. Yazisiz V, Arslan G, Ozbudak IH et al. Lung involvement in patients with primary Sjögren’s syndrome: what are the predictors? Rheumatol Int 2010; 30(10): 1317–1324. Dostupné z DOI: <http://dx.doi.org/10.1007/s00296–009–1152–8>.
17. Davidson BK, Kelly CA, Griffiths ID. Ten year follow up of pulmonary function in patients with primary Sjögren‘s syndrome. Ann Rheum Dis 2000; 59(9): 709–712.
18. Fertig N, Domsic RT, Rodriguez-Reyna T et al. Anti-U11/U12 RNP antibodies in systemic sclerosis: a new serologic marker associated with pulmonary fibrosis. Arthritis Rheum 2009; 61(7): 958–965. Dostupné z DOI: <http://dx.doi.org/10.1002/art.24586>.
19. Hill MB, Phipps JL, Cartwright RJ et al. Antibodies to membranes of endothelial cells and fibroblasts in scleroderma. Clin Exp Immunol 1996; 106(3): 491–497.
20. Mimori T, Imura Y, Nakashima R et al. Autoantibodies in idiopathic inflammatory myopathy: an update on clinical and patophysiological significance. Curr Opin Rheumatol 2007; 19(6): 523–529.
21. Love LA, Leff RL, Fraser DD et al. A new approach to the classification of idiopathic inflammatory myopathy: myositis-specific autoantibodies define useful homogeneous patient groups. Medicine (Baltimore) 1991; 70(6): 360–374.
22. Mimori T, Nakashima R, Shida N et al. Interstitial lung diseases in myositis: clinical subsets, biomarkers, and treatment. Curr Rheumatol Rep 2012; 14(3): 264–274. Dostupné z DOI: <http://dx.doi.org/10.1007/s11926–012–0246–6>.
23. Hamaguchi Y, Fujimoto M, Mathushita T et al. Common and distinct of clinical features in adult patients with anti-aminoacyl-tRNA synthetase antibodies: heterogeneity within the syndrome. PLos One 2013; 8(4): e60442. Dostupné z DOI: <http://dx.doi.org/10.1371/journal.pone.0060442>.
24. Teruya A, Kawamura K, Ichikado K et al. Successful polymyxin B hemoperfusion treatment associated with serial reduction of serum anti-CADM-140/MDA5 antibody levels in rapidly progressive interstitial lung disease with amyopathic dermatomyositis. Chest 2013; 144(6): 1934–1936. Dostupné z DOI: <http://dx.doi.org/10.1378/chest.13–0186>.
25. Cavagna L, Nuño L, Scirè CA et al. [AENEAS (American, European NEtwork of Antisynthetase Syndrome) collaborative group]. Clinical Spectrum Time Course in Anti Jo-1 Positive Antisynthetase Syndrome: Results From an International Retrospective Multicenter Study. Medicine (Baltimore) 2015; 94(32): e1144. Dostupné z DOI: <http://dx.doi.org/10.1097/MD.0000000000001144>.
26. Hoshino K, Muro Y, Sugiura K et al. Anti-MDA5 and anti-TIF1-gamma antibodies have clinical significance for patients with dermatomyositis. Rheumatology 2010; 49(9): 1726–1733. Dostupné z DOI: <http://dx.doi.org/10.1093/rheumatology/keq153>.
27. Bodolay E, Szekanecz Z, Dévényi K et al. Evaluation of interstitial lung diseasse in mixed connective tissue disease (MCTD). Rheumatology 2005; 44(5): 656–661.
28. Homma Y, Ohtsuka Y, Tanimura K et al. Can interstitial pneumonia as the sole presentation of collagen vascular diseases be differentiated from idiopathic interstitial pneumonia? Respiration 1995; 62(5): 248–251.
29. Corte TJ, Copley SJ, Desai SR et al. Significance of connective tissue disease features in idiopathic interstitial pneumonia. Eur Respir J 2012; 39(3): 661–668. Dostupné z DOI: <http://dx.doi.org/10.1183/09031936.00174910>.
30. Cottin V. Idiopathic interstitial pneumonias with connective tissue diseases features: A review. Respirology 2016; 21 : 245–258.
31. Fischer A, Antoniou KM, Brown KK et al. An official European Respiratory Society/American Thoracic Society research statement: interstitial pneumonia with autoimmune features. Eur Respir J 2015; 46(4): 976–987. Dostupné z DOI: <http://dx.doi.org/10.1183/13993003.00150–2015> a <http://www.ers-education.org/guidelines.aspx>. [19. 5. 2016].
32. Jennette JC, Falk RJ, Andrassy K et al. Nomenclature of systemic vasculitides. Proposal of an international consensus conference. Arthritis Rheum 1994; 37(2): 187–192.
33. Jennette JC, Falk RJ, Bacon PA et al. 2012 Revised international Chapel Hill Consensus Conference Nomenclature of Vasculitides. Artrhitis Rheum 2013; 65(1): 1–11. Dostupné z DOI: <http://dx.doi.org/10.1002/art.37715>.
34. Davies DJ, Moran JE, Niall JF et al. Segmental necrotising glomerulonephritis with antineutrophil antibody: possible arbovirus aetiology? Br Med J (Clin Res Ed) 1982; 285(6342): 606.
35. Van der Woude FJ, Rasmussen N, Lobatto S et al. Autoantibodies against neutrophils and monocytes: tool for diagnosis and marker of disease activity in Wegener’ granulomatosis. Lancet 1985; 1(8426): 425–429.
36. Basu N, Watts R, Bajema I et al. EULAR points to consider in the development of classification and diagnostic criteria in systemic vasculitis. Ann Rheum Dis 2010; 69(10): 1744–1750. Dostupné z DOI: <http://dx.doi.org/10.1136/ard.2009.119032>.
37. Griffith ME, Coulthart A, Pusey CD. T cell responses to myeloperoxidase (MPO) and proteinase 3 (PR3) in patients with systemic vasculitis. Clin Exp Immunol 1996; 103(2): 253–258.
38. Kallenberg CG. Pathogenesis and treatment of ANCA-associated vasculitides. Clin Exp Rheumatol 2015; 33(4 Suppl 92): S11-S14.
39. Pendergraft WF, Preston GA, Shah RR et al. Autoimmunity is triggered by cPR-3(105–201), a protein complementary to human autoantigen proteinase-3. Nat Med 2004; 10(1): 72–79.
40. Harper L, Cockwell P, Adu D et al. Neutrophil priming and apoptosis in anti-neutrophil cytoplasmic autoantibody-associated vasculitis. Kidney Int 2001; 59(5): 1729–1738.
41. Radford DJ, Savage CO, Nash GB. Treatment of rolling neutrophils with antineutrophil cytoplasmic antibodies causes conversion to firm integrin-mediated adhesion. Arthritis Rheum 2000; 43(6): 1337–1345.
42. Lyons PA, Rayner TF, Trivedi S et al. Genetically distinct subsets within ANCA-associated vasculitis. N Engl J Med 2012; 367(3): 214–223. Dostupné z DOI: <http://dx.doi.org/10.1056/NEJMoa1108735>.
43. Savige J, Davies D, Falk RJ et al. Antineutrophil cytoplasmic antibodies and associated diseases: a review of the clinical and laboratory features. Kidney Int 2000; 57(3): 846–862.
44. Savige JA, Paspaliaris B, Silvestrini R et al. Immunofluorescent patterns associated with antineutrophil cytoplasmic antibodies (ANCA) and their differentiation from other antibodies. J Clin Pathol 1998; 51(8): 568–575.
45. Savige J, Gillis D, Benson E et al. International consensus statement on testing and reporting of antineutrophil cytoplasmic antibodies (ANCA). Am J Clin Pathol 1999; 111(4): 507–513.
46. Radice A, Sinico RA. Antineutrophil cytoplasmic antibodies (ANCA). Autoimmunity 2005; 38(1): 93–103.
47. Radice A, Bianchi L, Sinico RA. Anti-neutrophil cytoplasmic autoantibodies: methodological aspects and clinical signifikance in systemic vasculitis. Autoimmunity Rev 2013; 12(4): 487–495. Dostupné z DOI: <http://dx.doi.org/10.1016/j.autrev.2012.08.008>.
48. Savige J, Dimech W, Fritzler M et al. [International group for Consensus Statement on Testing and Reporting of Antineutrophil Cytoplasmic Antibodies (ANCA)]. Addendum to the International Consensus Statement on testing and reporting of neutrophil cytoplasmatic antibodies. Quality control guidelines, comments, and recommendations for testing in other autoimmune diseases. Am J Clin Path 2003; 120(3): 312–318.
49. Shulte-Pelkum J, Radice A, Norman GL et al. Novel clinical and diagnostic aspects of antineutrophil cytoplasmatic antibodies. J Immunol Res 2014; 2014 : 185416. Dostupné z DOI: <http://dx.doi.org/10.1155/2014/185416>.
50. Csernok E, Moosing F. Current and emerging techniques for ANCA detection in vasculitis. Nat Rev Rheumatol 2014; 10(8): 494–501. Dostupné z DOI: <http://dx.doi.org/10.1038/nrrheum.2014.78>.
51. Sinico RA, Di Toma L, Maggiore U et al. Prevalence and clinical significance of antineutrophil cytoplasmic antibodies in Churg-Strauss syndrome. Arthritis Rheum 2005; 52(9): 2926–2935.
52. Katsumata Y, Kawaguchi Y, Yamanaka H. Interstitial lung disease with ANCA-associated vasculitis. Clin Med Insights Circ Respir Pulm Med 2015; 9(Suppl 1): 51–56. Dostupné z DOI: <http://dx.doi.org/10.4137/CCRPM.S23314>.
53. Kagiyama N, Takayanagi N, Kanauchi T et al. Antineutrophil cytoplasmic antibody-positive conversion and microscopic polyangiitis development in patients with idiopathic pulmonary fibrosis. BMJ Open Respir Res 2015; 2(1): e000058. Dostupné z DOI: <http://dx.doi.org/10.1136/bmjresp-2014–000058>.
54. Arulkumaran N, Periselneris N, Gaskin G et al. Interstitial lung disease and ANCA-associated vasculitis: a retrospective observational cohort study. Rheumatology 2011; 50(11): 2035–2043. Dostupné z DOI: <http://dx.doi.org/10.1093/rheumatology/ker236>.
55. Wiesner O, Russell KA, Lee AS et al. Antineutrophil cytoplasmic antibodies reacting with human neutrophil elastase as a diagnostic marker for cocaine-induced midline destructive lesions but not auto-immune vasculitis. Arthritis Rheum 2004; 50(9): 2954–2965.
56. Lally L, Spiera R. Current landscape of antineutrophil cytoplasmic antibody-associated vasculitis. Rheum Dis Clin N Am 2015; 41(1): 1–19, vii. Dostupné z DOI: <http://dx.doi.org/10.1016/j.rdc.2014.09.003>.
57. Kain R, Tadema H, McKinney EF et al. High prevalence of autoantibodies to hLAMP2 in anti-neutrophil cytoplasmic antibody-associated vasculitis. J Am Soc Nephrol 2012; 23(3): 556–566. Dostupné z DOI: <http://dx.doi.org/.1681/ASN.2011030273>.
58. Finkielman JD, Merkel PA, Schroeder D et al. [WGET Research Group]. Antiproteinase 3 antineutrophil cytoplasmic antibodies and disease activity in Wegener granulomatosis. Ann Intern Med 2007; 147(9): 611–619.
59. Birck R, Schmitt WH, Kaelsch IA et al. Serial ANCA determinations for monitoring disease aktivity in patients with ANCA-Associated vasculitis: systematic review. Am J Kidney Dis 2006; 47(1): 15–23.
60. Kerr GS, Fleisher TA, Hallahan CW et al. Limited prognostic value of changes in antineutrophil cytoplasmic antibody titer in patients with Wegener’s granulomatosis. Arthritis Rheum 1993; 36(3): 365–371.
61. Tomasson G, Grayson PC, Mahr AD et al. Value of ANCA measurements during remission to predict a relapse of ANCA-associated vasculitis--a meta-analysis. Rheumatology (Oxford) 2012; 51(1): 100–109. Dostupné z DOI: <http://dx.doi.org/10.1093/rheumatology/ker280>.
62. Boomsma MM, Stegeman CA, van der Leij MJ et al. Prediction of relapses in Wegener’s granulomatosis by measurement of antineutrophil cytoplasmic antibody levels: a prospective study. Arthritis Rheum 2000; 43(9): 2025–2033.
63. Verstockt B, Bossuyt X, Vanderschueren S et al. There is no benefit in routinely monitoring ANCA titres in patiens with granulomatosis with polyangiitis. Clin Exp Rheumatol 2015; 33(2 Suppl 89): S72-S76.
64. Lionaki S, Blyth ER, Hogan SL et al. Classification of antineutrophil cytoplasmic autoantibody vasculitides. The role of ANCA specifity for myeloperoxidase or proteinase 3 in disease recognition and prognosis. Arthritis Rheum 2012; 64(10): 3452–3462. 10.1002/art.34562
65. Walsh M, Flossmann O, Berden A et al. [European Vasculitis Study Group]. Risk factors for relapse of antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheum 2012; 64(2):542–548.
66. Mahr A, Katsahian S, Varet H et al. [French Vasculitis Study Group (FVSG) and the European Vasculitis Society (EUVAS)]. Revisiting the classification of clinical phenotypes of anti-neutrophil cytoplasmic antibody associated vasculitis – a cluster analysis. Ann Rheum Dis 2013; 72(6): 1003–1010. Dostupné z DOI: <http://dx.doi.org/10.1136/annrheumdis-2012–201750>.
Štítky
Diabetologie Endokrinologie Interní lékařstvíČlánek vyšel v časopise
Vnitřní lékařství

2017 Číslo 2
- Inovace v hojení ran: krytí Zetuvit Plus Silicone Border pro optimální management exsudátu z ran
- Ivabradin zlepšuje kvalitu života starších pacientů se srdečním selháním
- Fixní kombinace kandesartan/amlodipin v terapii arteriální hypertenze
- Clopidogrel je v prevenci kardiovaskulárních příhod přínosnější než kyselina acetylsalicylová
Nejčtenější v tomto čísle
- Autoprotilátky u systémových onemocnění pojiva a ANCA asociovaných vaskulitid, jejich vztah k intersticiálním plicním procesům a prognóze
- Karcinóm prištítneho telieska
- Dlouhodobě působící inzuliny v léčbě diabetu 2. typu a jejich postavení v rámci současného léčebného algoritmu
- Akútne obličkové poškodenie: aktuálny komplexný prehľad
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy