
Castlemanova choroba – jedno jméno, mnoho tváří
Castleman disease – one name, many faces
Castleman disease (CD) is a mesmerising group of disorders mainly affecting lymph nodes sharing some morphological features but with heterogeneous aetiology, clinical presentation and therapeutic approaches. Morphologically, hyaline-vascular (or hypervascular), plasmacytic, and mixed types of changes are distinguished. Confirmation of the diagnosis and subtype of Castleman disease involves meeting or excluding several clinical criteria and therefore requires close cooperation with a clinician. Unicentric Castleman disease involves usually a solitary enlarged lymph node with mild symptoms and excision surgery is often curative. Multicentric forms of Castleman disease affect multiple groups of lymph nodes and are associated with varying degrees of systemic clinical symptoms. Multicentric Castleman disease is either idiopathic or associated with human herpesvirus 8 infection or POEMS syndrome (polyneuropathy, organomegaly, endocrinopathy, M-protein, and skin changes). Idiopathic multicentric Castleman disease is further divided into a variant associated with TAFRO syndrome (thrombocytopenia, anasarca, fever, reticulin fibrosis / renal dysfunction, and organomegaly), idiopathic plasmacytic lymphadenopathy type, and not otherwise specified variant. The treatment of multicentric forms of Castleman disease is complex and depends on etiological factors, including biological therapy, chemotherapy, or interleukin-6 activity inhibition. The aim of this educational text is to present the current view of Castleman disease and provide a comprehensive description of the morphological changes and clinical characteristics of the individual subtypes of Castleman disease.
Keywords:
Castleman disease – POEMS syndrome – TAFRO syndrome – unicentric Castleman disease – idiopathic multicentric Castleman disease – HHV-8-positive multicentric Castleman disease – hyalinne-vascular morphology – hypervascular morphology – plasma cell type
Autoři:
Kateřina Kamarádová 1,2; Václav Stejskal 1; Dominika Écsiová 3
Působiště autorů:
Fingerlandův ústav patologie, Lékařská fakulta Univerzity Karlovy a Fakultní nemocnice Hradec Králové, Hradec Králové
1; Unilabs Pathology k. s., Laboratoř Praha, Praha
2; IV. interní hematologická klinika, Lékařská fakulta Univerzity Karlovy a Fakultní nemocnice Hradec Králové, Hradec Králové
3
Vyšlo v časopise:
Čes.-slov. Patol., 62, 2026, No. 1, p. 17-34
Kategorie:
Přehledový článek
Souhrn
Castlemanova choroba je fascinující skupina onemocnění postihující především lymfatické uzliny, které sdílí některé morfologické znaky, ale liší se etiologií, klinickou prezentací i terapeutickými přístupy. Morfologicky se rozlišuje hyalinně-vaskulární (respektive hypervaskulární), plazmocytární a smíšený typ změn. Potvrzení diagnózy a určení podtypu Castlemanovy choroby zahrnuje také splnění či vyloučení řady klinických kritérií a vyžaduje tedy úzkou spolupráci s klinickým lékařem. Unicentrická forma Castlemanovy choroby postihuje obvykle jednu lymfatickou uzlinu s minimem klinických příznaků a chirurgická léčba je obvykle kurativní. Multicentrické formy Castlemanovy choroby postihují více skupin lymfatických uzlin a jsou spojeny s různě závažnými systémovými klinickými příznaky. Multicentrická Castlemanova choroba je buď idiopatická nebo je asociovaná s infekcí lidským herpes virem 8 nebo s klinickým syndromem POEMS (polyneuropatie, organomegalie, endokrinopatie, M-protein a kožní změny). Idiopatická multicentrická Castlemanova choroba se dále dělí na variantu spojenou se syndromem TAFRO (trombocytopenie, anasarka, horečka, retikulinová fibrotizace / renální dysfunkce a organomegalie), typ s idiopatickou plazmocytární lymfadenopatií a blíže nespecifikovaný typ. Terapie multicentrických forem Castlemanovy choroby je komplexní a odvislá od etiologických faktorů a zahrnuje biologickou léčbu, chemoterapii či inhibici aktivity interleukinu-6. Cílem tohoto doškolovacího textu je ukázat současný pohled na Castlemanovu chorobu a poskytnout přehledný popis morfologických změn i klinických charakteristik jednotlivých typů Castlemanovy choroby.
Klíčová slova:
Castlemanova choroba – POEMS syndrom – unicentrická Castlemanova choroba – idiopatická multicentrická Castlemanova choroba – HHV-8-pozitivní multicentrická Castlemanova choroba – TAFRO syndrome – hyalinně-vaskulární morfologie – plazmocytární morfologie
Diagnostika Castlemanovy choroby (Castleman disease, CD) je vzrušující, ale zároveň extrémně komplikovaná, přestože histomorfologický obraz, především u rozvinuté hyalinně-vaskulární varianty, je vizuálně vděčný a může být diagnosticky vysoce sugestivní. Zároveň se nejedná o jednu nemoc, ale o skupinu minimálně 4 onemocnění, která jsou velice vzácná, a která kromě morfologických kritérií vyžadují také splnění četných klinických parametrů a vyloučení řady onemocnění, které mohou CD připomínat klinicky nebo i morfologicky. Patolog bude vždy potřebovat mezioborovou pomoc a korelaci s výsledky dalších vyšetření. Někdy bude na základě morfologické suspekce iniciátorem, který nasměruje kliniky k dalšímu dovyšetření pacienta a předání do adekvátní, obvykle hematoonkologické péče. A protože je Castlemanova choroba dělena do jednotlivých variant na základě rozsahu onemocnění a dalších klinických projevů, tak diagnózu Castlemanovy choroby, včetně zařazení do některého z podtypů, neudělá patolog nikdy sám.
V roce 1954 vyšel v New England Journal of Medicine, v sekci Týdenní klinickopatologická cvičení, popis případu mediastinálního tumoru u čtyřicetiletého muže s morfologií nápadné hyperplázie lymfatických uzlin s cévní proliferací (1). Případ prezentoval Dr. Benjamin Castleman jako disputaci se svými kolegy z dalších oborů. O dva roky později již Castleman publikoval sérii třinácti případů s obdobným klinickým zasazením a podrobným histopatologickým popisem, které dle dnešních kritérií odpovídají hyalinně-vaskulární unicentrické formě onemocnění a Castlemanovo jméno tak dalo tomuto onemocnění název (2). Kromě hyalinně-vaskulární morfologie byly v průběhu 70. let popsány i případy plazmocytárního typu, který byl spojen se systémovými příznaky a také případy s multicentrickou lymfadenopatií spojené s konstitucionálními symptomy a histologickými charakteristikami podobnými unicentrickému typu CD (UCD) (3-7).
Na začátku 80. let byl také popsán syndrom polyneuropatie, organomegalie, endokrinopatie, M-proteinu a kožních změn (POEMS), jehož součástí je také lymfadenopatie s morfologií Castlemanovy choroby (8,9). Na konci 80. let pak japonští autoři detekovali vysoké hladiny interleukinu-6 (IL-6) přímo v uzlinách pacientů s CD a představili ho jako klíčový mediátor klinické manifestace Castlemanovy choroby (10,11). O deset let později byl objeven nový lidský herpes virus spojený s Kaposiho sarkomem (herpes virus asociovamý s Kaposiho sarkomem neboli lidský herpes virus 8, KSHV/HHV-8). (12,13) HHV-8 byl následně identifikován v primárních exsudativních lymfomech (PEL) a v multicentrické CD (MCD), zejména u osob pozitivních na virus lidské imunodeficience (HIV) (14-17).
O 70 let později víme, že Castlemanova choroba (CD) nepředstavuje jedno onemocnění, ale skupinu minimálně čtyř vzácných, převážně uzlinových, lymfoproliferativních poruch s různorodou etiologií, heterogenní klinickou prezentací a prognózou, které vyžadují odlišný terapeutický přístup. Základní rozdělení Castlemanovy choroby vychází z rozsahu uzlinového postižení a přítomnosti klinických symptomů (18-22). Základem je dělení na unicentrickou (UCD) formu s postižením jedné izolované zvětšené lymfatické uzliny či skupiny uzlin jedné oblasti a na multicentrickou (MCD) formu, která postihuje 2 a více uzlinových oblastí. Multicentrická Castlemanova choroba se pak dále dělí na několik podtypů, a to na POEMS-MCD, HHV-8-pozitivní MCD a idiopatickou MCD (iMCD). Tzv. POEMS-MCD je podtyp asociovaný s klinickým syndromem s tímto akronymem s projevy polyneuropatie, organomegalie, endokrinopatie, s přítomností M-proteinu (při monoklonální plazmocytární poruše) a s přidruženými kožními změnami. MCD asociovaná s lidským herpes virem 8 postihuje jednak pacienty infikované virem HIV, tak i pacienty HIV-negativní. Idiopatická MCD (iMCD) představuje HHV-8 negativní typ MCD bez jasné infekční či neoplastické příčiny (19-21,23). iMCD se rozděluje na blíže nespecifikovaný typ (iMCD-NOS, not otherwise specified) a na idiopatickou MCD spojenou s klinickými a laboratorními znaky syndromu, který byl popsán japonskými autory v desátých letech 21. století a který je známý pod akronymem (iMCD-TAFRO) (24-27). TAFRO syndrom zahrnuje trombocytopenii, anasarku, horečku, retikulinovou fibrotizaci v kostní dřeni či renální dysfunkce a organomegalii a kromě iMCD může být spojen i s jinými komorbiditami (27,28). Blíže o tomto podtypu viz článek v tomto čísle od prof. Arianny di Napoli. Koncept HHV-8 negativní (idiopatické) MCD byl představen v roce 2014. (29) Pro zlepšení záchytu a diagnostiky idiopatické formy MCD vydala v roce 2017 skupina autorů z Castleman Disease Collaborative Network (CDCN) konsenzuální diagnostická kritéria (30).
Recentně pak byly představeny nové podtypy CD definované na základě specifických klinických projevů. Asymptomatičtí pacienti s limitovaným postižením obvykle sousedících uzlinových oblastí jsou klasifikováni jako oligocentrická varianta CD (oligoCD) a bezpříznakoví pacienti s postižením více uzlinových oblastí jako asymptomatická MCD (aMCD), ta ale může přestavovat velmi časné stadium iMCD (31-33). V rámci iMCD pak byla definována nová forma s obrazem tzv. idiopatické plazmocytární lymfadenopatie (iMCD-IPL), spojená s vysokou koncentrací imunoglobulinů, chronickými zánětlivými projevy a překvapivě indolentním chováním. (34) IPL byla původně popsána v 80. letech minulého století japonskými autory a historicky představovala předobraz iMCD-NOS s nejmírnějšími klinickými příznaky (35,36). Na základě nové definice nicméně zařazují někteří autoři tuto fenotypickou variantu iMCD s plazmocytární morfologií jako samostatný podtyp, a to i v rámci aktualizace diagnostických kritérií (32,33,37,38). Castlemanova choroba si také vysloužila svoje místo ve WHO klasifikaci hematolymfoidních tumorů v kapitole tumor-like lézí s převahou B-buněk (Tumour-like lesions with B-cell predominance), čímž, především v systémové variantě, upevnila svoji příslušnost do skupiny onemocnění spadajících do péče hematologů a hematoonkologů (39). Dělení CD včetně zařazení nových podtypů a vztah k morfologickým variantám zobrazuje přehledové schéma na obrázku 1.

EPIDEMIOLOGIE CASTLEMANOVY CHOROBY
Přesná epidemiologická data ke Castlemanově chorobě stále chybí. Důvodem je především vzácnost onemocnění a také chybějící diagnostický kód (alespoň v České republice) pro lepší detekci onemocnění a další statistická zhodnocení. Celosvětová incidence je neznámá a odhady incidence se liší geograficky i na základě použité metodiky. Starší studie předpokládaly incidenci zhruba 21-25 případů Castlemanovy choroby na 1 milion osob za rok, přičemž většina případů byla zařazena v unicentrické formě (70–75 %) (40). Recentní americká studie z roku 2022, která využila nově zavedený ICD-10 kód pro Castlemanovu chorobu (D47.Z2, v ČR není tento kód evidován), pracuje s nižší incidencí. Incidence všech případů CD se v této studii pohybovala ve zkoumaných letech mezi 5,5–5,8 / 1 milion obyvatel / rok a UCD tvořila v této studii pouze 35 % případů (41). V České republice by takovýto odhad incidence CD znamenal záchyt mezi 50–250 pacienty s kterýmkoliv typem CD ročně. Castlemanova choroba však nadále zůstává poddiagnostikovaná a část pacientů je patrně skryta pod diagnózami systémových autoimunitních či imunitně podmíněných onemocnění (42,43).
ETIOLOGIE A PATOGENEZE DLE VARIANT CASTLEMANOVY CHOROBY
UCD
Mechanismy stojící za vznikem UCD historicky zahrnovaly virovou, nádorovou i reaktivní zánětlivou etiologii (19,44). Vícečetné studie nejvíce podporují nádorovou etiologii UCD průkazem monoklonálního původu onemocnění s nejpravděpodobnějším původem ve stromálních buňkách, a to především ve folikulárních dendritických buňkách (FDC). Tuto teorii podporují především případy pacientů s UCD s navazujícím FDC sarkomem ve stejné uzlinové oblasti, u kterých byla zároveň prokázána vysoká exprese chemokinu CXCL13 (45,46). Mezi rekurentní genetické změny patří mutace v genu PDGFRB prokázané v CD45-negativních stromálních elementech v UCD (47). Nejnovější proteomické a transkriptomické studie ukazují, že se jedná o mnohem komplexnější mechanismus, který zahrnuje komunikaci více typů stromálních elementů s produkcí růstových faktorů a cytokinů zodpovědných za typickou morfologii (48,49). Podobně jako u ostatních typů CD se jedná především o IL-6 či vaskulární endoteliální faktor (VEGF) či o aktivaci JAK/ STAT, TGF-b či MAPK dráhy (11,48).
HHV-8-pozitivní MCD
Hlavním etiologickým faktorem u HHV-8-pozitivní MCD je infekce lidským herpes virem 8. U imunokompromitovaných pacientů dochází k replikaci HHV-8 v plazmablastech v lymfatické uzlině a zároveň dochází k transkripci a produkci virového homologu interleukinu 6 (vIL-6), který je zodpovědný za klinické příznaky i za morfologické změny v lymfatických uzlinách. Zároveň dochází k aktivaci dalších cytokinů včetně lidského IL-6. HHV-8 je také schopen potencovat diferenciaci IgM-pozitivních naivních B-buněk do plazmablastů i bez průchodu zárodečným centrem a tyto plazmablasty u HHV-8 MCD jsou pak lokalizovány do oblasti folikulárních plášťů (44,50-52). Jedním z predisponujících faktorů imunosuprese je infekce virem HIV. Téměř u všech pacientů s HIV a Castlemanovou chorobou se vyskytuje HHV-8-pozitivní MCD varianta. U HIV negativních pacientů je pak výskyt HHV-8-pozitivní MCD odvislý od endemické zátěže tímto virem a kolísá mezi 2-50 %. (19,53). Molekulárně je pak prokázána např. upregulace jaderného faktoru NF-kB či aktivace VEGF signalizace (44).
POEMS-MCD
V případě multicentrické Castlemanovy choroby asociované se syndromem POEMS jsou hlavním etiologickým faktorem klonální nádorové plazmocyty. Známky CD-like změn či rozvinutý obraz MCD je přítomen u cca 11–30 % pacientů s dokumentovanou plazmocytární neoplázií. Hlavními uznávanými faktory pro rozvoj CD změn jsou zvýšená produkce VEGF a také interleukinu 6 a interleukinu-12 (IL-12) nádorovými plazmocyty na podkladě somatických mutací (19,54,55). Studie zaměřená na genetické pozadí syndromu POEMS pak popsala mutace v sedmi rekurentně mutovaných genech, jmenovitě KLHL6, LTB, EHD1, EML4, HEPHL1, HIPK1 a PCDH10. Genetické změny v plazmocytech POEMS syndromu jsou odlišné od mutačního profilu mnohočetného myelomu, ale sdílí některé chromozomální abnormality s monoklonální gamapatií neurčitého významu (MGUS), které by mohly představovat parciální překryv v časné fázi vývoje MGUS a POEMS (19,56).
iMCD
Etiologie iMCD jednoznačně ozřejmena není, nicméně hypotézy počítají s více možnými mechanismy rozvoje onemocnění. Mezi hlavní možné cesty rozvoje iMCD patří autoimunitní či autoinflamatorní mechanismy (např. přítomnost autoprotilátek či germinální genomické alterace v drahách pro zánětlivé reakce), paraneoplastické projevy (včetně somatických mutací v klonálních nádorových buňkách) nebo infekční etiologie spojená s jinými viry, než je HHV-8 (29). Poslední hypotéza je však málo pravděpodobná, protože vícečetné studie neprokázaly přítomnost RNA či nukleotidové sekvence žádných potenciálních patogenů (19,57). Je možné, že vícečetné dráhy v etiologii iMCD kulminují do obrazu cytokinové bouře spojené s podobnou klinickou prezentací (44,58,59). Centrální roli v rozvoji symptomů i části morfologických změn hraje opět IL-6 a jeho nadprodukce na podkladě některé z uvedených hypotéz. Zvýšené hladiny IL-6 nejsou přítomny u všech případů a zhruba polovina pacientů tak neodpovídá na terapeutickou inhibici IL-6 (42,60). Mezi další patogenetické markery patří zvýšení produkce vaskulárního endoteliálního faktoru (VEGF), které je ale menší než u POEMS-MCD (19). Mezi nověji popsané patogenní faktory patří chemokiny a chemotaktické proteiny jako CXCL13 (produkovaný periferními pomocnými T-lymfocyty), CCL21 a CCL19 (59,61,62). Důležitou roli hrají také signalizační dráhy spojené s prozánětlivou signalizací přes IL-6, jako je JAK/STAT a PI3K/AKT (63,64). Do popředí se dostává i studium aktivace dráhy mTOR, především u iMCD-TAFRO, kde zvyšuje produkci IGFBP-1 (insulin-like growth factor binding protein-1), který by se mohl stát diagnostickým i prognostickým biomarkerem a zároveň otevírá možnost terapeutického zásahu mTOR inhibitory (49,65,66).
Role interleukinu-6 v rozvoji klinických a zčásti i morfologických změn u CD je zcela zásadní. Interleukin-6 je prozánětlivý cytokin s širokou škálou účinků produkovaný buňkami imunitního systému (T/B-lymfocyty, makrofágy), ale také endoteliemi či fibroblasty (11,67). Hlavní účinky IL-6 představují zvýšení produkce reaktantů akutní fáze (CRP, sérový amyloid A, transferrin apod.) či účast na maturaci B-lymfocytů a stimulace syntézy a sekrece imunoglobulinů vedoucí k hypergamaglobulinémii. IL-6 také ovlivňuje diferenciaci pomocných T-lymfocytů (CD4+ TH) především k TH17 typu a potencuje tak produkci dalších prozánětlivých cytokinů a chronický zánět. V hepatocytech IL-6 také suprimuje produkci albuminu s následou hypoalbuminémií a rozvojem otoků. Podpora tvorby VEGF pak vede k angiogenezi i zvýšené vaskulární permeabilitě. IL-6 je schopen ovlivňovat i sympatický i centrální nervový systém a vést k symptomům jako je horečka a únava (11,67).
HISTOPATOLOGICKÉ CHARAKTERISTIKY CASTLEMANOVY CHOROBY
Diagnostika kteréhokoliv typu Castlemanovy choroby vyžaduje potvrzení přítomnosti histomorfologických změn typických pro CD, a to mandatorně z excize celé lymfatické uzliny. Stanovení diagnózy z jehlové biopsie je de facto nemožné, jelikož jehlová biopsie neumožňuje hodnocení celkové architektoniky lymfatické uzliny a rozsahu změn v uzlině a není tedy obecně doporučováno (30,61,68). Původní Castlemanův popis hyalinně-vaskulární morfologie a následné doplnění o plazmocytární variantu definovalo na dlouhou dobu rozdělení morfologie CD na tyto dvě varianty. Později byl přidán třetí tzv. smíšený (intermediární) morfologický typ (30). To, že se jedná spíše o spektrum morfologických nálezů než tři odlišitelné varianty, se odrazilo především v rámci přípravy konsenzuálních kritérií pro diagnostiku idiopatické multicentrické Castlemanovy choroby (30). Tato kritéria zařazují změny s převažujícím nálezem regresivně změněných zárodečných center s výraznou vaskularizací na hyalinně-vaskulární, respektive hypervaskulární (hyperV) konec spektra a plazmocytózu s hyperplázií zárodečných center na plazmocytární konec spektra. Případy s překryvnou morfologií pak představují tzv. smíšenou variantu histomorfologických změn (30). V rámci konsenzuálních kritérií dochází pro účely diagnostiky iMCD ke změně morfologické terminologie z hyalinně-vaskulární (HV) na hypervaskulární (hyperV) typ z morfologických i diagnostických důvodů (viz níže) (20,30).
Hyalinně-vaskulární / hypervaskulární morfologie
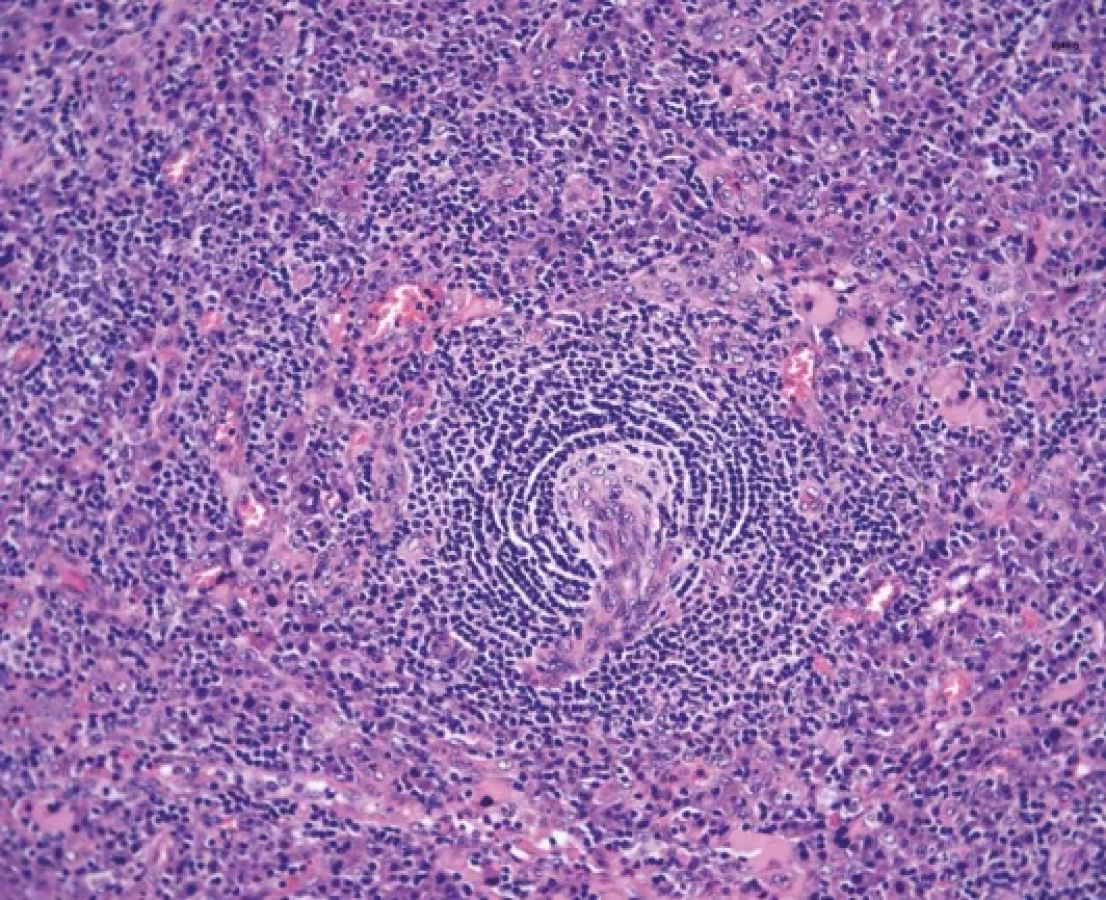
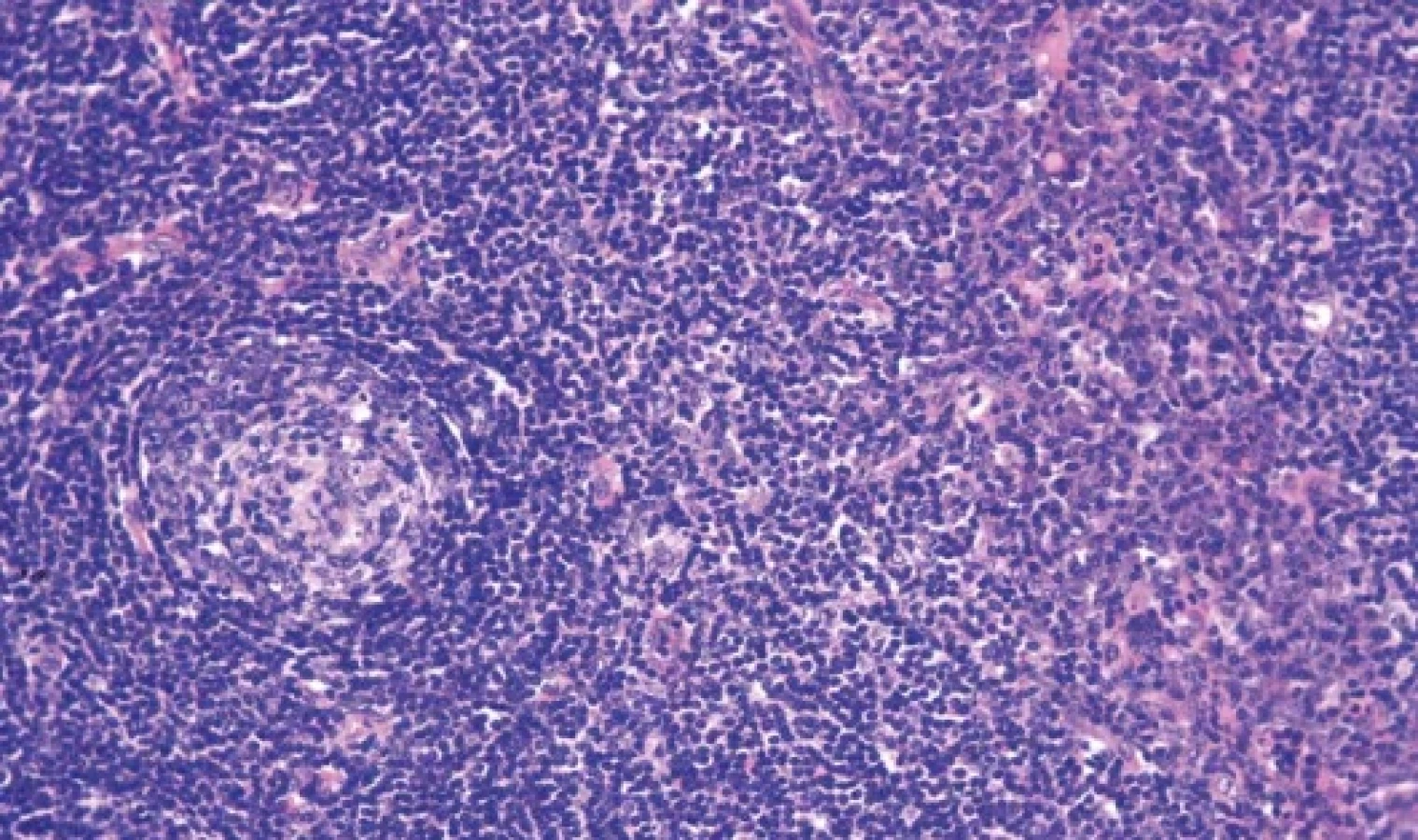
Hyalinně-vaskulární (HV) morfologie se nejčastěji vyskytuje u unicentrické formy Castlemanovy choroby (75-91 %). (21) Lymfatická uzlina má často rozšířené pouzdro s fibrózou a vazivové pruhy mohou od periferie vstupovat i do centra uzliny. Zároveň dochází k obliteraci až vymizení lymfatických sinusů. Lymfatické folikuly jsou zmnožené, ale typickým nálezem jsou regresivně změněná zárodečná centra (obr. 2). Častým nálezem je také přítomnost více než jednoho zárodečného centra pod společným pláštěm folikulu (twinning/budding) (obr. 3). Zárodečná centra vykazují úbytek B-lymfocytů a jsou tvořena převážně folikulárními dendritickými buňkami (FDC), které jsou pozitivní v průkazu CD21, CD23 nebo CXCL13. Reziduální lymfocyty v zárodečných centrech jsou pozitivní v průkazu CD10 a BCL6, ale jsou BCL2 negativní. Proliferace FDC, ale také fibroblastických retikulárních buněk (FRC) je součástí obrazu UCD a dendritické buňky mohou až u ¼ případů vykazovat dysplastické rysy. Ty jsou charakterizovány zvětšenými nepravidelnými jádry, někdy zdvojenými, s prominentními jadérky nebo s chromatinem vzhledu pomačkaného papíru. Dysplastické FDC jsou zastiženy jak v centru zárodečných center, tak interfolikulárně, mohou být i mnohojaderné a připomínat Reedové-Sternbergovy (RS) nebo Warthin-Finkledeyovy buňky (69-71) (obr. 4A). Záměna za buňky klasického Hodgkinova lymfomu (CHL) je možná, ale ověření FDC fenotypu a nevýrazná příměs eozinofilů mohou zamezit případné záměně (21,72). Jednou z vlastností FDC, kterou lze také v rámci CD pozorovat, je emperipoléza malých lymfocytů (73) (obr. 4B). Dalším nálezem je penetrace sklerotické cévy či více cév přes plášť do regresivně změněného zárodečného centra (tzv. obraz lízátka, anglický termín “lollipop lesion”) (obr. 5). Cévy jsou často sklerotické až hyalinizované, někdy s kalcifikacemi a v centru folikulu mohou být zastižena také hyalinní (PAS pozitivní) depozita. Pláště jsou naopak rozšířené s koncentricky uspořádanými menšími lymfocyty s tzv. cibulovitým uspořádáním (anglický termín “onion skinning”) (obr. 3-5). Někdy naopak nabývají změny ve folikulech až obrazu, který je podobný progresivní transformaci zárodečných center (PTGC-like) (19,21,30). Interfolikulárně je patrná proliferace postkapilárních venul s dužnatými endoteliemi (obr. 6). Postkapilární venuly mohou být obklopeny plazmocytoidními dendritickými buňkami (PDC) a proliferujícím stromatem. (obr. 7A) Proliferaci cév lze ozřejmit expresí vaskulárních markerů CD31 a CD34. Pro identifikaci plazmocytoidních dendritických buněk lze využít expresi CD123 (obr. 7B). Součástí interfolikulárního infiltrátu jsou imunoblasty, eosinofily i plazmocyty, ty však netvoří nápadnější plachtovité formace. Příměs TdT pozitivních lymfoblastů je obvykle minimální, ale existují případy s interfolikulární hyperplázií TdT-pozitivních buněk. Tato proliferace neklonálních lymfoblastů nevykazuje aberantní fenotyp a spadá do kategorie nenádorové indolentní T-lymfoblastické proliferace (20,72,74-76).




Hypervaskulární morfologie (hyperV) je termín používaný především v rámci diagnostiky idiopatické formy MCD a je zahrnuta i v rámci konsenzuálních kritérií pro iMCD (30). Zavedení tohoto termínu má dva důvody. První důvod odráží fakt, že pojem hyalinně-vaskulární je pro některé diagnostiky historicky spojen výhradně s projevem unicentrické CD a je tedy snaha vyhnout se potenciální záměně benigní léze za klinicky agresivnější onemocnění. Druhý důvod je čistě morfologický, který poukazuje na to, že u pacientů s iMCD nemusí být typický, plně rozvinutý, obraz HV morfologie přítomný. Dysplázie dendritických buněk či sklerotické cévy jsou u iMCD zastiženy méně často a zároveň jsou v uzlinách zachované struktury sinusů (19,30,61). Kombinace přítomnosti regresivních zárodečných center s hypervaskularitou bez plazmocytózy má u iMCD tedy spíše HV-like charakter, a proto je v současné literatuře pro tuto morfologii v iMCD upřednostňován právě termín hypervaskulární morfologie (hyperV). (19,61) (obr. 8) Tento typ hypervaskulární morfologie převažuje především u případů iMCD-TAFRO (21,26). Zároveň však neplatí, že by např. UCD byla spojena výlučně s HV morfologií nebo iMCD s výlučně hyperV nebo PC morfologií (20).

Plazmocytární morfologie
Plazmocytární typ morfologie je nejčastěji přítomen u případů HHV-8-pozitivní MCD, iMCD-IPL, iMCD-NOS a u 60 % případů POEMS-MCD. (19,34,77) Naopak případy UCD vykazují tuto morfologii méně často (9 – 26 %). (21) PC morfologie se vyznačuje kombinací hyperplázie polarizovaných zárodečných center ve zvětšených folikulech a plachovitými infiltráty plazmocytů v interfolikulárních zóně. (obr. 9A,B) Ojedinělá zárodečná centra mohou být naopak regresivně změněná a může být přítomna i mírná vaskularizace (30,61). Plazmocyty jsou převážně polytypické, ale až u třetiny případů mohou být plazmocyty monotypické, nejčastěji s expresí řetězců lambda (19,20,78,79). V interfolikulární oblasti může být podobně jako u HV typu patrná proliferace postkapilárních venul a také příměs eozinofilů a mastocytů, která ale nepřevyšuje výrazný plazmocytární infiltrát. Ojedinělý výskyt koncentricky až cibulovitě uspořádaných plášťů či céva penetrující do zárodečného centra nevylučuje PC morfologii. Narozdíl od klasické HV-morfologie jsou u PC varianty zachované uzlinové splavy (80).

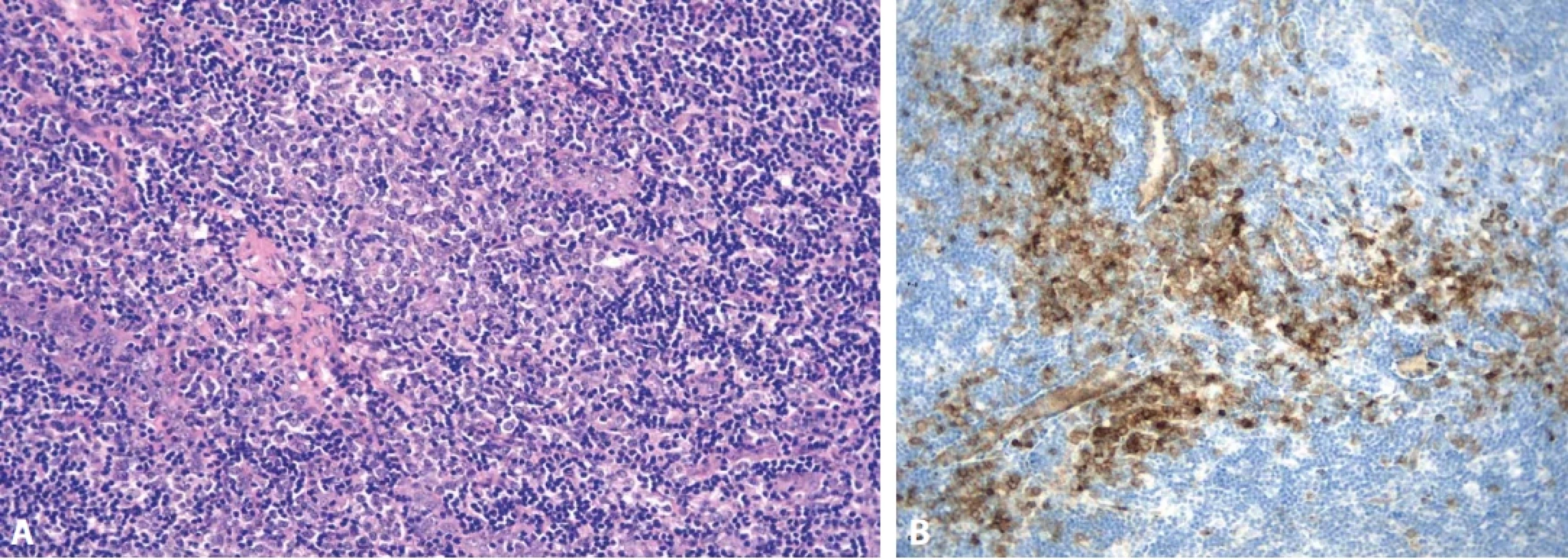
Jistá specifika, nepřítomná u ostatních typů MCD, vykazuje HHV-8-pozitivní MCD. Některé reaktivní folikuly mají hůře definované pláště, ve kterých jsou přítomny velké imunoblasty a plazmablasty, často pozitivní v imunohistochemickém průkazu latentního nukleárního antigenu (LANA/HHV-8) a v IL-6 (obr. 10 A, B). Historicky se tato morfologie označovala jako plazmablastický typ. Tyto virem infikované elementy mohou exprimovat monotypicky IgM-lambda, ale zároveň zůstávají polyklonální. (obr. 10 C,D) (19,51,81) Plazmablasty pak mohou vytvářet malé skupiny či klastry buněk odpovídající “mikrolymfomu” či přejít do plně rozvinutého lymfomu s plachtovitými infiltráty plazmablastů (51,69,81,82).

Smíšená morfologie
Smíšená morfologie vykazuje přítomnost morfologických změn jak z HV/hyperV tak z PC spektra. Nejvíce se kombinují regresivně změněná zárodečná centra se zvýšenou vaskularizací kombinovaná s plazmocytózou. (obr. 11) Naopak proliferace folikulárních dendritických buněk nebo hyperplázie zárodečných center je méně nápadná (19,21,30).
Součástí diagnostiky CD je nejen potvrzení přítomnosti typických histopatologických změn, ale také vyloučení řady onemocnění, pro které je nutné využití ancilárních vyšetření včetně imunohistologie a případně i molekulárních metod. Některá tato vyšetření zároveň mohou podpořit diagnózu CD nebo pomoci zařazení do specifického podtypu (např. HHV-8-pozitivní MCD). Základní přehled imunohistochemických markerů a dalších vyšetření využitelných v rámci diagnostiky Castlemanovy choroby je uveden v tab. 1.

ZÁKLADNÍ KLINICKÉ CHARAKTERISTIKY A ROZDĚLENÍ CASTLEMANOVY CHOROBY
Unicentrická CD
Unicentrická CD je nejčastějším podtypem CD a tvoří zhruba 60–70 % případů. UCD je obvykle benigní onemocnění, které typicky postihuje jednu lymfatickou uzlinu nebo více uzlin v jedné uzlinové oblasti. Nejčastěji postižené oblasti zahrnují mediastinum, intraabdominální oblast s převahou mesenteria, retroperitonea a krční lymfatické uzliny. Velikost postižené uzliny je až 5 cm a uzliny jsou obecně větší než u multicentrických forem (21,71). UCD postihuje pacienty v jakémkoliv věku s mediánem okolo 40 let a s mírnou predominancí mužů. Klinické příznaky obvykle chybí nebo jsou mírné případně zahrnují příznaky z útlaku okolních struktur.
Morfologicky převažuje u případů UCD rozvinutá hyalinně - -vaskulární morfologie (80 – 90 %) (71,72,76). UCD s plazmocytární morfologií je méně obvyklá, ale častěji symptomatická s horečkami, nočními poty, únavou a abnormálními laboratorními změnami jako je vyšší sedimentace, anémie nebo hypergamaglobulinémie. Klinické projevy obvykle vymizí po chirurgické excizi postižené lymfatické uzliny (21,71,72). Mezi komplikace, které postihují až 18 % pacientů s UCD, patří paraneoplastický pemphigus, bronchiolitis obliterans, amyloidóza AA typu nebo rozvoj malignity typu lymfomu nebo sarkomu z folikulárních dendritických buněk (FDCS) (19,20,72,83).
Oligocentrická CD – nováček na scéně
Případy pacientů s hraničním nálezem mezi UCD a limitovanou iMCD byly opakovaně popsány ve větších souborech pacientů (60,84). Obvykle se jednalo o pacienty, kteří měli buď zvětšené vícečetné uzliny v jedné lokalizaci („clustered nodes“) nebo s postižením uzlin v sousedící či přilehlé oblasti (např. bilaterální krční uzliny nebo krční a axilární uzliny unilaterálně). Tito pacienti zároveň vykazovali mírné systémové příznaky jako anémie, vysoké hladiny CRP a IL-6 a nízké hladiny albuminu, ale nevykazovali elevaci hladin imunoglobulinů. (60) Recentně publikovaná data vycházející z registru ACCELARATE umožnila tuto skupinu pacientů lépe charakterizovat a definovat jako nový podtyp, tzv. oligocentrickou Castlemanovu chorobu (oligoCD) (32). V souladu s předchozími popisy jsou postižené uzlinové oblasti ve vzájemné blízkosti a obvykle na jedné straně bránice. Pacienti vykazují průměrně nižší věk v době diagnózy (34 let), častěji se jedná o ženy (až 71 %) a počet postižených uzlinových oblastí se pohybuje mezi 2-5 (median 3) (32). OligoCD pak sdílí klinickou podobnost s UCD, což by se do budoucna mělo odrazit i v adekvátním terapeutickém přístupu k pacientům.
Multicentrická CD
Společným znakem MCD jsou zvětšení lymfatické uzliny (≥ 1 cm v krátké ose) s postižením ≥ 2 uzlinových oblastí a přítomnost klinických projevů. Asymptomatická MCD je vzácná, ale recentně popsaná (31). Všechny formy MCD jsou charakterizovány přítomností různě závažných klinických příznaků systémového zánětu spojených s orgánovými dysfunkcemi a laboratorními abnormalitami. Tyto příznaky běžně rychle progredují, ale vzhledem ke vzácnosti onemocnění je jejich klinické rozpoznání a správné zařazení mnohdy opožděno. Mezi klíčové příznaky patří horečka, hubnutí, otoky až anasarka, generalizovaná lymfadenopatie a organomegalie. Zmnožené lymfatické uzliny mají obvykle menší průměrnou velikost než u unicentrické formy. Časté jsou i cytopenie, zvýšené parametry zánětu jako sedimentace či hladiny C-reaktivního proteinu (CRP) a hypoalbuminémie. Zánětlivé příznaky mohou mít epizodický charakter, ale u těžkých případů mohou přetrvávat a progredovat až do život ohrožující cytokinové bouře (31).
POEMS-MCD
POEMS je akronym pro klinický syndrom polyneuropatie, organomegalie, endokrinopatie, M-proteinu a kožních změn, který vzniká na podkladě klonální plazmocytární poruchy (téměř výlučně s produkcí řetězců lambda), a která je spolu s obvykle demyelinizující polyneuropatií povinným kritériem pro tuto diagnózu (8,19). Klasický POEMS je vzácný a často asociován s osteosklerotickým myelomem. Dalšími hlavními kritérii jsou Castlemanova choroba, elevace hladin VEGF a sklerotické kostní léze. Mezi malá kritéria pak patří organomegalie, edém, výpotky či ascites, endokrinopatie, kožní změny jako hyperpigmentace, hypertrichóza, glomeruloidní hemangiomy a také edém papily očního nervu a trombocytóza či polycytémie (9,19). Diagnostická kritéria pro POEMS syndrom jsou přehledně uvedena v tab. 2. Pacienti s POEMS-MCD jsou obvykle středního věku, průměrně okolo 50 let. Vlastní monoklonální populace plazmocytů u POEMS je zhruba u 2/3 pacientů přítomna v kostní dřeni, kdežto u 1/3 se projevuje lymfadenopatií s CD-like morfologií.

Tato kritéria nemusí být splněna všechna, což znamená, že existují i případy POEMS syndromu bez přítomnosti MCD. Zároveň existují případy s CD-like morfologií, které nesplní např. kritérium rozvinuté polyneuropatie nebo dokonce nevykazují ani přítomnost plazmocytárního klonu, ale splní vícečetná další kritéria pro POEMS. Tyto případy by se měly klasifikovat jako tzv. CD varianta POEMS (19,21). Dalšími nálezy v kostní dřeni jsou lymfoidní agregáty lemované plazmocyty a hyperplázie megakaryocytů, která připomíná myeloproliferativní onemocnění, ale průkaz mutace JAK2V617F obvykle chybí (85). Megakaryocytární hyperplázie se však může vyskytovat i u iMCD-TAFRO. (30,85,86) Biopsie kostní dřeně je pak doporučována u všech případů MCD k vyloučení plazmocytární neoplázie a odlišení POEMS-MCD a ostatních podtypů včetně iMCD (30).
HHV-8-pozitivní MCD
HHV-8-pozitivní MCD je nejčastěji diagnostikovaná u pacientů s infekcí virem HIV, a to i u kontrolované HIV infekce s nízkou virovou náloží, ale může se vyskytovat u jinak imunokompromitovaných pacientů. Průměrný věk rozvoje onemocnění je 42 let pro HIV+ pacienty a 65 let pro HIV-negativní pacienty a převažuje výskyt u mužů (53). Častěji jsou postiženy periferní lymfatické uzliny. Většina pacientů je v době diagnózy symptomatická s horečkami, splenomegalií, edémy a výpotky, respiračními příznaky či s hemofagocytárním syndromem. U více než 50 % pacientů pak může být zároveň v době diagnózy přítomen i Kaposiho sarkom. Hlavní laboratorní změny zahrnují anémii, trombocytopenii, zvýšené CRP a hypergamaglobulinémii. Zhruba třetina pacientů může vykazovat i známky monoklonální gamapatie. (53) Virová nálož HHV-8 v periferní krvi pak obvykle koreluje se vzplanutím zánětlivých symptomů (20).
Idiopatická MCD (iMCD)
Idiopatická multicentrická Castlemanova choroba je klinicky velice podobná HHV-8-pozitivní MCD, ale průkaz virové asociace s HHV-8 je negativní. iMCD představuje třetinu až polovinu ze všech MCD a může postihnout pacienty v každém věku s mediánem 50 let (2–80 let) (18). iMCD představuje vzácné a potenciálně život ohrožující onemocnění, které se projevuje systémovými příznaky zánětu, polyklonální lymfoproliferací a dysfunkcí více orgánových systémů způsobených cytokinovou bouří často zahrnující IL-6. Stanovení správné diagnózy je obtížné, protože překryv klinických příznaků s infekčními, autoimunitními i maligními onemocněními je značný. Hlavními příznaky jsou horečka, noční poty, ascites, lymfadenopatie, hepatosplenomegalie, elevace CRP, hypoalbuminémie a anémie (20,30,33,42). V současné době se rozlišují tři klinické podtypy s rozdílnou prezentací – iMCD-TAFRO, iMCD-IPL a iMCD-NOS.
iMCD-TAFRO (Castlemanova-Kojimova choroba)
Pacienti s iMCD-TAFRO se prezentují těžkým syndromem cytokinové bouře, která se může podobat akutnímu průběhu hemofagocytární lymfohistiocytózy (HLH). Klinický obraz doplňuje organomegalie, trombocytopenie s rizikem krvácení, renální dysfunkce a retence tekutin s edémy, výpotky a anasarkou. Pacienti s TAFRO syndromem mají typicky normální hladiny imunoglobulinů. Průměrný věk pacientů je mezi 50-59 lety, ale existují i případy z mladších věkových skupin (14-22 let), bez rozdílu výskytu mezi pohlavími (19,20). Lymfadenopatie je vícečetná, ale velikostně menší než u jiných variant s průměrnou velikostí uzlin do 1,5 cm (26). Nejčastěji je iMCD-TAFRO spojeno se smíšeným nebo hypervaskulárním či HV typem morfologie. Vyšetření kostní dřeně je součástí diagnostického algoritmu. V kostní dřeni je patrná retikulinová fibrotizace a hyperplázie megakaryocytů (27). TAFRO syndrom nemusí být vždy spojen s iMCD, ale může se vyskytovat v souvislosti s jinými autoimunitami, infekcemi či malignitami (21,27,28,64).
iMCD-IPL
Idiopatická plazmocytární lymfadenopatie jako podtyp iMCD byla definována relativně nedávno a představuje klinicky nejpříznivější formu iMCD. (37) Pacienti mají typicky lymfadenopatii, anémii, známky těžkého zánětu a výraznou polyklonální hypergamaglobulinémii. Histologicky dominuje v uzlinách nápadná plachtovitá plazmocytóza a hyperplázie zárodečných center. Oproti ostatním typům iMCD je také přítomna signifikantní trombocytóza a zvýšené hladiny IgG a IgG4 (37,87). Naopak je méně častý výskyt pleurálních výpotků nebo ascites. Pacienti obecně splní kritéria pro iMCD, ale obvykle vykazují indolentní průběh a dobrou léčebnou odpověd na anti-IL6 terapii. Hlavní diferenciální diagnóza je pak oproti lymfadenopatii při IgG4 asociované chorobě s iMCD-like morfologií, která však nevykazuje známky systémového zánětu s horečkou a anémií. Rozpoznání tohoto podtypu je tedy důležité i pro volbu terapie a další prognózu pacientů (33,34,37,87).
iMCD-NOS
V kategorii blíže nespecifikované iMCD v tuto chvíli zůstavají pacienti, kteří nesplní kritéria pro iMCD-IPL a iMCD-TAFRO. iMCD-NOS je obvykle spojena s mnohočetnou lymfadenopatií, uzliny jsou středně velké (< 3 cm) častěji s plazmocytárním nebo smíšeným typem morfologie. Opět dominují známky zánětu se zvýšením CRP a sedimentací. Kromě zánětlivých symptomů je iMCD-NOS často spojena i se známkami autoimunity včetně plicního intersticiálního postižení. Polyklonální hypergamaglobulinémie je obvykle mírná. (22,30,33) Průběh onemocnění je variabilní od indolentních případů až po rapidní progresi a kritický, potenciálně fatální průběh (33,88).
DIAGNOSTICKÁ DOPORUČENÍ PRO iMCD A GRADING MORFOLOGICKÝCH ZMĚN
Právě skupina iMCD, spojená se systémovými příznaky a možným těžkým průběhem onemocnění si vyžaduje včasné odhalení a odlišení jednak od indolentní UCD, tak od MCD spojených se specifickou etiologií (POEMS-MCD, HHV-8-pozitivní MCD). Pro tyto účely vznikla v roce 2017 konsenzuální kritéria skupiny CDCN postavená na sadě kritérií, která je nutno splnit nebo naopak vyloučit (tab. 3) (30). Tato kritéria by měla umožnit odlišit jednak jiné typy MCD (například na základě potvrzení infekce HIV a HHV-8), tak odlišit iMCD od onemocnění, které ji napodobují buď klinicky nebo i morfologicky

Tato kritéria jsou rozdělena do 3 skupin – velká kritéria, malá kritéria rozdělená na klinickou a laboratorní část a tzv. vylučující kritéria. Pro dg. iMCD je potřeba splnit tři velká kritéria (Major Criteria), kterými jsou typická histomorfologie CD, postižení více než 2 uzlinových oblastí se zvětšením uzlin ≥ 1 cm v kratší ose a negativní imunohistochemický průkaz KSHV/ HHV8 LANA (30,39,61). Slezina není do postižených oblastí zahrnuta. Rozsah postižení by měl být hodnocen na podkladě vyšetření celotělovou počítačovou tomografií (CT) nebo s využitím pozitronové emisní tomografie a CT po aplikaci deoxyglukózy značené radioaktivním fluorem, 18F-fluorodeoxyglukózou (FDG-PET/CT). FDG-PET/CT potenciálně umožňuje odlišit iMCD a HHV-8-pozitivní MCD od high-grade lymfomu na podkladě odlišného vychytávání značené glukózy (89-91). Dále je potřeba splnit alespoň dvě malá kritéria (Minor Criteria) z celkového počtu 11 parametrů, čítající alespoň jednu laboratorní abnormalitu. Splnění těchto požadavků nemá pouze diagnostický význam, ale také význam prediktivně terapeutický, a to včetně odpovědi na terapii anti-IL6 protilátkami (viz níže) (30).
Dále je nutné vyloučit různé maligní a lymfoproliferativní poruchy, které jsou součástí onemocnění v tzv. vylučujících kritériích (Exclusion Criteria). Samostatně je pak uvedena skupina podpůrných parametrů, které ale nejsou pro diagnózu iMCD povinné. Sem patří zvýšené hladiny IL-6, VEFG, IgA, LDH, beta2-mikroglobulinu aj., retikulinová fibróza kostní dřeně (pro pacienty s TAFRO syndromem) a další poruchy spojené s iMCD jako paraneoplastický pemphigus, autoimunitní cytopenie, polyneuropatie (u pacientů bez POEMS), nefropatie a jiné (30,61,92).
Pro posouzení histopatologických změn v uzlinách je pak v rámci konsenzuálních kritérií uveden grading odrážející stupeň rozvoje či míru postižení lymfatické uzliny danou morfologickou změnou. Parametry hodnocené v kritériích z roku 2017 a použité i při přípravě kritérií pro WHO klasifikaci jsou regresivně změněná zárodečná centra, prominence folikulárních dendritických buněk a vaskularita (pro HV/hyperV morfologii) a hyperplastická zárodečná centra a plazmocytóza (pro PC morfologii). Pro splnění morfologického velkého kritéria je nutné splnění alespoň grade 2-3 pro regresivní zárodečná centra nebo pro plazmocytózu. (30,39) Důvodem je fakt, že do různé míry vyjádřené, podobné, tzv. Castleman-like změny, provází i jiné lymfadenopatie, které jsou pak součástí exkluzních kritérií. Patří sem především lymfadenopatie u autoimunitních onemocnění jako revmatoidní artridita či systémový lupus erytematodes, IgG4 lymfadenopatie a dále maligní lymfomy s CD - -like morfologií jako folikulární lymfom či klasický Hodgkinův lymfom (80,93).
iMCD – detaily gradingu morfologických změn
Autoři CDCN konsenzuálních kritérií z roku 2017 využivají pro grading jednotlivých morfologických parametrů škálu grade 0 – grade 3, kdy grade 0 znamená nepřítomnost daného parametru. Pro další stupně změn byly původně použity pouze slovní kvantifikátory jako málo-mnoho-většina (few-many-most) pro regresivně změněná zárodečná centra, mírně zvýšená-středně zvýšená-velmi zvýšená/plachtovitá (mild-moderate-very increased / sheet-like) pro plazmocytózu a grading byl publikován pouze ve formě vizuální škály. Přesná metodika rozdělení změn do jednotlivých grade skupin v rámci kritérií uvedena nebyla. Užití gradingu nicméně může umožnit spolehlivější zařazení lymfadenopatie do CD kategorie s vyšší jistou a lze využít i pro diagnostiku dalších typů včetně UCD. Nověji pak byly slovní kvantifikátory nahrazeny procentuálním zastoupením změn v lymfatické uzlině s hraničními hodnotami 0 % (grade 0), 10 - < 25 % (grade 1), 25 - < 50 % (grade 2) a ≥ 50 % (grade 3) pro jednotlivé parametry. (61,72) Nový postup pro grading morfologických změn přidává do parametrů také příznak zdvojení zárodečných center ve folikulech (twinning). Hodnocení pak probíhá ve třech fázích a různých zvětšeních. Na malém zvětšení jsou hodnoceny folikulární parametry – twinning, regresivní anebo hyperplastická zárodečná centra. Hodnoceno by mělo být alespoň 10 folikulů. Posuzováno je procento folikulů s danou změnou a přiřazen grade. Na středním zvětšení se hodnotí vaskularita v interfolikulárním prostoru a stanovuje se jako procento plochy, resp. buněčnosti, kterou představují proliferující cévy. Ve velkém zvětšení je hodnocena interfolikulární plazmocytóza (jako procento plochy, kterou zaujímají plazmatické buňky) a dále prominence FDC v zárodečných centrech jako procento celularity daného zárodečného centra. Počet hodnocených zárodečných center stanoven není (94). Histologický grading CD je přehledně zobrazen v tab. 4.

Recentně pak jako přídatný diagnostický parametr byla využita také přítomnost hemosiderinových depozit či vírovitý charakter cév v zárodečných centrech („whirpool vessel“), a to ve studii využívající hodnocení parametrů v rámci strojového učení (61,95).
Použití výše uvedeného gradingu se čím dál častěji objevuje i ve studiích zaměřených i na jiné podtypy CD než jen iMCD, pro které byl původně vytvořen. To je důležité především v případě primodiagnostiky nové lymfadenopatie bez znalosti rozsahu postižení a dalších klinických příznaků. Patolog může jednotlivé parametry zhodnotit a zařadit na škále jednotlivých gradů a vyjádřit tím i míru jistoty s postižením uzliny při CD. Naopak parciální vyjádření změn nižšího stupně může podpořit postižení v rámci jiné diagnózy nebo vést k pečlivému dovyšetření pacienta či odběru nové lymfatické uzliny v čase. Pacient se suspekcí na multicentrickou Castlemanovou chorobu by měl ideálně být předán do péče hemato-onkologa, a protože patolog obvykle v době diagnostiky potenciální Castlemanovy choroby nemá dostatek klinických informací, může na základě gradingu vyjádřit různou míru jistoty, resp. suspekce z diagnózy Castlemanovy choroby. Například skupina italských autorů využila grading a CDCN kritéria k retrospektivní analýze případů lymfadenopatií s Castleman-like změnami a u 60 % případů potvrdila diagnózu CD, což zdůrazňuje jednak nutnost klinicko-patologické spolupráce a zároveň potvrzuje nakolik je CD mnohdy poddiagnostikovaná (43).
SEKUNDÁRNÍ MALIGNITY SPOJENÉ S CASTLEMANOVOU NEMOCÍ
Sekundární malignity u pacientů s CD jsou vcelku časté. Pacienti s UCD mají zvýšené riziko rozvoje FDC sarkomu i lymfomů Hodgkinova i non-Hodgkinova typu. Riziko rozvoje lymfomu je zvýšeno i u HIV pozitivních pacientů s HHV-8-pozitvní MCD, a to až 15x oproti běžné populaci. Až 50 % pacientů s HHV-8-pozitivní MCD rozvine Kaposiho sarkom, a u pacientů s iMCD je riziko rozvoje malignity až třikrát vyšší.
Vztah UCD a klasického Hodgkinova lymfomu (CHL) je komplikovaný a odlišení obou lézí může být obtížné. Castlemanova choroba může vykazovat Hodgkin-like rysy především díky atypickým dendritickým buňkám nebo i díky přítomnosti CD30-pozitvních imunoblastů. Stejně tak CHL se může prezentovat CD-like morfologií (80,96). Klasický Hodgkinův lymfom byl opakovaně popsán u pacientů s UCD a většina případů UCD a CHL byla zachycena v jedné lymfatické uzlině. Nádorové elementy CHL navíc produkují IL-6 a mohou tedy indukovat rozvoj morfologických CD-like změn a odlišit reaktivní CD-like pozadí od pravého projevu UCD může být obtížné (obr. 12) (80,96).

Klasický Hodgkinův lymfom zachycený společně Castlemanovou chorobou se často prezentuje jako interfolikulární varianta (až 2/3 případů) a obvykle je spojen s plazmocytární morfologií CD. Fenotyp nádorových buněk CHL je obvyklý s koexpresí CD30/CD15 a slabou expresí PAX5. Část případů vykazuje i přítomnost EBV a IL-6 (96).
Non-Hodgkinské lymfomy jsou naopak častěji spojené s HV-UCD a vyskytují se obvykle v uzlinách jiné oblasti (72 %) a jedná se více o B-buněčné lymfomy. Mezi nejčastější typy patří difúzní velkobuněčný B-lymfom (DLBCL) včetně KSHV/HHV-8 pozitivní varianty, další HHV-8 asociované lymfomy jako primární exsudativní lymfom (PEL) či lymfom z buněk pláště nebo plazmocytární myelom (78,80,97). Nutné je zároveň odlišit lymfomy s CD-like morfologií (např. folikulární lymfom, viz níže) bez přímé vazby na Castlemanovu chorobu (80,93). Zhruba 10-20 % pacientů s HV-UCD rozvine sarkom z folikulárních dendritických buněk (FDCS), někteří na podkladě proliferace interfolikulárně lokalizovaných dysplastických FDC buněk (obr. 13). FDCS může na UCD navazovat jako sekundární malignita nebo může být diagnostikován souběžně s diagnózou UCD. (76,98) FDCS je tvořen fascikulární, storiformní až nodulární proliferací ovoidních až vřetenitých elementů, které exprimují FDC markery. Popsány jsou i vzácnější tumory vznikající z FRC buněk a nově byl popsán také neobvyklý stromální tumor s prokázanou přestavbou genu ALK kombinující rysy HV-CD a inflamatorního myofibroblatického tumoru (IMT) (99,100).

Kaposiho sarkom (KS) je KSHV/HHV-8 asociovaný low-grade vaskulární tumor, který postihuje častěji pacienty s HIV-pozitivní HHV-8-pozitivní MCD (až 75 %), méně často pak pacienty s HIV - -negativní HHV-8-pozitivní MCD (cca 13 %) (101). KS se nejčastěji vyskytuje v kůži, ale může postihovat i sliznice či gastrointestinální trakt. U pacientů s HHV-8-pozitivní MCD je pak navíc KS často (až v 63 %) zachycen ve stejné lymfatické uzlině, která vykazuje známky MCD, kde postihuje buď endotelie v oblasti pouzdra nebo sept (76,101) (obr. 14 A, B).

CASTLEMAN-LIKE MORFOLOGIE A KLINICKO-PATOLOGICKÁ DIFERENCIÁLNÍ DIAGNÓZA CD
Morfologické rysy připomínající histopatologické změny při Castlemanově chorobě (CD-like) se mohou objevit v lymfatických uzlinách i na podkladě jiné etiologie. CD-like změny se mohou vyskytovat jak v nenádorových lymfadenopatiích, např. v terénu autoimunitního onemocnění nebo na podkladě infekcí jiných, než je HHV8 anebo se jedná o nádorové lymfoproliferace případně proliferace ze stromálních buněk. Ze stejných skupin jsou i onemocnění, které byly zahrnuty do exkluzních kritérií pro iMCD, která navíc zahrnují i onemocnění mimikující iMCD na základně podobných klinických příznaků (tab. 2). Nicméně s ohledem na obvyklou neznalost klinických projevů a rozsahu onemocnění jsou pro patologickou diagnostiku nejdůležitější léze mimikující CD morfologicky, které navíc při nesprávném rozpoznání mohou následně vést k odlišnému terapeutickému postupu.
Nenádorové léze podobné CD
Hlavní diferenciální diagnózou především pro plazmocytární variantu CD je lymfadenopatie při IgG4-asociované chorobě (IgG4-related disease, IgG4-RD). IgG4-RD je chronické imunitně podmíněné multisystémové onemocnění spojené obvykle s tumorózním zvětšením postižených orgánů při fibroinflamatorních změnách se zvýšenou účastí IgG4 pozitivních plazmocytů a se zvýšenými hladinami sérového IgG4. Postižení lymfatických uzlin, na rozdíl od jiných orgánů, není spojeno se storiformní fibrózou a flebitidou, ale zahrnuje pět morfologických podtypů: multicentrický Castleman-like typ (I), typ s reaktivní folikulární hyperplázii (II), interfolikulární expanzi s imunoblasty (III), progresivní tranformaci zárodečných center (PTGC-typ, IV) a změny charakteru zánětlivého pseudotumoru (typ V) (102-106). Postižena může být jedna i více lymfatických uzlin. CD-like varianta IgG4 lymfadenopatie pak nejvíce připomíná plazmocytární nebo smíšenou morfologii CD s interfolikulárními infiltráty plazmocytů. (obr. 15) Lymfatické folikuly jsou častěji normální velikosti nebo hyperplastické, ale může být přítomen znak lízátka (céva vstupující do zárodečného centra) a rozšíření plášťů folikulů (103,107).

Odlišení IgG4-lymfadenopatie typu I a multicentrické CD, především iMDC-IPL, může být na morfologické úrovni obtížné. Obě onemocnění spojují vyšší hladiny IgG4 v séru a přítomnost IgG4 pozitivních plazmocytů v interfolikulární oblasti. Počet IgG4 pozitivních plazmocytů u iMCD-IPL je však průměrně nižší a pouze některé případy splní početní kritéria pro IgG4-RD (106). Navíc, exprese IgG4 u IgG4-RD je indukována interleukinem-4 a interleukinem-10. V případě Castlemanovy choroby indukuje produkci imunoglobulinů IL-6 a dochází ke zvýšené produkci i jiných tříd imunoglobulinů včetně IgA. Mezi parametry, které favorizují dg. IgG4 lymfadenopatie patří vyšší věk pacientů (průměrně 5.–6. dekáda), nízké hladiny CRP a IgA v séru, atopická historie a přítomnost extranodálního postižení (108,109). Na morfologické úrovni lze využít přítomnost směsné populace zralých i méně zralých plazmocytů, imunoblastů, malých lymfoctyů a eosinofilních granulocytů, které jsou častější v IgG4-RD a naopak vyšší přítomnost IgA pozitivních buněk a výrazná depozita hemosiderinu v makrofázích v MCD (87,102,106,108-112).
Mezi další imunitně podmíněná a autoimunitní onemocnění, jejichž součástí je lymfadenopatie s možnými Castleman-like změnami, patří revmatoidní atritida (RA), juvenilní idiopatická artritida, Stillova choroba dospělých, systémový lupus erytematodes (SLE) a autoimunitní lymfoproliferativní syndrom (ALPS) (80,113). Infekce, které pak mohou vykazovat CD-like změny, jsou především pozdní fáze HIV-asociované lymfadenitidy, toxoplazmóza či infekce virem Epstein-Barrové (EBV) a cytomegalovirem (CMV). Vyloučení výše uvedených chorob je také součástí klinických exkluzních kritérií (30,80).
Nádorové proliferace podobné CD
B-buněčné non-Hodgkinské lymfomy
Morfologickou podobnost s CD, obvykle ve formě hyalinně-vaskulárních změn s expanzí folikulů a rozšířením plášťové zóny, vykazují lymfomy s nodulárním typem růstu vznikající z některé části lymfatického folikulu. Jedná se nejčastěji o klasický folikulární lymfom (cFL), lymfom z buněk pláště (MCL) a nodální lymfom z buněk marginální zóny (nMZL) (93,114). V případě cFL se jedná obvykle o plně rozvinutý obraz s folikulárním růstem a adekvátním fenotypem v imunohistochemickém vyšetření (CD10/BCL6/BCL2), kdy morfologie HV-CD morfologie je vyjádřena pouze v části lymfatické uzliny (114) (obr. 16 A,B,C). Nicméně nádorové folikuly jsou více buněčné a hranice mezi zárodečnými centry a rozšířenými plášti je u FL neostrá. U plně rozvinutého MCL je prominentní především rozšíření plášťů a přítomnost atretických zárodečných center (093). (obr. 16 D,E,F) MCL ale může být zastižen i ve formě in situ proliferace, kdy věrně imituje cibulovitě uspořádané plášťové zóny HV-CD a existuje i varianta MCL s interfolikulární expanzí plazmocytů připomínající PC-CD. (115,116) Imunohistochemický průkaz aberantního fenotypu s koexpresí CD20, CD5 a cyklinuD1 je pak opět důležitý pro správné zařazení do lymfomové kategorie. Plazmocytárně diferencované lymfomy jako je MZL nebo lymfoplazmocytický lymfom (LPL) pak mohou úspěsně mimikovat plazmocytární variantu CD včetně monotypické exprese lehkého řetězce imunoglobulinů (80,93).

T-buněčné non-Hodgkinské lymfomy
Především skupina nodálních T-buněčných lymfomů z folikulárních pomocných lymfocytů (nTFHL) včetně angioimunoblastického typu může vykazovat CD-like změny s atretickými zárodečnými centry, zvýšenou vaskularitou interfolikulárních prostor a plazmocytární příměsí. Na rozdíl od CD je však u těchto lymfomů přítomna hyperplázie sítí FDC, nicméně zapojení spektra imunohistochemických markerů a případného molekulárního vyšetření je pro vyloučení lymfomu kruciální (80).
KSHV/HHV-8-asociovaná germinotropní lymfoproliferativní porucha
Samostatnou zmínku si zaslouží také plazmablastická proliferace popsaná v rámci studia lézí spojených s infekcí HHV-8. Tato léze je spojená s agregáty HHV-8 pozitivních plazmablastů v zárodečných centrech lymfatických folikulů často se současnou pozitivitu při průkazu infekce virem EBV (117-119). Tato léze dostala název KSHV/HHV-8 asociovaná germinotropní lymfoproliferativní porucha a postihuje obvykle imunokompetentní pacienty bez průkazu infekce HIV. Přestože může vykazovat některé morfologické rysy MCD, tak je v současné době zařazena v rámci WHO klasifikace jako samostatná jednotka v kategorii B-buněčných lymfoproliferací a lymfomů asociovaných s KSHV/ HHV-8 a od Castlemanovy choroby ji odlišuje právě ko-infekce s EBV (39,119-121).
TERAPEUTICKÉ MOŽNOSTI A PROGNÓZA CD
Primárním terapeutickým zásahem v případě UCD je kompletní chirurgická excize postižené lymfatické uzliny. Rekurence jsou vzácné, bez ohledu na morfologický podtyp. Asymptomatičtí pacienti s neresekovatelnou UCD mohou být sledováni, v případě kompresních příznaků je možná imunochemoterapie (s navazující resekcí při redukci velikosti léze, radioterapie či embolizace). Pacienti s inflamatorními projevy a neresekovatelným onemocněním mohou podstoupit anti-IL-6 terapii nebo alternativně imunoterapii s možnou kombinací s kortikosteroidy (83).
Prognóza pacientů s UCD je obvykle excelentní a celkové pětileté se pohybuje mezi 95–98 % (21,122). Pro případy regionální resp. oligocentrické CD je upřednostňován spíše konzervativní přístup s primární chirurgickou terapií či debulkingem oproti systémové chemoterapii. Nicméně pacienti s inflamatorními symptomy budou spíše profitovat z terapeutického algoritmu pro iMCD (32,83).
Terapie MCD se liší podle jednotlivých podtypů. V případě HHV-8-pozitivní MCD je na prvním místě biologická léčba anti-CD20 protilátkou (rituximab) nebo kombinace s kortikosteroidy či chemoterapeutiky (68,123). Léčba MCD-POEMS cílí na eliminaci vyvolávající plazmocytární neoplázie, pokud je taková nalezena a diagnostikována. Základem terapie iMCD je potlačení aktivity interleukinu-6 pomocí cílené biologické léčby (124 - 126). Anti-IL-6 preparáty siltuximab a tocilizumab neutralizují biologickou aktivitu IL-6 přímou vazbu na IL-6 resp. blokováním receptoru pro IL-6. Obvyklá je kombinovaná terapie s rituximabem nebo se systémovou chemoterapií, a to podle zhodnocení tíže onemocnění. iMCD-TAFRO spadá mezi těžké onemocnění s potenciálně fulminantním průběhem a terapie tedy zahrnuje anti-IL-6 terapii nebo systémovou kombinovanou chemoterapii (42,60,61,68,127).
Před zavedením anti-IL-6 terapie byla diagnóza iMCD spojena s relativně špatnou prognózou a pětileté celkové přežití dosahovalo zhruba 65 % (21). Zavedení multimodální terapie zahrnující anti-IL-6 terapii, kortikosteroidy, rituximab, chemoterapii a další typy imunosupresivních a imunomodulačních léčiv přežití pacientů postupně zlepšuje a celkové pětileté přežití se blíží k 95 % (21,42,128). Především pacienti s iMCD jsou nicméně zatíženi dalšími komorbiditami či opakovaným vzplanutím onemocnění. (129) V současné době se pak výzkum soustředí na identifikaci prediktivních markerů jako je CXCL13 u iMCD, jehož pokles sérových hladin CXCL13 po zahájení IL-6 terapie predikuje léčebnou odpověď na anti-IL6 terapii v dalších cyklech (130).
ZÁVĚR
Pojem Castlemanova choroba v současné době zastřešuje skupinu několika onemocnění s podobnou morfologií, ale rozdílnou etiologií, klinickou prezentací i terapeutickými přístupy. Cílem tohoto přehledového sdělení bylo poukázat na současné pojetí diagnózy Castlemanovy choroby a komplexní přístup k diagnostice jednotlivých podtypů. Přestože se jedná o skupinu vzácných onemocnění, je nutné mít CD na paměti, především při diagnostice rozličných lymfadenopatií. Morfologické podezření na Castlemanovu chorobu může být sice vyjádřeno již ze základního barvení, nutné je ale doplnění imunohistochemického vyšetření k vyloučení široké škály chorob včetně některých nádorových lymfoproliferací. Případně je vhodné doplnění těchto metod ve spolupráci s pracovištěm, které se zabývá diagnostikou hematologických malignit. Pro potvrzení diagnózy a přesné určení typu Castlemanovy choroby je kruciální spolupráce s klinickými lékaři s předáním suspektního pacienta do péče hematologů či hematoonkologů.
PODĚKOVÁNÍ A DEDIKACE
Tento výstup vznikl v rámci programu Cooperatio, vědní oblasti DIAG a byl podpořen projektem BBMRI-CZ LM2023033 a MZ ČR – RVO (FNHK, 00179906).
PROHLÁŠENÍ
Autor práce prohlašuje, že v souvislosti s tématem, vznikem a publikací tohoto článku není ve střetu zájmů a vznik ani publikace článku nebyly podpořeny žádnou farmaceutickou firmou. Toto prohlášení se týká i všech spoluautorů.
Zdroje
1. Castleman B, Towne VW. Case records of the Massachusetts General Hospital: Case No. 40011. N Engl J Med 1954; 250(1): 26-29
2. Castleman B, Iverson L, Menendez VP. Localized mediastinal lymph node hyperplasia resembling thymoma. Cancer 1956; 9 : 822–830.
3. Keller AR, Hochholzer L, Castleman B. Hyaline-vascular and plasma-cell types of giant lymph node hyperplasia of the mediastinum and other locations. Cancer 1972; 29(3): 670 - 683.
4. Frizzera G, Banks PM, Massarelli G, et al. A systemic lymphoproliferative disorder with morphologic features of Castleman’s disease. Pathological findings in 15 patients. Am J Surg Pathol 1983; 7 : 211–231.
5. Gaba AR, Stein RS, Sweet DL, et al. Multicentric giant lymph node hyperplasia. Am J Clin Pathol 1978; 69 : 86–90.
6. Frizzera G, Peterson BA, Bayrd ED, et al. A systemic lymphoproliferative disorder with morphologic features of Castleman’s disease: clinical findings and clinicopathologic correlations in 15 patients. J Clin Oncol 1985; 3 : 1202–1216.
7. Weisenburger DD, Nathwani BN, Winberg CD, et al. Multicentric angiofollicular lymph node hyperplasia: a clinicopathologic study of 16 cases. Hum Pathol 1985; 16 : 162–172.
8. Bardwick PA, Zvaifler NJ, Gill GN, et al. Plasma cell dyscrasia with polyneuropathy, organomegaly, endocrinopathy, M protein, and skin changes: the POEMS syndrome. Report on two cases and a review of the literature. Medicine 1980; 59(4): 311-322.
9. Dispenzieri A. POEMS syndrome: 2021 Update on diagnosis, risk-stratification, and management. Am J Hematol 2021; 96(7): 872 - 888.
10. Yoshizaki K, Matsuda T, Nishimoto N, et al. Pathogenic significance of interleukin-6 (IL6/BSF-2) in Castleman‘s disease. Blood 1989; 74(4): 1360-1367.
11. Yoshizaki K, Murayama S, Ito H, et al. The Role of Interleukin-6 in Castleman Disease. Hematol Oncol Clin North Am 2018; 32(1): 23 - 36.
12. Chang Y, Cesarman E, Pessin MS, et al. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science 1994; 266 : 1865–1869.
13. Moore PS, Chang Y. Detection of herpesvirus-like DNA sequences in Kaposi’s sarcoma in patients with and without HIV infection. N Engl J Med 1995; 332 : 1181–1185.
14. Cesarman E, Chang Y, Moore PS, et al. Kaposi’s sarcoma associated herpesvirus-like DNA sequences in AIDS-related body-cavity - -based lymphomas. N Engl J Med 1995; 332 : 1186–1191.
15. Soulier J, Grollet L, Oksenhendler E, et al. Kaposi’s sarcomaassociated herpesvirus-like DNA sequences in multicentric Castleman’s disease. Blood 1995; 86 : 1276–1280.
16. Dupin N, Gorin I, Deleuze J, et al. Herpes-like DNA sequences, AIDS-related tumors, and Castleman’s disease. N Engl J Med 1995; 333 : 798.
17. Powles T, Stebbing J, Bazeos A, et al. The role of immune suppression and HHV-8 in the increasing incidence of HIV-associated multicentric Castleman‘s disease. Ann Oncol 2009; 20(4): 775-779.
18. Liu AaY, Nabel CS, Finkelman BS, et al. Idiopathic multicentric Castleman‘s disease: a systematic literature review. Lancet Haematol 2016; 3(4): e163-e175.
19. Dispenzieri A, Fajgenbaum DC. Overview of Castleman disease. Blood 2020; 135(16): 1353-1364.
20. Carbone A, Borok M, Damania B, et al. Castleman disease. Nat Rev Dis Primers 2021; 7(1): 84.
21. Nishimura MF, Nishimura Y, Nishikori A, et al. Historical and pathological overview of Castleman disease. J Clin Exp Hematop 2022; 62(2): 60-72.
22. Hoffmann C, Oksenhendler E, Littler S, et al. The clinical picture of Castleman disease: a systematic review and meta-analysis. Blood Adv 2024; 8(18): 4924-4935.
23. Yu L, Tu M, Cortes J, et al. Clinical and pathological characteristics of HIV - and HHV - -8-negative Castleman disease. Blood 2017; 129(12): 1658-1668.
24. Takai K, Nikkuni K, Shibuya H et al. Thrombocytopenia with mild bone marrow fibrosis accompanied by fever, pleural effusion, ascites and hepatosplenomegaly. Japanese J Clin Hematol 2010; 51 : 320-325.
25. Kawabata H, Takai K, Kojima M, et al. Castleman-Kojima disease (TAFRO syndrome): a novel systemic inflammatory disease characterized by a constellation of symptoms, namely, thrombocytopenia, ascites (anasarca), microcytic anemia, myelofibrosis, renal dysfunction, and organomegaly : a status report and summary of Fukushima (6 June, 2012) and Nagoya meetings (22 September, 2012). J Clin Exp Hematop 2013; 53(1): 57-61.
26. Iwaki N, Fajgenbaum DC, Nabel CS, et al. Clinicopathologic analysis of TAFRO syndrome demonstrates a distinct subtype of HHV - -8-negative multicentric Castleman disease. Am J Hematol 2016; 91(2): 220-226.
27. Nishimura Y, Fajgenbaum DC, Pierson SK, et al. Validated international definition of the thrombocytopenia, anasarca, fever, reticulin fibrosis, renal insufficiency, and organomegaly clinical subtype (TAFRO) of idiopathic multicentric Castleman disease. Am J Hematol 2021; 96(10): 1241-1252.
28. Masaki Y, Kawabata H, Takai K, et al. 2019 Updated diagnostic criteria and disease severity classification for TAFRO syndrome. Int J Hematol 2020; 111(1): 155-158.
29. Fajgenbaum DC, van Rhee F, Nabel CS. HHV-8-negative, idiopathic multicentric Castleman disease: novel insights into biology, pathogenesis, and therapy. Blood 2014; 123(19): 2924-2933.
30. Fajgenbaum DC, Uldrick TS, Bagg A, et al. International, evidence-based consensus diagnostic criteria for HHV-8-negative/idiopathic multicentric Castleman disease. Blood 2017; 129(12): 1646-1657.
31. Zhang L, Liu QH, Zhou H, et al. Asymptomatic multicentric Castleman disease: a potential early stage of idiopathic MCD. Blood Adv 2024; 8(21): 5598-5602.
32. Pierson SK, Brandstadter JD, Torigian DA, et al. Characterizing the heterogeneity of Castleman disease and oligocentric subtype: findings from the ACCELERATE registry. Blood Adv 2025; 9(8): 1952-1965.
33. Chen LYC, Zhang L, Fajgenbaum DC. Expert Perspective: Diagnosis and Treatment of Castleman Disease. Arthritis Rheumatol. Published online June 2, 2025.
34. Gao YH, Liu YT, Zhang MY, et al. Idiopathic multicentric Castleman disease (iMCD)-idiopathic plasmacytic lymphadenopathy: A distinct subtype of iMCD-not otherwise specified with different clinical features and better survival. Br J Haematol 2024; 204(5): 1830-1837.
35. Mori S., Mohri N., Uchida T. et al. [Idiopathic plasmacytic lymphadenopathy with polyclonal hyperimmunoglobulinemia: a syndrome related to giant lymph node hyperplasia of plasma cell type]. J Jpn Soc Res 1980; 20(suppl): 55-65.
36. Kojima M, Nakamura N, Otuski Y, et al. Pulmonary lesion of idiopathic plasmacytic lymphadenopathy with polyclonal hyperimmunoglobulinemia appears to be a cause of lymphoplasmacytic proliferation of the lung: a report of five cases. Pathol Res Pract 2008; 204(3): 185-190.
37. Nishikori A, Nishimura MF, Nishimura Y, et al. Idiopathic Plasmacytic Lymphadenopathy Forms an Independent Subtype of Idiopathic Multicentric Castleman Disease. Int J Mol Sci 2022; 23(18): 10301.
38. Nishikori A, Nishimura MF, Nishimura Y, et al. Distinct interleukin-6 production in IPL and TAFRO subtypes of idiopathic multicentric Castleman disease. Haematologica Published online September 11, 2025.
39. Judith AF, Naresh KN, Chadburn A et al. Tumour-like lesions with B-cell predominance – Castleman disease In: WHO Classification of Tumours Editorial Board. Haematolymphoid tumours [Internet]. Lyon (France): International Agency for Research on Cancer; 2024 [cited 2025/10/15]. (WHO classification of tumours series, 5th ed.; vol. 11). Available from: https://tumourclassification.iarc.who. int/chapters/XX.
40. Munshi N, Mehra M, van de Velde H et al. Use of a claims database to characterize and estimate the incidence rate for Castleman disease. Leuk Lymphoma 2015; 56(5): 1252-1260.
41. Mukherjee S, Martin R, Sande B et al. Epidemiology and treatment patterns of idiopathic multicentric Castleman disease in the era of IL-6-directed therapy. Blood Adv 2022; 6(2): 359-367.
42. Lang E, van Rhee F. Idiopathic multicentric Castleman disease: An update in diagnosis and treatment advances. Blood Rev 2024; 64 : 101161.
43. Pelliccia S, Rogges E, Cardoni A, et al. The application of a multidisciplinary approach in the diagnosis of Castleman disease and Castleman-like lymphadenopathies: A 20-year retrospective analysis of clinical and pathological features. Br J Haematol 2024; 204(2): 534-547.
44. Fajgenbaum DC, Shilling D. Castleman Disease Pathogenesis. Hematol Oncol Clin North Am 2018; 32(1): 11-21.
45. Vermi W, Lonardi S, Bosisio D, et al. Identification of CXCL13 as a new marker for follicular dendritic cell sarcoma. J Pathol 2008; 216(3): 356-364.
46. Chang KC, Wang YC, Hung LY, et al. Monoclonality and cytogenetic abnormalities in hyaline vascular Castleman disease. Mod Pathol 2014; 27(6): 823-831.
47. Li Z, Lan X, Li C, et al. Recurrent PDGFRB mutations in unicentric Castleman disease. Leukemia 2019; 33(4): 1035-1038.
48. Smith D, Eichinger A, Fennell É, et al. Spatial and single cell mapping of castleman disease reveals key stromal cell types and cytokine pathways. Nat Commun 2025; 16(1): 6009.
49. Horna P, King RL, Jevremovic D, et al. The lymph node transcriptome of unicentric and idiopathic multicentric Castleman disease. Haematologica 2023; 108(1): 207-218.
50. Oksenhendler E, Carcelain G, Aoki Y, et al. High levels of human herpesvirus 8 viral load, human interleukin-6, interleukin-10, and C reactive protein correlate with exacerbation of multicentric castleman disease in HIV-infected patients. Blood 2000; 96(6): 2069-2073.
51. Dupin N, Diss TL, Kellam P, et al. HHV-8 is associated with a plasmablastic variant of Castleman disease that is linked to HHV-8-positive plasmablastic lymphoma. Blood 2000; 95(4): 1406-1412.
52. Chadburn A, Hyjek EM, Tam W, et al. Immunophenotypic analysis of the Kaposi sarcoma herpesvirus (KSHV; HHV-8)-infected B cells in HIV+ multicentric Castleman disease (MCD). Histopathology 2008; 53(5): 513-524.
53. Oksenhendler E, Boutboul D, Fajgenbaum D, et al. The full spectrum of Castleman disease: 273 patients studied over 20 years. Br J Haematol 2018; 180(2): 206-216.
54. Kanai K, Sawai S, Sogawa K, et al. Markedly upregulated serum interleukin-12 as a novel biomarker in POEMS syndrome. Neurology 2012; 79(6): 575-582.
55. Wang C, Huang XF, Cai QQ, et al. Remarkable expression of vascular endothelial growth factor in bone marrow plasma cells of patients with POEMS syndrome. Leuk Res 2016; 50 : 78-84.
56. Nagao Y, Mimura N, Takeda J, et al. Genetic and transcriptional landscape of plasma cells in POEMS syndrome. Leukemia 2019; 33(7): 1723-1735.
57. Miller I, Mumau MD, Shyamsundar S, et al. No evidence for active viral infection in unicentric and idiopathic multicentric Castleman disease by Viral-Track analysis. Sci Rep 2025; 15(1): 1676.
58. Fajgenbaum DC, June CH. Cytokine Storm. N Engl J Med 2020; 383(23): 2255-2273.
59. Harada T, Kikushige Y, Miyamoto T, et al. Peripheral helper-T-cell-derived CXCL13 is a crucial pathogenic factor in idiopathic multicentric Castleman disease. Nat Commun 2023; 14(1): 6959.
60. van Rhee F, Voorhees P, Dispenzieri A, et al. International, evidence-based consensus treatment guidelines for idiopathic multicentric Castleman disease. Blood 2018; 132(20): 2115-2124.
61. Alnoor F, Spies NC, Kumar J, et al. The Evolution and Recent Advances in Diagnostic Criteria for Idiopathic Multicentric Castleman Disease. Am J Hematol 2025; 100(11): 2064 - 2073.
62. Wing A, Xu J, Meng W, et al. Transcriptome and unique cytokine microenvironment of Castleman disease. Mod Pathol 2022; 35(4): 451-461.
63. Pierson SK, Shenoy S, Oromendia AB, et al. Discovery and validation of a novel subgroup and therapeutic target in idiopathic multicentric Castleman disease. Blood Adv 2021; 5(17): 3445-3456.
64. Caballero JC, Conejero N, Solan L et al. Unraveling TAFRO Syndrome: An In-Depth Look at the Pathophysiology, Management, and Future Perspectives. Biomedicines 2024; 12(5): 1076.
65. Arenas DJ, Floess K, Kobrin D, et al. Increased mTOR activation in idiopathic multicentric Castleman disease. Blood 2020; 135(19): 1673-1684.
66. Sumiyoshi R, Koga T, Kawakami A. Biomarkers and Signaling Pathways Implicated in the Pathogenesis of Idiopathic Multicentric Castleman Disease/Thrombocytopenia, Anasarca, Fever, Reticulin Fibrosis, Renal Insufficiency, and Organomegaly (TAFRO) Syndrome. Biomedicines 2024; 12(6): 1141.
67. Milota T, Střížová Z, Sobotková M, Bartůňková J. Úloha interleukinu-6 v patogenezi a léčbě Castlemanovy choroby – pohled imunologa. Cas Lek Cesk 2023; 162(2-3): 106-111.
68. NCCN Clinical Practice Guidelines in Oncology – Castleman disease, verze 2.2025
69. DM, Warnke RA. Castleman disease: an update on classification and the spectrum of associated lesions. Adv Anat Pathol 2009; 16(4): 236-246.
70. Medina EA, Fuehrer NE, FR et al. Dysplastic follicular dendritic cells in hyaline-vascular Castleman disease: a rare occurrence creating diagnostic difficulty. Pathol Int 2016; 66(9): 535-539.
71. Nishimura MF, Nishimura Y, Nishikori A, et al. Clinical and Pathological Characteristics of Hyaline-Vascular Type Unicentric Castleman Disease: A 20-Year Retrospective Analysis. Diagnostics (Basel) 2021; 11(11): 2008.
72. Alnoor F, Rangel A, Luo M, et al. Unicentric Castleman Disease: Updates and Novel Insights Into Spindle Cell Proliferations and Aggressive Forms of a Localized Disease. Int J Lab Hematol 2025; 47(1): 26-35.
73. Murro D, Agab M, Brickman A et al. Cytological features of Castleman disease: a review. J Am Soc Cytopathol 2016; 5(2): 100-106.
74. Ohgami RS, Zhao S, Ohgami JK, et al. TdT+ T-lymphoblastic populations are increased in Castleman disease, in Castleman disease in association with follicular dendritic cell tumors, and in angioimmunoblastic T-cell lymphoma. Am J Surg Pathol 2012; 36(11): 1619 - 1628.
75. Saglam A, Singh K, Gollapudi S, et al. Indolent T-lymphoblastic proliferation: A systematic review of the literature analyzing the epidemiologic, clinical, and pathologic features of 45 cases. Int J Lab Hematol 2022; 44(4): 700-711.
76. Gasljevic G, Bonometti A, Anagnostopoulos I, et al. The morphological spectrum of Castleman disease and related disorders: a report from the Lymphoma Workshop of the 22nd Meeting of the European Association of Hematopathology. Virchows Arch 2025; 487(2): 253-273.
77. Rodriguez-Merino L, Montes-Moreno S. Castleman disease-type histopathological patterns of lymph nodes in patients with plasma cell neoplasia and POEMS syndrome. Ann Diagn Pathol 2025; 74 : 152414.
78. Zhou T, Wang HW, Pittaluga S et al. Multicentric Castleman disease and the evolution of the concept. Pathologica 2021; 113(5): 339 - 353.
79. Wu D, Lim MS, Jaffe ES. Pathology of Castleman Disease. Hematol Oncol Clin North Am 2018; 32(1): 37-52.
80. Zhang X, Niyazi S, Guo H et al. Mimickers and Associated Neoplasms of Castleman Disease. J Clin Transl Pathol 2025; 5(1): 20-29.
81. Du MQ, Liu H, Diss TC, et al. Kaposi sarcoma-associated herpesvirus infects monotypic (IgM lambda) but polyclonal naive B cells in Castleman disease and associated lymphoproliferative disorders. Blood 2001; 97(7): 2130-2136.
82. Dargent JL, Lespagnard L, Sirtaine N et al. Plasmablastic microlymphoma occurring in human herpesvirus 8 (HHV-8)-positive multicentric Castleman‘s disease and featuring a follicular growth pattern. APMIS 2007; 115(7): 869-874.
83. van Rhee F, Oksenhendler E, Srkalovic G, et al. International evidence-based consensus diagnostic and treatment guidelines for unicentric Castleman disease. Blood Adv 2020; 4(23): 6039-6050.
84. Beckham TH, Yang JC, Chau KW et al. Excellent Outcomes with Surgery or Radiotherapy in the Management of Castleman Disease Including a Case of Oligocentric Disease. Clin Lymphoma Myeloma Leuk 2020; 20(10): 685 - 689.
85. Dao LN, Hanson CA, Dispenzieri A et al. Bone marrow histopathology in POEMS syndrome: a distinctive combination of plasma cell, lymphoid, and myeloid findings in 87 patients. Blood 2011; 117(24): 6438-6444.
86. Belyaeva E, Rubenstein A, Pierson SK, et al. Bone marrow findings of idiopathic Multicentric Castleman disease: A histopathologic analysis and systematic literature review. Hematol Oncol 2022; 40(2): 191-201.
87. Nishikori A, Nishimura MF, Fajgenbaum DC, et al. Diagnostic challenges of the idiopathic plasmacytic lymphadenopathy (IPL) subtype of idiopathic multicentric Castleman disease (iMCD): Factors to differentiate from IgG4-related disease. J Clin Pathol 2025; 79(1): 43-49.
88. Fajgenbaum DC, Pierson SK, Kanhai K, et al. The disease course of Castleman disease patients with fatal outcomes in the ACCELERATE registry. Br J Haematol 2022; 198(2): 307 - 316.
89. Han EJ, O JH, Jung SE, et al. PET/CT Findings of Castleman Disease Assessed by Histologic Subtypes and Compared with Laboratory Findings. Diagnostics (Basel) 2020; 10(12): 998.
90. Koa B, Borja AJ, Aly M, et al. Emerging role of 18F-FDG PET/CT in Castleman disease: a review. Insights Imaging 2021; 12(1): 35.
91. Koukalová R, Selingerová I, Řehák Z et al. FDG-PET/ CT v diagnostice a hodnocení léčebné odpovědi Castlemanovy choroby - retrospektivní studie 29 případů z jednoho centra. Klin Onkol 2021; 34(2): 120-127.
92. Blommers M, Selegean S, Wood RK, et al. Idiopathic multicentric Castleman disease with marrow fibrosis and extramedullary hematopoiesis. Eur J Haematol 2024; 113(6): 833-841.
93. Siddiqi IN, Brynes RK, Wang E. B-cell lymphoma with hyaline vascular Castleman disease-like features: a clinicopathologic study. Am J Clin Pathol 2011; 135(6): 901-914.
94. https://www.castlemandiseaseeducation. com/grading 2025_11_05 12 : 58
95. Nishimura MF, Haratake T, Nishimura Y, et al. International Consensus Histopathological Criteria for Subtyping Idiopathic Multicentric Castleman Disease Based on Machine Learning Analysis. Am J Hematol 2025; 100(9): 1502-1512.
96. Lyapichev KA, You MJ, Vega F et al. Classic Hodgkin lymphoma and Castleman disease: an entity appears to be emerging. Virchows Arch 2020; 477(3): 437-444.
97. Hsi ED, Lorsbach RB, Fend F, Dogan A. Plasmablastic lymphoma and related disorders. Am J Clin Pathol 2011; 136(2): 183-194.
98. Facchetti F, Simbeni M, Lorenzi L. Follicular dendritic cell sarcoma. Pathologica 2021; 113(5): 316-329.
99. Andriko JW, Kaldjian EP, Tsokos M et al. Reticulum cell neoplasms of lymph nodes: a clinicopathologic study of 11 cases with recognition of a new subtype derived from fibroblastic reticular cells. Am J Surg Pathol 1998; 22(9): 1048-1058.
100. Yagi H, Satou A, Enomoto Y, et al. ALK-Rearranged Mesenchymal Neoplasm With Hyaline-Vascular Castleman Disease-Like Features: A Case Report. Pathol Int 2025; 75(10): 538-543.
101. Naresh KN, Rice AJ, Bower M. Lymph nodes involved by multicentric Castleman disease among HIV-positive individuals are often involved by Kaposi sarcoma. Am J Surg Pathol 2008; 32(7): 1006-1012.
102. Sato Y, Kojima M, Takata K, et al. Systemic IgG4-related lymphadenopathy: a clinical and pathologic comparison to multicentric Castleman‘s disease. Mod Pathol 2009; 22(4): 589-599.
103. Cheuk W, Bledsoe JR. IgG4-related lymphadenopathy. Semin Diagn Pathol 2024; 41(2): 108-115.
104. Jo JH, Park YS, Jeon YK et al. Comparison of plasma cell type of Castleman‘s disease and IgG4-related sclerosing disease: a histopathological and immunohistochemical study. Pathobiology 2011; 78(4): 227-232.
105. Umehara H, Okazaki K, Kawa S, et al. The 2020 revised comprehensive diagnostic (RCD) criteria for IgG4-RD. Mod Rheumatol 2021; 31(3): 529-533.
106. Nishikori A, Nishimura MF, Nishimura Y, et al. Investigation of IgG4-positive cells in idiopathic multicentric Castleman disease and validation of the 2020 exclusion criteria for IgG4-related disease. Pathol Int 2022; 72(1): 43-52.
107. Bledsoe JR, Ferry JA, Neyaz A, et al. IgG4-related Lymphadenopathy: A Comparative Study of 41 Cases Reveals Distinctive Histopathologic Features. Am J Surg Pathol 2021; 45(2): 178-192.
108. Sasaki T, Akiyama M, Kaneko Y, et al. Distinct features distinguishing IgG4-related disease from multicentric Castleman‘s disease. RMD Open 2017; 3(1): e000432.
109. Nijim S, Fajgenbaum DC. Identifying Castleman disease from non-clonal inflammatory causes of generalized lymphadenopathy. Hematology Am Soc Hematol Educ Program 2024; 2024(1): 582-593.
110. Manabe A, Igawa T, Takeuchi M et al. Immunohistochemical analysis of IgA expression differentiates IgG4-related disease from plasma cell-type Castleman disease. Med Mol Morphol 2017; 50(1): 34-41.
111. Otani K, Inoue D, Fujikura K, et al. Idiopathic multicentric Castleman‘s disease: a clinicopathologic study in comparison with IgG4-related disease. Oncotarget 2018; 9(6): 6691-6706.
112. Han Y, Igawa T, Ogino K, et al. Hemosiderin deposition in lymph nodes of patients with plasma cell-type Castleman disease. J Clin Exp Hematop 2020; 60(1): 1-6.
113. González García A, Fernández-Martín J, Robles Marhuenda Á. Idiopathic multicentric Castleman disease and associated autoimmune and autoinflammatory conditions: practical guidance for diagnosis. Rheumatology (Oxford) 2023; 62(4): 1426-1435.
114. Pina-Oviedo S, Wang W, Vicknair E et al. Follicular lymphoma with hyaline-vascular Castleman disease-like follicles and CD20 positive follicular dendritic cells. Pathology 2017; 49(5): 544-547.
115. Dobrea C, Mihai M, Dănăilă E et al. „In situ“ mantle cell lymphoma associated with hyaline-vascular Castleman disease. Rom J Morphol Embryol 2011; 52(3 Suppl): 1147-1151.
116. Igawa T, Omote R, Sato H, et al. A possible new morphological variant of mantle cell lymphoma with plasma-cell type Castleman disease-like features. Pathol Res Pract 2017; 213(11): 1378-1383.
117. Du MQ, Diss TC, Liu H, et al. KSHV - and EBV - -associated germinotropic lymphoproliferative disorder. Blood 2002; 100(9): 3415-3418.
118. Peker D, Alkan S, Zhang L et al. HIV-associated plasmablastic multicentric Castleman disease with microlymphoma coinfected with HHV8 and EBV. J Hematopathol 2013; 6, 109–114.
119. Bhavsar T, Lee JC, Perner Y, et al. KSHV-associated and EBV-associated Germinotropic Lymphoproliferative Disorder: New Findings and Review of the Literature. Am J Surg Pathol 2017; 41(6): 795-800.
120. Gonzalez-Farre B, Martinez D, Lopez-Guerra M, et al. HHV8-related lymphoid proliferations: a broad spectrum of lesions from reactive lymphoid hyperplasia to overt lymphoma. Mod Pathol 2017; 30(5): 745-760.
121. Zanelli M, Zizzo M, Bisagni A, et al. Germinotropic lymphoproliferative disorder: a systematic review. Ann Hematol 2020; 99(10): 2243-2253.
122. Boutboul D, Fadlallah J, Chawki S, et al. Treatment and outcome of Unicentric Castleman Disease: a retrospective analysis of 71 cases. Br J Haematol 2019; 186(2): 269-273.
123. Lurain K, Yarchoan R, Uldrick TS. Treatment of Kaposi Sarcoma Herpesvirus-Associated Multicentric Castleman Disease. Hematol Oncol Clin North Am 2018; 32(1): 75-88.
124. van Rhee F, Wong RS, Munshi N, et al. Siltuximab for multicentric Castleman‘s disease: a randomised, double-blind, placebo-controlled trial. Lancet Oncol 2014; 15(9): 966-974.
125. Deisseroth A, Ko CW, Nie L, et al. FDA approval: siltuximab for the treatment of patients with multicentric Castleman disease. Clin Cancer Res 2015; 21(5): 950-954.
126. Adam Z, Zeman D, Chodacki A, et al. Léčba Castlemanovy choroby siltuximabem – popis případu a přehled literatury. Klin Onkol 2023; 36(4): 320-329.
127. Lomas OC, Streetly M, Pratt G, et al. The management of Castleman disease. Br J Haematol 2021; 195(3): 328-337.
128. Ramaswami R, Lurain K, Polizzotto MN, et al. Characteristics and outcomes of KSHV - -associated multicentric Castleman disease with or without other KSHV diseases. Blood Adv 2021; 5(6): 1660-1670.
129. Bustamante MS, Pierson SK, Ren Y, et al. Longitudinal, natural history study reveals the disease burden of idiopathic multicentric Castleman disease. Haematologica 2024; 109(7): 2196-2206.
130. Pierson SK, Katz L, Williams R, et al. CXCL13 is a predictive biomarker in idiopathic multicentric Castleman disease. Nat Commun 2022; 13(1): 7236.
Štítky
Patologie Soudní lékařství ToxikologieČlánek vyšel v časopise
Česko-slovenská patologie

2026 Číslo 1
Nejčtenější v tomto čísle
- Testování exprese claudinu 18.2 u adenokarcinomu žaludku a gastroesofageální junkce: Současný stav a výhled do blízké budoucnosti
- Castlemanova choroba – jedno jméno, mnoho tváří
- Přednosti a limitace nového stagingového systému karcinomu endometria FIGO 2023 z pohledu klinika a patologa
- Cílené profilování genové exprese jako nástroj pro diagnostické určení buněčného subtypu a prognostickou stratifikaci u difuzního velkobuněčného B-lymfomu
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy