
Hematopoietic stem cell transplantation in patients with chronic granulomatous disease in the Czech Republic and Slovakia (2007–2019)
Objective: Chronic Granulomatous Disease (CGD) is a rare primary immunodeficiency with X-linked or autosomal recessive inheritance caused by defects in the genes encoding phagocyte oxidase subunits of the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase complex. This complex is responsible for the respiratory burst inside the cell, which leads to the killing of microbes ingested by phagocytic cells. NADPH oxidase also plays key role in regulating immunity and therefore CGD is also characterized by autoimmune and autoinflammatory manifestations. Hematopoietic stem cell transplant (HSCT) is the only definitive curative treatment. Here we present the outcome of patients with CGD who from 2007 till 2018 underwent allogeneic HSCT at pediatric transplant centers in Prague and Bratislava.
Methods: Fourteen patients with chronic granulomatous disease received first hematopoietic stem cell transplant at a medium age of 6.4 years (1.0–17.8). CYBB gene mutation (X-linked) was identified in thirteen of them. The stem cell source was bone marrow (n=10) or peripheral stem cells (n=4) from a human leukocyte antigen identical unrelated (n=13) or healthy sibling (n=1) donor.
Results: Primary engraftment was obtained in all 14 patients. Five patients, who later lost the primary graft, were successfully re-grafted following second HSCT. One patient died six months after HSCT due to cerebral bleeding following repeated surgery for cerebral aspergilloma, one developed secondary acute myeloid leukemia eight years after HSCT and 12 months ago underwent second transplant. 13/14 patients are alive with median follow-up 6.1 (range 0.1–12.2) years after the last HSCT.
Conclusions: Allogeneic HSCT were safely performed without serious transplant-related complications. Early indication, pre-transplant organ involvement, donor selection and conditioning regimens remain the key points with the aim to decrease the risk of late graft failure and need for re-HSCT.
Keywords:
Quality of life – infection – chronic granulomatous disease – hematopoietic stem cell transplant
Autoři:
P. Sedláček 1; D. Dóczyová 2; A. Janda 3; P. Říha 1; R. Formánková 1; P. Keslová 1; I. Boďová 2; T. Sýkora 2; V. Urdová 2; T. Freiberger 4,5; P. Švec 2; P. Čižnár 6; J. Starý 1; J. Horáková 2
Působiště autorů:
Klinika dětské hematologie a onkologie 2. LF UK a FN Motol, Praha
1; Klinika detskej hematológie a onkologie, Národný ústav detských chorôb, Bratislava, Slovensko
2; Ústav imunologie 2. LF UK a FN Motol, Praha
3; Centrum kardiovaskulární a transplantační chirurgie, Brno
4; Ústav klinické imunologie a alergologie, Lékařská fakulta Masarykovy univerzity, Brno
5; Detská klinika, Národný ústav detských chorôb a LF Univerzity Komenského, Bratislava, Slovensko
6
Vyšlo v časopise:
Čes-slov Pediat 2020; 75 (3): 154-162.
Kategorie:
Původní práce
Souhrn
Cíl studie: Chronická granulomatózní nemoc (CGD) je vzácná primární imunodeficience s X-vázanou nebo autosomálně recesivní dědičností způsobená defekty v genech kódujících podjednotky fagocytárních oxidáz z komplexu nikotinamid adenin dinukleotid fosfát (NADPH) oxidázy. Tento komplex je v buňce zodpovědný za respirační vzplanutí, což vede k usmrcení mikrobů požitých fagocytovými buňkami. NADPH oxidáza také hraje klíčovou roli v regulaci imunity, a proto je CGD též charakterizována autoimunitními a autoinflamačními projevy. Transplantace hematopoetických kmenových buněk (HSCT) je jedinou definitivní kurativní léčebnou metodou. Uvádíme zde výsledky pacientů s CGD, kteří podstoupili v letech 2007–2019 alogenní HSCT v dětských transplantačních centrech v Praze a Bratislavě.
Metodika: Čtrnáct pacientů s chronickou granulomatózou podstoupilo první transplantaci hematopoetických kmenových buněk v mediánu věku 6,4 let (1,0–17,8). Genová mutace genu CYBB (X-vázaná dědičnost) byla identifikována u třinácti z nich. Zdrojem kmenových buněk byla kostní dřeň (n = 10) nebo periferní kmenové buňky (n = 4) HLA identického nepříbuzného dárce (n = 13) nebo zdravého HLA identického sourozence (n = 1).
Výsledky: U všech 14 pacientů po transplantaci došlo k primárnímu přihojení štěpu. Pět pacientů, kteří později odhojili primární štěp, úspěšně podstoupili druhou transplantaci. Jeden pacient zemřel šest měsíců po HSCT v důsledku cerebrálního krvácení po opakovaném chirurgickém zákroku pro aspergilom mozku, u jiného pacienta byla diagnostikována sekundární akutní myeloidní leukémie osm let po HSCT a před 12 měsíci podstoupil druhou transplantaci. 13/14 pacientů žije s mediánem sledování 6,1 (0,1–12,2) let po poslední HSCT.
Závěry: Alogenní HSCT byly provedeny bez závažnějších komplikací souvisejících s transplantací. Klíčovým bodem této terapie zůstává včasná indikace, stupeň postižení orgánů před transplantací a výběr vhodných dárců a přípravných režimů s cílem snížit riziko pozdních odhojení štěpů a potřebu re-transplantací.
Klíčová slova:
chronická granulomatózní nemoc – transplantace kmenových buněk krvetvorby – kvalita života – infekce
ÚVOD
Chronická granulomatózní nemoc (CGD) je vzácným vrozeným onemocněním, způsobeným geneticky podmíněným defektem některé z komponent enzymu NADPH-oxidázy, manifestujícím se zvýšeným výskytem hnisavých bakteriálních a plísňových infekcí a tvorbou granulomů. Mutace v genu CYBB je příčinou v Evropě a ve Spojených státech amerických nejčastější a nejzávažnější (X-vázané) formy CGD, změny v genech CYBA, NCF1, NCF2 a NCF4 vedou k autosomálně recesivní (AR-CGD) formě nemoci [1].
Na podkladě genetického defektu dochází ve fagocytujících buňkách k poruše tvorby reaktivních forem kyslíku a regulace pH s následnou nedostatečnou aktivací proteolytických enzymů. Důsledkem je snížená mikrobicidní aktivita a porucha schopnosti likvidovat fagocytovaný materiál. Pacienti trpící tímto onemocněním jsou proto náchylní k závažným život ohrožujícím infekcím. K nejčastějším původcům infekcí u pacientů s CGD patří Staphylococcus aureus, Aspergillus fumigatus, Nocardia spp., Burkholderia spp, Klebsiella spp., Salmonella spp a Serratia spp. [2].
Odhadovaná incidence CGD v České a Slovenské republice dle získaných údajů z retrospektivní analýzy období od roku 1956 do roku 2008 je 1 : 280 000 živě narozených dětí [3].
Onemocnění se obvykle projevuje už v prvních letech života. K častým příznakům patří přetrvávající rýma, zánět kůže, zánět sliznic úst, vzácněji se může vyskytnout chorioretinitida či ložiska subretinálních granulomů. Postižení gastrointestinálního traktu se projevuje průjmy, bolestí břicha a tvorbou perianálních abscesů. Děti jsou v té době často vyšetřovány pro podezření na Crohnovu nemoc či jiný nespecifický střevní zánět. CGD je spojena s rozvojem granulomatózních reakcí v kůži, plicích, kostech a lymfatických uzlinách. K dalším projevům patří infekce kostí, obstrukce močopohlavního traktu a/nebo trávicího traktu, tvorba jaterních a mozkových abscesů a opožděný růst [4].
K prevenci závažných infekcí musí pacienti s CGD celoživotně užívat profylakticky antibiotika a antimykotika. Díky účinné a cílené antimikrobní profylaxi se významně změnil přirozený průběh onemocnění i doba celkového přežívání. Kvalita života ale nadále s věkem klesá, navíc dochází ke zhoršování celkového zdravotního stavu i z jiných než bezprostředně infekčních příčin. Do popředí se dostávají autoinflamatorní a autoimunní projevy onemocnění, které probíhají nepříznivě především u X-vázané formy CGD [5].
Transplantace hematopoetických kmenových buněk (HSCT) je nadále jedinou definitivní a kauzální léčbou defektu imunity i autoimunních a autoinflamatorních projevů onemocnění [6]. Její provedení by nemělo být odkládáno, pokud je k dispozici vhodný identický dárce v rodině či v registrech dobrovolných dárců. V posledním desetiletí navíc dochází k významnému zlepšení prognózy transplantovaných pacientů vlivem lepšího výběru vhodných dárců a volbou vhodných předtransplantačních přípravných režimů [7–9].
Počáteční zkušenosti s užitím genové terapie u pacientů bez vhodného dárce k alogenní HSCT s použitím retrovirových vektorů byly neuspokojivé, především z důvodu vysokého rizika nahodilé integrace do chromosomální DNA s následnou up-regulací exprese proto-onkogenů. V současné době se využívá mnohem bezpečnějších lentivirových vektorů. Další velmi nadějnou metodou genové terapie ke korekci mutace genu je využití CRISPR/Cas9 (clustered regularly interspaced short palindromic repeats/CRISPR-associated protein 9) techniky. Zatím je ale genová terapie nadále považovaná za experimentální léčbu dostupnou v rámci klinických studií jen velmi omezené skupině pacientů, kteří nemají vhodného dárce k transplantaci [10, 11].
Mutace v genu CYBB, který leží na chromosomu X, je z důvodu nižšího výskytu příbuzenských sňatků příčinou v Evropě a ve Spojených státech amerických nejčastější formy CGD. Významnější delece v oblasti sousedních genů na chromosomu X může mít za následek tzv. Souvislý syndrom delece X-chromosomového genu (Contiguous X gene deletion syndrome). Mutace zasahující do těchto obklopujících genů mohou mít za následek Duchennovu svalovou dystrofii, Retinitis pigmentosa (X-vázaná), deficit ornitin transkarbamylázy, McLeodův syndrom a poruchy v jiných genech vrozeného imunitního systému, jako jsou geny komplementové dráhy [12].
Při intersticiální deleci u pacienta s X-CGD může dojít k deleci genu XK pro Kell antigen, což se projevuje absencí nebo velmi sníženou tvorbou a expresí Kell antigenů na erytrocytární membráně. McLeodův syndrom (MLS) je projev mutace genu XK ovlivňujícího funkci sekundárního podpůrného proteinu pro antigen Kell na povrchu červených krvinek. Souběžně s MLS se může projevit akantocytóza erytrocytů s vyšší tendencí k hemolýze (neuroakantocytóza). MLS je vzácně se vyskytující neurodegenerativní onemocnění periferních nervů i centrální nervové soustavy, krve a některých orgánů. Manifestuje se až mezi 18. – 61. rokem (nejčastěji okolo 35. roku) problémy s ovládáním končetin, ztrátou pohyblivosti, kardiomyopatií, poruchou srdečního rytmu, poruchou hlubokého čití. Postižený začne propadat psychotickým stavům, mění se jeho osobnost. Pacienti s MLS mají v době manifestace onemocnění významně zvýšenou sérovou hladinu kreatinkinázy. HSCT odstraní pouze hematologické projevy (hemolýza) spojené s akantocytózou, pokud je tato vůbec přítomna. Léčba MLS ani dnes není možná a HSCT nemá na manifestaci tohoto onemocnění žádný vliv. Neurologické obtíže se mohou objevit i u heterozygotních nosiček mutace [13–15].
METODY
K transplantaci kmenových buněk krvetvorby jsme v letech 2007 až 2016 indikovali čtrnáct chlapců s chronickou granulomatózou. Jejich klinické obtíže před transplantací shrnuje tabulka 1.

BCG – vakcinační kmen Bacille Calmette Guerin
IBD – zánětlivé střevní onemocnění
Průměrný věk v době diagnózy CGD byl 12 měsíců (rozmezí 6–160). Diagnostika se opírala kromě klinických projevů o funkční vyšetření fagocytární poruchy granulocytů pomocí NBT (nitroblue tetrazolium; mikroskopem pozorovaná redukce bezbarvé tetrazoliové soli na barevné formazány) testu a Burst testu s pomocí průtokové cytometrie, kterým lze posoudit kvantitativně schopnost stimulace fagocytózy granulocytů (po stimulaci E. coli nebo PMA). Následně jsme u všech pacientů v souboru indikovali molekulárně-genetické vyšetření. Téměř u všech (13/14) jsme prospektivně či retrospektivně prokázali přítomnost mutace v genu CYBB na chromosomu X, která je kauzální pro X-vázanou formu CGD. Pouze u jednoho pacienta jsme mutaci v genu CYBB vyloučili a považujeme tedy formu jeho CGD za autosomálně recesivní dědičnou formu. U jeho matky byl funkčně prokázán deficit potvrzující heterozygotní formu onemocnění s klinicky bezpříznakovým nosičstvím. Indikací k transplantaci v uvedeném období byla anamnéza závažných život ohrožujících infekcí a orgánového postižení ve shodě s publikovanými doporučeními [16–18].
První transplantaci hematopoetických kmenových buněk podstoupily děti 4–202 měsíců od diagnózy CGD (medián 3,9 let) v mediánu věku 6,4 roku (1,0–17,8). U některých pacientů byla transplantace provedena déle než 8 let od diagnózy, často z důvodu dlouhého odmítání indikace rodiči. Indikací k transplantaci byly závažné zdravotní obtíže a infekce (tab. 2, tab. 3).


* retransplantace proběhla recentně v říjnu 2019
Zdrojem kmenových buněk při první transplantaci byla kostní dřeň (n = 10) nebo periferní kmenové buňky (n = 4) HLA identického nepříbuzného dárce (n = 13) nebo zdravého HLA identického sourozence (n = 1), tab. 4.
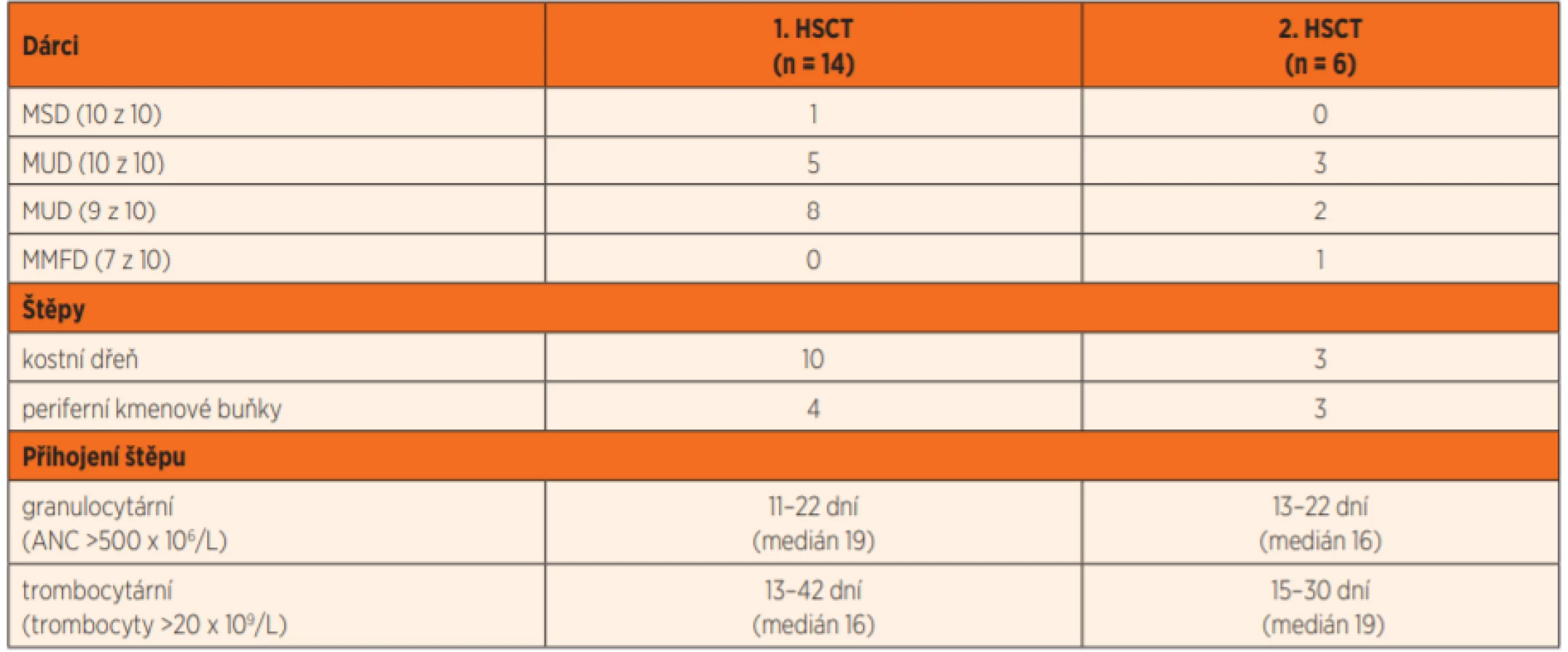
MUD – HLA identický nepříbuzný dárce
MMFD – HLA haploidentický rodinný dárce
HLA – antigeny hlavního histokompatibilního systému
HSCT – transplantace kmenových buněk krvetvorby
10 z 10; 9 z 10; 7 z 10 – míra shody v 10 (resp. v 9, 7) z 10 HLA antigenů I. a II. třídy mezi dárcem a příjemcem
ANC– absolutní počet neutrofilů
Šest pacientů podstoupilo druhou transplantaci v období od roku 2012 do konce roku 2019 ve věku 2–17,6 let (medián 9,7 roku). Indikací k re-transplantaci byla pozdní rejekce prvního štěpu (n = 5) 0,2 – 0,7 – 0,8 – 3,3 – 9,3 let od první transplantace a sekundární akutní myeloidní leukémie v první remisi (n = 1). Dárcem k druhé transplantaci byl HLA identický nepříbuzný dárce z registru (n = 5) a HLA haploidentický zdravý otec (n = 1). Zdrojem kmenových buněk při druhé transplantaci byla kostní dřeň (n = 3) nebo periferní kmenové buňky (n = 3). Charakteristiky přípravných režimů při první i druhé transplantaci jsou uvedeny v tabulce 5.

Flu – fludarabin; Alem – alemtuzumab (Mabcampath); Treo – treosulfan
TT – thiotepa; AML – akutní myeloidní leukémie; LGR – pozdní rejekce štěpu (late graft rejection)
HSCT – transplantace kmenových buněk krvetvorby
VÝSLEDKY
V době posledního sledování žije ve věku 5,9–5,4 (medián 13,5) let celkem 13 ze 14 transplantovaných pacientů 0,1–12,2 (medián 6,1) let od poslední transplantace (tab. 6, graf 1).

HSCT – transplantace kmenových buněk krvetvorby

K primárnímu přihojení trilineární dárcovské krvetvorby došlo řádně u všech pacientů, v granulocytární řadě v mediánu 19 dní (resp. 16 dní u druhé HSCT) a v trombocytární v mediánu 16 dní (resp. 19 dní po druhé HSCT) (tab. 4).
Poslední transplantací bylo dosaženo stabilního buněčného chimérismu v mediánu 5 let po transplantaci u 11/14 pacientů v rozmezí 95–100 % dárcovské krvetvorby. Dlouhodobě velmi nízký dárcovský buněčný chimérismus přetrvával u jednoho pacienta na úrovni 6 % dárcovské krvetvorby. Jedná se o jediného pacienta (P4) ze souboru s AR formou CGD. Opakované funkční testy potvrdily výrazně sníženou funkci granulocytů v pásmu jako při diagnóze. Pacient při farmakologické antimikrobní prevenci neměl žádné projevy autoimunní ani jiné projevy CGD, ale před nasazením profylaxe již začal být zvýšeně nemocný. V současné době jsme proto rozhodli o nutnosti re-transplantace 9,5 roku od první transplantace. Pacient je zatím krátce (1 měsíc) po druhé transplantaci, její efekt proto nemůžeme dosud hodnotit. Mírnější průběh onemocnění při selhávání dárcovské krvetvorby a plné autologní rekonstituci by mohl souviset s geneticky příznivější mutací (tab. 7).

U všech pěti pacientů s plnou autologní rekonstitucí a vymizením funkčních dárcovských granulocytů (36 %; potvrzeno i funkčními NBT a Burst testy) jsme úspěšně provedli druhou transplantaci. U dalšího pacienta (P12) se podařilo rejekci štěpu odvrátit opakovaným (4x) podáním infuze dárcovských lymfocytů (DLI). Transplantace byly komplikovány rozvojem středně závažné (stupeň II) akutní reakce štěpu proti hostiteli (GvHD) u 8 pacientů, u čtyř po léčbě imunosupresivy GvHD odezněla, ale u čtyř pacientů přešla do chronické formy. U jednoho pacienta (P12) jsme diagnostikovali limitovanou formu chronické GvHD až po opakovaném podání dárcovských lymfocytů s cílem předejít pozdní rejekci štěpu. Závažnou extenzivní formu chronické GvHD jsme diagnostikovali pouze u jednoho pacienta (P14), a to až po opakované transplantaci. Je jediným pacientem z celého souboru, který nadále užívá imunosupresi (tacrolimus v monoterapii) (tab. 8).

GvHD – reakce štěpu proti hostiteli
limit. – limitovaná, ext. – extenzivní
Necelých 8 let po první HSCT jsme u pacienta (P5) se stabilním 90% podílem dárcovské krvetvorby, bez infekčních či jiných komplikací, diagnostikovali akutní myeloidní leukémii s fúzním genem MLL/ENL původem z vlastních krvetvorných buněk. Po intenzivní chemoterapii s pomalejším ústupem leukémie dosáhl molekulárně-genetické remise a před dvanácti měsíci podstoupil druhou HSCT. Nyní rok po druhé transplantaci má vysazena imunosupresiva, je bez známek reakce štěpu proti hostiteli a detekujeme u něj pouze dárcovské krvinky. U mutace v genu CYBB není popisován žádný zvýšený výskyt leukémie, považujeme proto výskyt leukémie za náhodný, související možná s předchozí expozicí autologní krvetvorby přípravným režimem před první transplantací.
Jediný pacient (P3) v souboru, který zemřel 6,5 měsíce po HSCT, byl diagnostikován v šesti letech věku a transplantován jako sedmnáctiletý. Důvodem pro souhlas s HSCT po dlouho odkládaném rozhodnutí byla těžká forma zánětlivé enterokolitidy s četnými píštělemi, která by chirurgicky byla řešitelná jen za cenu významné resekce střeva a založením vývodu. U pacienta jsme před HSCT ještě indikovali parciální resekci abscesu jater, kde byla kultivačně potvrzena přítomnost Peptostreptococcus spp. Dlouhodobě v době před transplantací užíval preventivně itrakonazol. Po transplantaci v neutropenii i po přihojení užíval posakonazol v terapeutických hladinách. Při prospektivní monitoraci D+40 po transplantaci byla nově detekována pozitivita galaktomananu v krvi. Po několika dnech začal udávat bolesti hlavy. D+50 se manifestovaly generalizované křeče, vyšetřením na magnetické rezonanci byl v mozku zjištěn opouzdřený absces velikosti 21 x 20 x 14 mm. Přítomnost jiných ložisek mykózy jsme ani při extenzivním cíleném vyšetřování nikde neprokázali. D+63 byl proveden první chirurgický výkon, při kterém byl odstraněn absces, ve kterém byl mikroskopicky prokázán Aspergillus, dourčený metodou PCR jako A. ustus, který byl rezistentní na všechna azolová antimykotika. Zahájili jsme léčbu preparátem amfotericinu (Abelcet), ale bez efektu na hladiny galaktomananu. Léčba byla úspěšná až při změně léčby na lipozomální amfotericin B (Ambisome), což jsme dokumentovali mj. i vymizením pozitivity galaktomananu v krvi i v mozkomíšním moku. Pro sekundárně vzniklý hydrocefalus bylo nutno opakovaně zavádět zevní komorové drenáže. Před propuštěním do ambulantní péče 6,5 měsíce po transplantaci po změně zevní na vnitřní komorovou drenáž se po několika hodinách od výkonu manifestovalo devastující krvácení do CNS. Na sekci v CNS již přítomnost mykózy nebyla prokázána. Spekulujeme, že k infekci CNS muselo dojít již v době před transplantací, protože následně byl řadu týdnů až do manifestace izolován v pokoji s hepa-filtrací na sterilní části transplantační jednotky. Jak je známo u pacientů s CGD, mohou i při invazivní aspergilové infekci vykazovat trvale negativitu sérového galaktomananu. K manifestaci mohlo dojít právě až po přihojení funkčních dárcovských granulocytů [19].
V prvních měsících po transplantaci jsme u čtyř pacientů diagnostikovali imunní cytopenie (autoimunní hemolytická anémie 3x), u řady z nich došlo k reaktivaci virů (CMV, EBV, BKV) většinou s nutností zahájení virostatické léčby. V současné době jsou všichni žijící pacienti bez projevů zvýšené náklonosti k infekcím, bez projevů autoinflamatorních či autoimunních symptomů a bez známek pozdních následků.
V potenciálním riziku jinak velmi vzácného syndromu na sebe navazujících genů na X chromosomu jsou v našem souboru dle dostupných detailních popisů nalezených mutací v oblasti genu CYBB dva ze třinácti (15%) pacientů s X-vázanou formou CGD. U těchto dvou pacientů (P6 a P14) jsme prokázali rozsáhlou deleci postihující i gen XK. Podrobné genetické vyšetření probíhá u třetího pacienta (P4), výsledek není dosud k dispozici. Recentně vyšetřená sérová hladina kreatinkinázy je u nich v normě. Protože mají oba dárcovskou erytropoézu, nemohou mít příznaky akantocytózy. Ta u nich nebyla diagnostikována ani před transplantací, ale v té době po ní nebylo cíleně pátráno. V době posledního sledování (ve věku 8,5 a 14 let) nemají žádné projevy neurologického deficitu. Zřejmě až v průběhu delší doby sledování uvidíme, zda k elevaci sérové CK dochází ještě v asymptomatickém stadiu, nebo až v době prvních neurologických příznaků. Je možné, že v časné asymptomatické fázi CK elevována není. U obou totiž musíme počítat s rozvojem neurodegenerativních příznaků při MLC neuroakantocytóze v dospělosti, i když sama přítomnost mutace v genu XK (fenotypický McLeodův syndrom) neznamená jistotu manifestace neurodegenerativního onemocnění [20].
DISKUSE
Transplantace hematopoetických buněk je jedinou kauzální léčebnou metodou pro pacienty s CGD, která poskytuje dárcovské granulocyty s funkční produkcí NADPH a superoxidového aniontu. Jedná se však i nadále o vysoce rizikový postup, který může být spojen s významnými riziky morbidity a mortality. Příčinou úmrtí po transplantaci bývají komplikace spojené s rozvojem významné reakce štěpu proti hostiteli. Výsledky transplantace jsou značně ovlivněny pečlivou volbou vhodné předtransplantační přípravy, výběrem dárce podle míry shody v systému HLA, věkem a celkovým klinickým stavem pacienta. Dalšími rizikovými faktory jsou možné pozdní účinky chemoterapie a i v našem souboru především vysoké riziko časného i pozdního odhojení štěpu. Proto byla indikace k HSCT historicky vyhrazena pro pacienty s vysoce rizikovým onemocněním a odpovídajícím dárcem. Ale vzhledem k tomu, jak se vyvíjí pokroky v léčbě CGD a HSCT, posunula se indikace k HSCT štěpem vhodného, a to i nepříbuzného dárce ke skupině mladších pacientů dosud bez závažnějšího orgánového postižení, autoinflamatorních a autoimunních projevů či nezvládnutelné infekce [21].
Ve skupině 55 pacientů transplantovaných v letech 1998–2017 pro CGD v dětském transplantačním centru v Newcastle (UK) bylo dosaženo pětiletého celkového přežití (OS) 89 %, ale dokonce 100 % u těch, kteří byli transplantováni ve věku do 5 let. 82 % pacientů mělo prognosticky méně příznivou formu (X-CGD). Přežití pět let bez příhody (EFS) bylo 77%. V době sledování sedm z 55 pacientů (13 %) podstoupilo pro sekundární selhání štěpu druhou transplantaci [22]. Tato zkušenost podporuje časnou indikaci k transplantaci.
Čtyřicet pacientů v mediánu věku 16 let bylo transplantováno po přípravném režimu s alemtuzumabem a redukovanou intenzitou v letech 2007–2015 pro CGD v dětském transplantačním centru v Marylandu (USA). V této skupině bylo dosaženo 3,5letého celkového přežití (OS) 82,5 %. Autoři ale spekulují, že vyšší intenzita přípravného režimu a modifikace podávání alemtuzumabu by snížila riziko smíšeného chimérismu a tedy suboptimálního efektu transplantace [23]. Riziko smíšeného chimérismu, odhojení štěpu či autoimunních projevů po transplantaci, při použití redukované intenzity přípravného režimu v kombinaci s alemtuzumabem, je vyšší u starších dětí se závažnějšími zánětlivými projevy v době před HSCT.
Imunologické konsorcium ve Spojených státech v roce 2019 publikovalo retrospektivní soubor 507 pacientů s diagnózou CGD diagnostikovaných v letech 1953–2016. Pouze 50 z nich (cca 10 %) podstoupilo v letech 1982–2016 alogenní HSCT ve velmi širokém období od stanovení diagnózy (0–45,8 let). Celkové přežití transplantovaných a netransplantovaných se v této studii nelišilo. Výsledky transplantací byly ale výborné ve skupině pacientů transplantovaných ve věku do 14 let a při použití HLA identického sourozeneckého dárce. Významný byl pozitivní efekt transplantace na snížení incidence infekčních komplikací, na střední hodnotu skóre výkonnosti (mean performance score) s nižším výskytem invalidity (disability; 11 % vs. 52 % u netransplantovaných pacientů starších 15 let). Problémem netransplantovaných je postupně narůstající incidence zánětlivých reakcí autoimunního či autoinflamatorního původu. Imunosupresivní léčba těchto projevů dále prohlubuje poruchu imunity a zvyšuje incidenci infekcí a mortality. Na zhoršené kvalitě života se podílí významně i symptomatické zánětlivé postižení střev s tvorbou granulomů a píštělí a rozvoj poruchy plicních funkcí. Celkově horší výsledky přežití v celém souboru měli pacienti s X-vázanou formou CGD (mutace genu CYBB), kterých bylo v souboru 66 % [24].
Prostřednictvím standardizovaných dotazníků (PedsQL a SDQ score) se ve studii z Newcastle vyslovilo 47 rodičů dětí ve věku 3–15 let (21 dětí bylo po transplantaci). Medián věku dětí netransplantovaných byl 9 let, transplantovaných 10 let a ty byly 1–9 let (medián 3 roky) po HSCT. Přežití ve skupině transplantovaných bylo 90 %. Rodiče netransplantovaných dětí udávali signifikantní snížení kvality života ve srovnání se zdravými dětmi a vyšší emoční zátěž. Rodiče transplantovaných udávali parametry kvality života srovnatelné se zdravou populací [25].
Celonárodní francouzská studie se zaměřila na dlouhodobé sledování 80 pacientů s diagnózou CGD. U 71 z nich bylo diagnostikováno celkem 224 zánětlivých epizod, většinou v oblasti trávicího traktu (50 %). Ačkoliv se medián přežití netransplantovaných pacientů v čase významně prodloužil a v posledním desetiletí s účinnou antimykotickou prevencí přesahuje hranici 30 let, jejich kvalita života je nadále suboptimální, a to do významné míry především z důvodu zdravotních obtíží způsobených zánětlivými a autoimunními reakcemi, jejichž léčba vyžaduje dlouhodobé podávání imunosupresiv (kortikoidy, blokátory TNF alfa aj.). Nikoliv jejich incidence, ale především jejich závažnost stoupá s věkem. Porucha růstu je u pacientů s CGD multifaktoriální. Podílí se na ní dlouhodobá léčba kortikoidy, progredující plicní postižení, chronický zánět aj. Porucha růstu se méně projevuje u pacientů s reziduální antimikrobní aktivitou granulocytů. U třetiny dětí došlo k významné redukci školního vzdělání [26].
Imunní cytopenie sice nejsou po transplantacích od nepříbuzných dárců ničím nečekaným či nepředvídatelným, ale jejich četnost v našem souboru se přece jen zdá vyšší, což by mohlo být i ve shodě s publikovanými daty o vyšší incidenci imunních cytopenií v prvních měsících po transplantaci dětí s CGD v jiných centrech [27]. Nepochybně tento jev souvisí s pozvolnou eliminací vystupňované imunity z doby před transplantací a postupnou náhradou zdravým imunitním systémem dárce. I když v periferní krvi detekujeme pouze dárcovské lymfocyty, v orgánech a tkáních dlouho přetrvává signifikantní populace autologních lymfocytů.
V našem souboru zemřel jediný pacient. Domníváme se, že nepřímou příčinou infekční komplikace byla i léta odkládaná transplantace, která proběhla až v době, kdy byl celkový stav pacienta velmi nepříznivý a šance na úspěch transplantace byly úměrně tomu nižší.
Výsledky celkového přežití (OS) pacientů po transplantaci v našem souboru jsou plně srovnatelné s publikovanými daty, ale v našem souboru máme vysoký podíl pacientů, u kterých došlo k pozdnímu odhojení dárcovské krvetvorby a transplantaci bylo nutno opakovat. Riziko pozdního odhojení je u těchto pacientů vyšší, než jak se projevuje standardně například u pacientů transplantovaných pro akutní leukémie, a to navzdory tomu, že medián věku pacientů v době první transplantace je 6,4 let. Jak z našich dat vyplývá, riziko v našem malém souboru zřejmě nesouviselo s výběrem přípravného režimu ve smyslu jeho efektivity v eliminaci krvetvorby (myeloablace). Významný je především dlouhodobý efekt intenzity a trvání počáteční eliminace autologních lymfocytů (lymfoablace). Použití alemtuzumabu vede na jedné straně ke snížení rizika reakce štěpu proti hostiteli, ale na straně druhé zřejmě nadměrně oslabuje dárcovské lymfocyty a umožňuje znovunabytí převahy autologní imunity. Je proto jistě čas k přehodnocení strategie časování, dávkování a volby vhodného produktu séroterapie v době přípravy před podáním alogenního štěpu.
ZÁVĚRY
Včasná transplantace kmenových buněk krvetvorby s pečlivou volbou předtransplantační přípravy a vhodného HLA identického rodinného či nepříbuzného dárce z registru je metodou volby a v současné době přináší vysokou šanci na vyléčení imunodeficitu. Přinejmenším u pacientů s kompletním dárcovským trilineárním chimérismem je transplantace i účinnou prevencí i léčbou autoimunních či autoinflamatorních projevů. U všech pacientů s rozsáhlou delecí zahrnující začátek a/nebo konec genu CYBB je vhodné prospektivní sledování rizikových faktorů syndromu na sebe navazujících genů na X chromosomu.
Transplantace není schopna zabránit rozvoji dalších X-vázaných genetických poruch v blízkosti genu CYBB pro X-CGD. Možné projevy těchto nemocí je potřeba pečlivě diagnostikovat a nezaměňovat je s projevy pozdních následků po HSCT. U pacientů s X-CGD před plánovanou transplantací je vhodné vyloučit na X chromosom vázanou akantocytózu a pečlivě přistupovat k transfuzím erytrocytů u pacientů Kell negativních. Sledování všech pacientů především s X-vázanou formou CGD po i mimo transplantaci by mělo probíhat ve specializovaných centrech s patřičnými zkušenostmi.
Podpořeno projektem MZ ČR koncepčního rozvoje výzkumné organizace 00064203 (FN Motol).
Prvotní výsledky prvních pěti transplantovaných pacientů v ČR, jejichž data jsou zahrnuta i v této práci, již byly publikovány v Čes.-slov. Pediatrii v roce 2012.
Poděkování
Za grafické zpracování dat autoři děkují Aleši Lukšovi z Kliniky dětské hematologie a onkologie.
Došlo: 19. 11. 2019
Přijato: 13. 12. 2010
Prof. MUDr. Petr Sedláček, CSc.
Klinika dětské hematologie a onkologie
2. LF Univerzity Karlovy
a FN Motol
V Úvalu 84
150 06 Praha 5 – Motol
e-mail: petr.sedlacek@fnmotol.cz
Zdroje
1. Chiriaco M, Salfa I, Di Matteo G, et al. Chronic granulomatous disease: Clinical, molecular, and therapeutic aspects. Pediatr Allergy Immunol 2016; 27 (3): 242–253.
2. Slack MA, Thomsen IP. Prevention of infectious complications in patients with chronic granulomatous disease. J Pediatric Infect Dis Soc 2018; 7 (Suppl 1): S25–S30.
3. Janda A, Ciznar P, Dankova E, et al. Patients with chronic granulomatous disease in Czech and Slovak Republic. Alergie 2010; 12 (2): 112–120.
4. Mortaz E, Azempour E, Mansouri D, et al. Common infections and target organs associated with chronic granulomatous disease in Iran. Int Arch Allergy Immunol 2019; 179 (1): 62–73.
5. Magnani A, Brosselin P, Beauté J, et al. Inflammatory manifestations in a single-center cohort of patients with chronic granulomatous disease. J Allergy Clin Immunol 2014; 134 (3): 655–662.
6. Marsh RA, Leiding JW, Logan BR, et al. Chronic granulomatous disease-associated IBD resolves and does not adversely impact survival following allogeneic HCT. J Clin Immunol 2019 Aug 2. doi: 10.1007//s10875-019-00659-8. [Epub ahead of print].
7. Åhlin A, Fasth A. Chronic granulomatous disease – conventional treatment vs hematopoietic stem cell transplantation: an update. Curr Opin Hematol 2015; 22 (1): 41–45.
8. Seger RA, Gungor T, Belohradsky BH, et al. Treatment of chronic granulomatous disease with myeloablative conditioning and an unmodified hemopoietic allograft: a survey of the European experience, 1985–2000. Blood 2002; 100 (13): 4344–4350.
9. Morillo-Gutierrez B, Beier R, Rao K, et al. Treosulfan-based conditioning for allogeneic HSCT in children with chronic granulomatous disease: a multicenter experience. Blood 2016; 128 (3): 440–448.
10. Arnold DE, Heimall JR. A review of chronic granulomatous disease. Adv Ther 2017; 34 (12): 2543–2557.
11. Jafarian A, Shokri G, Shokrollahi Barough M et al. Recent advances in gene therapy and modeling of chronic granulomatous disease. Iran J Allergy Asthma Immunol 2019; 18 (2): 131–142.
12. Watkins CE, Litchfield J, Song E, et al. Chronic granulomatous disease, the McLeod phenotype and the contiguous gene deletion syndrome – a review. Clin Mol Allergy 2011 23; 9 : 13–18.
13. Danek A, Rubio JP, Rampoldi L, et al. McLeod neuroacanthocytosis: genotype and phenotype. Ann Neurol 2001; 50 (6): 755–764.
14. Klempir J, Mikulenkova D, Pisacka M, et al. Differential diagnosis of neuroacanthocytoses. Cesk Slov Neurol N 2009; 72 (1): 24–29.
15. Hönig M, Flegel WA, Schwarz K, et al. Successful hematopoietic stem-cell transplantation in a patient with chronic granulomatous disease and McLeod phenotype sensitized to Kx and K antigens. Bone Marrow Transplant 2010; 45 (1): 209–211.
16. Seger RA. Modern management of chronic granulomatous disease. Br J Haematol 2008; 140 (3): 255–266.
17. Seger RA. Hematopoietic stem cell transplantation for chronic granulomatous disease. Immunol Allergy Clin North Am 2010; 30 (2): 195–208.
18. Janda A, Keslova P, Formankova R, et al. Hematopoietic stem cell transplantation in five patients with CGD in Czech Republic. Čes-slov Pediat 2012; 67 (2): 81–88.
19. Falcone EL, Holland SM. Invasive fungal infection in chronic granulomatous disease: insights into pathogenesis and management. Curr Opin Infect Dis 2012; 25 (6): 658–669.
20. Walker RH, Danek A, Uttner I, et al. McLeod phenotype without the McLeod syndrome. Transfusion 2007 Feb; 47 (2): 299–305.
21. Connelly JA, Marsh R, Parikh S, et al. Allogeneic hematopoietic cell transplantation for chronic granulomatous disease: controversies and state of the art. J Pediatric Infect Dis Soc 2018; 7 (Suppl 1): S31–S39.
22. Lum SH, Flood T, Hambleton S, et al. Two decades of excellent transplant survival for chronic granulomatous disease: a supraregional immunology transplant center report. Blood 2019; 133 (23): 2546–2549.
23. Parta M, Kelly C, Kwatemaa N, et al. Allogeneic reduced-intensity hematopoietic stem cell transplantation for chronic granulomatous disease: A single-center prospective trial. J Clin Immunol 2017 Aug; 37 (6): 548–558.
24. Yonkof JR, Gupta A, Fu P, et al. Role of allogeneic hematopoietic stem cell transplant for chronic granulomatous disease (CGD): A report of the United States Immunodeficiency Network. J Clin Immunol 2019; 39 (4): 448–458.
25. Cole T, McKendrick F, Titman P, et al. Health related quality of life and emotional health in children with chronic granulomatous disease: a comparison of those managed conservatively with those that have undergone haematopoietic stem cell transplant. J Clin Immunol 2013 Jan; 33 (1): 8–13.
26. Dunogué B, Pilmis B, Mahlaoui N, et al. Chronic granulomatous disease in patients reaching adulthood: A Nationwide Study in France. Clin Infect Dis 2017; 64 (6): 767–775.
27. Yanir AD, Hanson IC, Shearer WT, et al. High incidence of autoimmune disease after hematopoietic stem cell transplantation for chronic granulomatous disease. Biol Blood Marrow Transplant 2018 Aug; 24 (8): 1643–1650.
Štítky
Neonatologie Pediatrie Praktické lékařství pro děti a dorostČlánek vyšel v časopise
Česko-slovenská pediatrie

2020 Číslo 3
- Efektivita kartáčku Sonicare For Kids u dětí předškolního věku
- OZP pečuje o zdraví svých pojištěnců již 30 let. V čem je lepší a jaké výhody nabízí?
- Cytomegalovirové infekce u novorozenců a dětí
- Cytomegaloviróza a spalničky u dětí
Nejčtenější v tomto čísle
- Krvácení z rodidel v dětském věku z pohledu dětského gynekologa
- Bolesti břicha u dívek v dětském a pubertálním období
- Vrozené vývojové vady dělohy a pochvy – poruchy vývoje Müllerových vývodů a jejich derivátů – ultrazvuková diagnostika u dětí a dospívajících
- Eisenmengerův syndrom a jeho komplikace v dětském věku
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy