
Pětapadesátiletý muž s trombocytopenií a život ohrožujícím difuzním alveolárním krvácením: kazuistika
Fifty-five year old man with thrombocytopaenia and life threatening diffuse alveolar haemorrhage. Case report
This case report presents a patient with acute respiratory failure due to life-threatening haemoptysis resulting from severe and refractory thrombocytopenia. It presents the diagnostics, differential diagnostics and management of such illness.
KEYWORDS:
thrombocytopaenia – haemoptysis – respiratory failure – intensive care
Autoři:
T. Karvunidis 1; M. Harazím 1
![]() ; J. Raděj 1; P. Salaj 2; J. Horák 1; I. Novák 1; M. Matějovič 1
; J. Raděj 1; P. Salaj 2; J. Horák 1; I. Novák 1; M. Matějovič 1
Působiště autorů:
Jednotka intenzivní péče, I. interní klinika FN Plzeň a LF v Plzni, Univerzita Karlova v Praze
1; Ústav hematologie a krevní transfuze, Praha
2
Vyšlo v časopise:
Anest. intenziv. Med., 27, 2016, č. 3, s. 170-176
Kategorie:
Intenzivní medicína - Kazuistika
Souhrn
Kazuistika prezentuje případ nemocného s respiračním selháním při život ohrožující hemoptýze se současnou těžkou a refrakterní trombocytopenií, související diagnostiku, diferenciálně diagnostickou rozvahu a léčebný postup.
Klíčová slova:
trombocytopenie – hemoptýza – respirační selhání – intenzivní péče
KAZUISTIKA
Pětapadesátiletý muž, dřívějším povoláním obráběč kovů, s kontaktní dermatitidou, bez dalších zásadních chronických onemocnění, byl přijat na jednotku intenzivní péče (JIP) pro akutní respirační selhání s hemoptýzou. Tento stav byl vyústěním přibližně čtyři měsíce trvající postupné progrese námahové dušnosti a únavnosti, které byly následovány častější tvorbou hematomů. Již při primárním zajištění nemocného v regionální nemocnici před odesláním na naše pracoviště byla nezbytná orotracheální intubace a zahájení umělé plicní ventilace (UPV).
Bezprostředně po přijetí byla provedena bronchoskopie (BSK) se sanací dýchacích cest – odstraněním krve a koagul. Zdroj krvácení nebyl identifikován. Iniciálnímu vyšetření dominovalo postižení plic s difuzně zhrubělým poslechovým nálezem a četnými fenomény bronchostázy (krvácení). Po BSK byla přítomna již jen mírná porucha oxygenace a mírná retence CO2. Nebylo zjevné pokračující krvácení do dýchacích cest. Laboratorní screening odhalil těžkou trombocytopenii (11 x 109/l). Nebyla přítomna koagulopatie ani známky intravaskulární hemolýzy a i další laboratorní výsledky byly normální (tab. 1). Byly odebrány vzorky k laboratorní diagnostice případných autoimunitních onemocnění, testu přítomnosti trombocytárních autoprotilátek a kompletnímu mikrobiologickému screeningu včetně virologie. Bylo provedeno CT vyšetření plic ukazující difuzní oboustranně alární postižení plicního intersticia charakteru „crazy paving“ nejasné etiologie se současným difuzním alveolárním krvácením (difuse alveolar hemorrhage – DAH) (obr. 1). V rámci diferenciální diagnózy bylo z tohoto možné uvažovat o alergické (eozinofilní) granulomatózní angiitidě (syndromu Churg-Straussové), akutní intersticiální plicní fibróze (syndromu Hamman--Rich), difuzním bronchoalveolárním karcinomu či virové pneumonitidě. Další širší diferenciální diagnostiku a etiologii DAH shrnuje tabulka 1. Podle uvedených výsledků vyšetření však byla nejpravděpodobnější pracovní diagnózou imunitní (idiopatická) trombocytopenická purpura (ITP). Vzhledem k život ohrožujícímu DAH a současnému těžkému intersticiálnímu postižení plic s respiračním selháním byl bezprostředně po intubaci podán trombokoncentrát a zahájena systémová kortikoterapie vysokodávkovaným metylprednisolonem (HDMP; 500 mg/den s následným snížením dávky na 125 mg/den po pěti dnech). Následně došlo k zástavě krvácení, zlepšení mechaniky dýchání i výměny krevních plynů a třetí den hospitalizace bylo možné ukončit UPV a nemocného extubovat. Mikrobiologická vyšetření včetně virologických neidentifikovala žádné infekční agens způsobující uvedené symptomy a stav. Bylo provedeno i vyšetření kostní dřeně, které prokázalo fyziologickou hematopoezu ve všech řadách, bez známek primárního hematologického onemocnění či jiné patologické infiltrace kostní dřeně. Další vybrané výsledky vstupních vyšetření jsou sumarizovány v tabulce 2.


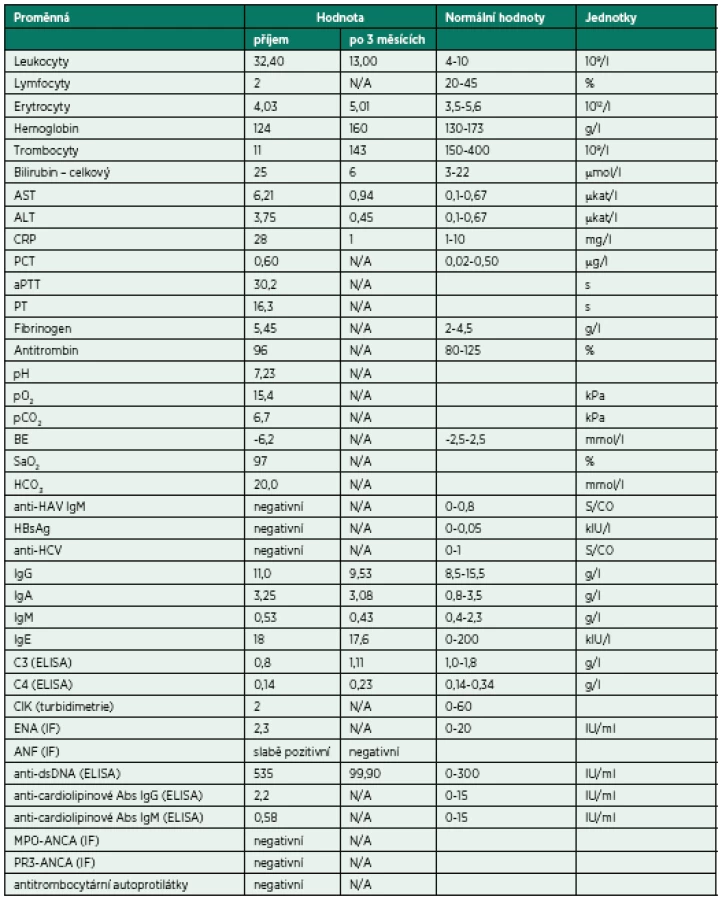
Pátý den hospitalizace došlo k recidivě krvácení do dýchacích cest, respektive DAH při perzistující těžké trombocytopenii (9 x 109/l) – obrázek 2. Opět byla nezbytná intubace, UPV, BSK sanace dýchacích cest a byla zahájena léčba intravenózními humánními polyvalentními imunoglobuliny (IVIG, 1 g/kg/den, celkem 2 dny). Detekovaná slabá pozitivita autoprotilátek proti dvojspirálové DNA (anti-ds-DNA), mírné snížení C3 složky komplementu a relativní lymfocytopenie vedla autory k zamyšlení, zda se v tomto případě nejedná o koincidenci ITP se systémovým lupusem (SLE), případně, zda trombocytopenie není sekundárním projevem SLE. Vzhledem k uvedenému a současné jasné absenci terapeutické odpovědi na zavedenou léčbu (HDMP + IVIG) byla sedmý den hospitalizace na JIP imunosupresivní léčba rozšířena o azatioprin.

Osmý den hospitalizace byl nemocný s trvající závažnou trombocytopenií (11 x 109/l) přeložen v relativně dobrém a stabilizovaném stavu na standardní oddělení a bylo provedeno kontrolní CT vyšetření plic ukazující významné zlepšení postižení plicního parenchymu, nicméně při trvajícím vážném reziduálním nálezu (obr. 3). Po dalších devíti dnech kombinované imunosupresivní léčby (kortikoid a azatioprin) relabovala těžká trombocytopenie (4 x 109/l) se současným krvácením do dýchacích cest (opět charakteru DAH) s respirační insuficiencí, respektive selháním. Nemocný byl opět přijat na JIP. Vzhledem k život ohrožujícímu stavu nebylo možné vyčkávat potenciálního efektu případné léčby cyklofosfamidem či plazmaferézou. Po zvážení všech možností, rizik a s ohledem na předchozí selhání léčby, byla indikována splenektomie jako postup, který může efektivně zabránit další destrukci trombocytů, a tím snížit riziko recidivy život ohrožujícího alveolárního krvácení. Po komplikované přípravě podáním množství trombokocentrátů byla dvacátý den hospitalizace nekomplikovaně provedena splenektomie. Následně došlo k vzestupu počtu trombocytů (20–30 x 109/l) a dramaticky klesla potřeba substituce trombocyty. Histologické vyšetření sleziny normální velikosti (14 x 8 x 8 cm) neodhalilo žádnou patologii. Současně se splenektomií byl zahájen druhý cyklus podání IVIG (0,5 g/kg/den, celková dávka 125 g v 5 dnech). Uvedený postup vedl k dalšímu vzestupu počtu krevních destiček (až 125 x 109/l) v následujících pěti dnech (viz obr. 2). Nemocný byl dále 27. den hospitalizace přeložen na hematologicko-onkologické oddělení a dále po několika dnech propuštěn do domácí a ambulantní péče již s téměř normálním krevním obrazem a bez dalších krvácivých komplikací na imunosupresivní léčbě kortikoidem (metylprednisolon 16 mg/den). Kontrolní CT plic prokázalo významné zlepšení postižení plicního intersticia (obr. 4).


DISKUSE
Imunitní (idiopatická) trombocytopenická purpura (ITP) je u dospělých relativně častým získaným onemocněním. Incidence je se zlepšující se diagnostikou v čase rostoucí a odhaduje se na 1–3 nové případy na 100 000 obyvatel za rok, použijeme-li jako diskriminační hodnotu počtu trombocytů 50 x 109/l [1, 2]. U dospělých má ve většině případů chronický průběh. U dětí naopak akutní a sebelimitující. Přibližně 70 % postižených tvoří ženy a asi 72 % z nich je mladších 40 let [3].
Etiologie ITP není zcela jasná; svůj podíl mají jistě faktory genetické i environmentální, vyvolávající aberantní imunitní odpověď či chování organismu.
V patogenezi se uplatňuje kombinace autoprotilátkami zprostředkované zvýšené destrukce trombocytů společně s inhibicí jejich produkce megakaryocyty v kostní dřeni. Tyto IgG autoprotilátky produkované B-lymfocyty hostitele jsou nejčastěji namířeny proti membránovým glykoproteinům trombocytů, např. GPIIb/IIIa a GPIb/IX [4, 5]. Nápadná je relativně častá (až 20% v dětské populaci) souvislost s proběhlou či probíhající systémovou infekcí hostitele, nejčastěji virovou [6, 7]. ITP může být součástí systémového autoimunitního onemocnění (literárně až ve 12 % případů) [5] nebo provázet solidní (karcinom prsu) i hematologická (low-grade lymfomy) maligní onemocnění [8]. Tyto formy ITP klasifikujeme jako sekundární. Zajímavá, i když vzácná, je asociace ITP a aplikace vakcín [9]. Mechanismus indukce imunitní trombocytopenie může být několikerý: molekulární mimikry s indukcí autoprotilátek s aktivací komplementu či T-lymfocyty zprostředkovaná imunitní dysregulace [5]. Zvláštní skupinou jsou léky indukované imunitní trombocytopenie (drug-induced thrombocytopenia, DITP). V těchto případech vazba farmaka na povrch trombocytů akceleruje jejich (protilátkami zprostředkovanou) destrukci [10].
Klinické projevy ITP jsou značně variabilní. První projevy onemocnění jsou většinou nenápadné, výjimečně se lze setkat s abruptním průběhem manifestovaným významným život ohrožujícím krvácením při těžké trombocytopenii. Právě krvácivé projevy bývají pro ITP typické. Jejich rozsah může být značně variabilní, od drobného nenápadného petechiálního krvácení, přes častější tvorbu hematomů až po zmíněné významné krvácivé projevy. Krvácení u ITP bývá typicky popisováno jako mukokutánní, tedy odlišné od vaskulitického či krvácení při koagulopatiích. Prezentace jako DAH je velmi raritní [11, 12]. Klinické projevy trombocytopenie při ITP se mění i s věkem postižených. Starší nemocní se prezentují mnohem častěji významným krvácením, často do gastrointestinálního traktu nebo intrakraniálně, nejspíše v důsledku dalších komorbidit (např. hypertenze) a medikace (např. antiagregancia, antikoagulancia apod.) [11, 12]. Korelace mezi tíží trombocytopenie a rizikem krvácení je obecně slabá.
Difuzní alveolární krvácení (DAH) jako život ohrožující situace vyžaduje neodkladné zajištění a monitorování vitálních funkcí a jejich podporu. Podle situace může být nezbytné zajištění dýchacích cest intubací, nejlépe endotracheální kanylou většího průměru, která umožní případné následné instrumentální (bronchoskopické) vyšetření a ošetření. Současně by měla probíhat náhrada a korekce případných poruch koagulacea/nebo trombocytů podle aktuálních doporučených postupů (Doporučený postup pro život ohrožující krvácení – mezioborové konsenzuální stanovisko; odborné společnosti České lékařské společnosti JEP). Vzhledem k povaze DAH není na rozdíl od krvácení do dýchacích cest z jediného nebo několika málo zdrojů možno bronchoskopicky provést hemostázu; tato slouží spíše ke zprůchodnění dýchacích cest odsátím krve a koagul. Obdobně nelze využít ani radiointervenční postupy či chirurgické ošetření. Zástava krvácení spočívá v optimalizaci podmínek krevní srážlivosti a samozřejmě v současné léčbě vyvolávajícího onemocnění. Při selhání standardních postupů je na místě zvážit „záchranné“ opatření v podobě podání rekombinantního aktivovaného faktoru VII (rFVIIa), případně i antifibrinolytik např. kyseliny tranexamové (rovněž lze vycházet z výše uvedených doporučených postupů). Tyto postupy však nikdy nebyly testovány v řádných klinických studiích, existují jen kazuistická či observační sdělení. Podobně otevřenou otázkou je použití extrakorporální membránové oxygenace (ECMO) jako život zachraňující intervence u kritické hypoxémie. Jsou popsány případy jeho využití i bez systémové antikoagulace po omezenou dobu. Antikoagulaci lze zahájit bezprostředně po dosažení kontroly hemostázy.
Diagnostika ITP je postavena na vyloučení jiných příčin trombocytopenie (per exclusionem) a současně identifikaci stavů potenciálně asociovaných s ITP. Ve většině případů je dostatek anamnestických, klinických i laboratorních vodítek ke správnému stanovení diagnózy. Stanovení specifických trombocytárních autoprotilátek je negativní až ve 40–50 % případů a není v současnosti obecně doporučováno v primární diagnostice ITP [4, 13].
Základním cílem léčby ITP je minimalizace rizika závažného krvácení, nikoliv normalizace počtu trombocytů. Je třeba mít na paměti, že případná léčba ITP je spojena s řadou potenciálně nežádoucích účinků a komplikací (imunosuprese). Měla by tedy být vyhrazena pro nemocné s těžkou trombocytopenií (< 10 x 109/l) a významným rizikem krvácení a/nebo již krvácejících [14]. Riziko závažného krvácení vzrůstá s věkem a s anamnézou již proběhlého krvácení (10–30krát) [15]. Do rozhodování o indikaci a formě léčby ITP je vhodné zapojit hematologa. Léčba nemocných s ITP prezentujících se akutním významným krvácením se sestává ze substituce trombokoncentráty a současné aplikace IVIG (1 g/kg dva dny po sobě) s kortikoidem (metylprednisolon 1 g/kg/den) [16, 17, 18]. Samozřejmostí zůstává adekvátní zajištění vitálních funkcí nemocných, případné chirurgické ošetření krvácení a terapeutické postupy zajištění hemostázy v případě život ohrožujícího krvácení, jak je zmíněno i výše. Základem imunosupresivní léčby první linie ITP jsou tedy kortikoidy a intravenózní imunoglobuliny. Při jejím selhání – perzistenci těžké trombocytopenie s krvácivými projevy lze zvážit splenektomii, aplikaci rituximabu (anti-CD20 monoklonální protilátka) či jiné/další aditivní „kortikoid-šetřící“ imunosupresivní léčby (cyklofosfamid, azatioprin aj.) [17, 18, 19]. Podrobná diskuse o léčbě, léčbě specifických skupin nemocných, výběru a kombinaci léčebných modalit je dobře dokumentována literárně a je nad rámec tohoto sdělení.
Nemocní s ITP mohou až v 10 % dosáhnout spontánní remise onemocnění [20, 21], zhruba 1/3 až 2/3 nemocných mají po léčbě bezpečný a stabilní počet trombocytů [20]. Mortalita nemocných s ITP je překvapivě obdobná či pouze marginálně vyšší v porovnání s obecnou populací [20, 22].
ITP či obecně trombocytopenie může být jedním z prvních příznaků systémového autoimunitního onemocnění, jako je např. SLE. Literárně se odhaduje, že u 3–15 % nemocných s ITP se v dalším průběhu diagnostikuje SLE [23]. Kombinace ITP a autoimunitní hemolytické anémie (Evansův syndrom) také relativně často předchází plnému rozvoji SLE. Těžkou trombocytopenii (< 50 x 109/l) nacházíme cca u 10 % nemocných se SLE [24, 25]. Vzhledem ke stejnému mechanismu rozvoje sekundární ITP je princip léčby obdobný [26].
V případě naší kazuistiky můžeme ITP s velkou pravděpodobností také označit za sekundární, asociovanou se SLE. Rozvoj difuzního alveolárního krvácení, které je v souvislosti s – zejména primární – ITP vzácné, lze přisoudit současnému poškození plic SLE indukovanou plicní kapilaritidou a těžké trombocytopenii (tab. 2). Právě systémové autoimunitní onemocnění spolu s perzistující těžkou trombocytopenií bylo důvodem kombinované imunosupresivní léčby (kortikoid a azatioprin).
ZÁVĚR
Popsaná kazuistika prezentuje případ ITP s velkou pravděpodobností asociované se SLE s opakovaným život ohrožujícím krvácením. Poukazuje na často obtížnou samotnou diagnostiku tohoto onemocnění a komplexnost jeho léčby. Rozlišení primární a sekundární formy ITP je spíše formální, význam spočívá více v tom, že je potřeba na možnou souvislost se systémovými onemocněními myslet v rámci diferenciální diagnostiky (virová onemocnění, autoimunitní onemocnění, lymfo/myeloproliferativní onemocnění aj.). Neméně důležitá je i těsná mezioborová spolupráce intenzivisty a hematologa jak v samotné diagnostice, tak v léčbě. Vždy je potřeba dobře vážit přínos a možná rizika zvolených postupů. Jak bylo popsáno výše, zejména provedení splenektomie je v situaci akutní těžké trombocytopenie potřeba dobře rozvážit. Má dobrý léčebný potenciál, ale příprava k jejímu bezpečnému průběhu včetně pooperační péče je velmi náročná. Je potřeba udržet dostatečný počet trombocytů k prevenci krvácivých perioperačních komplikací. Autoři současně doporučují kombinovat samotnou splenektomii s pokračující či extendovanou imunosupresivní léčbou k dosažení maximálního kurabilního potenciálu.
Podpora
- Program rozvoje vědních oborů Univerzity Karlovy (PRVOUK – projekt P36)
- MZ ČR – RVO (Fakultní nemocnice Plzeň – FNPl, 00669806)
- Národní program udržitelnosti (NPU I) No. LO1503 poskytnutý MŠMT
Adresa pro korespondenci:
MUDr. Thomas Karvunidis, Ph.D.
JIP, I. interní klinika FN a LF v Plzni, UK v Praze a Biomedicínské centrum v Plzni
Alej Svobody 80
304 60 Plzeň
email: karvunidist@fnplzen.cz
Zdroje
1. Abrahamson, P. E., Hall, S. A., Feudjo-Tepie, M., Mitrani-Gold, F. S., Logie, J. The incidence of idiopathic thrombocytopenic purpura among adults: a population-based study and literature review. Eur. J. Haematol., 2009, 83, 2, p. 83–89.
2. Terrell, D. R., Beebe, L. A., Vesely, S. K., Neas, B. R., Segal, J. B., George, J. N. The incidence of immune thrombocytopenic purpura in children and adults: A critical review of published reports. Am. J. Hematol., 2010, 85, 3, p. 174–180.
3. Schoonen, W. M., Kucera, G., Coalson, J., Li, L., Rutstein, M., Mowart, F., Fryzek, J., Kaye, J. A. Epidemiology of immune thrombocytopenic purpura in General Practice Research Database. Br. J. Haematol., 2009, 145, 5, p. 235–244.
4. Cines, D. B., Blanchette, V. S. Immune thrombocytopenic purpura. N. Engl. J. Med., 2002, 346, 19, p. 995–1008.
5. Cines, D. B., Liebman, H., Stasi, R. Pathobiology of secondary immune thrombocytopenia. Semin. Hematol., 2009, 46, 1, Suppl. 2, p. 2–14.
6. DiMaggio, D., Anderson, A., Bussel, J. B. Cytomegalovirus can make immune thrombocytopenic purpura refractory. Br. J. Haematol., 2009, 146, 1, p. 104–112.
7. Zhang, W., Nardi, M. A., Borkowsky, W., Li, Z., Karpatkin, S. Role of molecular mimicry of hepatitis C virus with platelet GPIIIa in hepatitis C-related immunologic thrombocytopenia. Blood, 2009, 113, 7, p. 4086–4093.
8. Ustun, C., Dainer, P., Hendricks, L., Bruker, C. T., Burgess, R. Association of breast cancer and immune thrombocytopenic purpura. South Med. J., 2002, 95, 1, p. 1335–1337.
9. Perricone, C., Ceccarelli, F., Nesher, G., Borella, E., Odeh, Q., Conti, F., Schoenfeld, Y., Valesini, G. Immune thrombocytopenic purpura (ITP) associated with vaccinations: a review of reported cases. Immunol. Res., 2014, 60, 2–3, p. 226–235.
10. Aster, R. H., Curtis, B. R., McFarland, J. G., Bougie, D. W. Drug-induced immune thrombocytopenia: pathogenesis, diagnosis, and management. J. Thromb. Haemost., 2009, 7, 6, p. 911–918.
11. Neunert, C., Noroozi, N., Norman, G., Buchanan, G. R., Goy, J., Nazi, I., Kelton, J. G., Arnold, D. M. Severe bleeding events in adults and children with primary immune thrombocytopenia: a systematic review. J. Thromb. Haemost., 2015, 13, 3, p. 457–464.
12. Moulis, G., Palmaro, A., Montastruc, J. L. et al. Epidemiology of incident immune thrombocytopenia: a nationwide population-based study in France. Blood, 2014, 124, 22, p. 3308–3315.
13. McMillan, R. The role of antiplatelet autoantibody assays in the diagnosis of immune thrombocytopenic purpura. Curr. Hematol. Rep., 2005, 4, 2, p. 160–165.
14. Toltl, L. J., Arnold, D. M. Pathophysiology and management of chronic immune thrombocytopenia: focusing on what matter. Br. J. Haematol., 2011, 152, 1, p. 52–60.
15. Cortelazzo, S., Finazzi, G., Buelli, M., Molteni, A., Viero, P., Barbiu, T. High risk of severe bleeding in aged patients with chronic idiopathic thrombocytopenic purpura. Blood, 1991, 77, 1, p. 31–33.
16. Goel, R., Ness, P. M., Takemoto, C. M., Krishnamurti, L., King, K. E., Tobian, A. A. Platelet transfusions in platelet consumptive disorders are associated with arterial thrombosis and in-hospital mortality. Blood, 2015, 125, 9, p. 1470–1476.
17. Ghanima, W., Godeau, B., Cines, D. B., Bussel, J. B. HowI treat immune thrombocytopenia: the choice between splenectomy or a medical therapy as a second-line treatment. Blood, 2012, 120, 5, p. 960–969.
18. George, J. N. Management of patients with refractory immune thrombocytopenic purpura. J. Thromb. Haemost., 2006, 4, 8, p. 1664–1672.
19. Braendstrup, P., Bjerrum, O. W., Nielsen, O. J. et al. Rituximab chimeric anti-CD20 monoclonal antibody treatment for adult refractory idiopathic thrombocytopenic purpura. Am. J. Hematol., 2005, 78, 4, p. 275–280.
20. Neylon, A. J., Saunders, P. W., Howard, M. R., Proctor, S. J., Taylor, P. R. Clinically significant newly presenting autoimmune thrombocytopenic purpura in adults: a prospective study of a population-based cohort of 245 patients. Br. J. Haematol., 2003, 122, 6, p. 966–974.
21. McMillan, R., Durette, C. Long-term outomes in adults with chronic ITP after splenectomy failure. Blood, 2004, 104, 4, p. 956–960.
22. Portielje, J. E., Westendrop, R. G., Kliun-Nelemans, H. C., Brand, A. Morbidity and mortality in adults with idiopathic thrombocytopenic purpura. Blood, 2001, 97, 9, p. 2549–2554.
23. Karpatkin, S. Autoimmune thrombocytopenic purpura. Blood, 1980, 56, 3, p. 329–343.
24. Keeling, D. M., Isenberg, D. A. Haematological manifestation of systemic lupus erythematosus. Blood Rev., 1993, 7, 4, p. 199–207.
25. Newman, K., Owlia, M. B., El-Hemaidi, I., Akhtari, M. Management of immune cytopenias in patients with systemic lupus erythematosus – old and new. Autoimmun. Rev., 2013, 10, 1, p. 784–791.
26. Liebman, H. Other immune thrombocytopenias. Semin. Hematol., 2007, 44, 4, Suppl. 5, p. 24–34.
Štítky
Anesteziologie a resuscitace Intenzivní medicínaČlánek vyšel v časopise
Anesteziologie a intenzivní medicína

2016 Číslo 3
- Neodolpasse účinně snižuje pooperační bolest
- Neodolpasse jako ideální volba tam, kde je bolest provázena spasmem kosterního svalstva
- Optimalizace léčby pooperační bolesti snižuje nároky na zdravotní péči
- Použití Neodolpasse v indikaci pooperační bolesti
- Neodolpasse je bezpečný přípravek v krátkodobé léčbě bolesti
Nejčtenější v tomto čísle
- Pleurální výpotek v intenzivní péči
- Poškodenie priedušnice ako následok punkčnej dilatačnej tracheostómie – kazuistiky a prehľad literatúry
- Perioperační péče o pacienty s diabetes mellitus
- Echokardiografické vyšetření při onemocnění mitrální chlopně
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy