
Morbus Dowling-Degos a morbus Galli-Galli. Popis případu
Morbus Dowling-Degos and Morbus Galli-Galli. Case Report
The authors present a case of 60-year-old woman, followed-up for melanoma removed eight years ago, with incidental findings of years-long numerous pigmented macules on the trunk and limbs. Histology suggested the diagnosis of morbus Dowling-Degos, with less common finding of horn cysts. The presence of acantholysis during the revision of biopsy led to the final diagnosis of morbus Galli-Galli, which is considered to be a subtype of genodermatosis morbus Dowling-Degos. The review provides current knowledge about this disease.
Keywords:
genodermatosis – Dowling-Degos disease – reticulate pigmentary disorders – Galli-Galli disease – clinical-pathological correlation
Autoři:
P. Kňažeková 1; L. Lacina 1,2,3; M. Petráčková 1; J. Štork 1
Působiště autorů:
Dermatovenerologická klinika 1. LF UK a VFN, přednosta prof. MUDr. Jiří Štork, CSc.
1; Anatomický ústav 1. LF UK, přednosta prof. MUDr. Karel Smetana, DrSc.
2; BIOCEV – Biotechnologické a biomedicínské centrum Akademie věd a Univerzity Karlovy ve Vestci u Prahy, vedoucí laboratoře/senior researcher: prof. MUDr. Karel Smetana, DrSc.
3
Vyšlo v časopise:
Čes-slov Derm, 94, 2019, No. 6, p. 233-237
Kategorie:
Kazuistika
Souhrn
Autoři popisují případ 60leté pacientky v preventivních kontrolách po excizi melanomu před osmi lety, s náhodně zjištěným vedlejším nálezem roky trvajících četných pigmentovaných makul na trupu a končetinách. Histologicky nález odpovídal diagnóze morbus Dowling-Degos s méně obvyklým nálezem rohových pseudocyst. Nález akantolýzy při revizi biopsie vedl k závěrečné diagnóze morbus Galli-Galli, považované za variantu genodermatózy morbus Dowling-Degos. V přehledu jsou uvedeny současné poznatky o tomto onemocnění.
Klíčová slova:
genodermatóza – morbus Dowling-Degos – retikulární pigmentovaná anomálie flexur – morbus Galli-Galli – klinickopatologická korelace
ÚVOD
Morbus Dowling-Degos (MDD) a morbus Galli-Galli (MGG) jsou vzácné, převážně autosomálně dominantní genodermatózy patřící do skupiny retikulárních pigmentových poruch kůže [5]. Přesná incidence nemoci není známa, v literatuře bylo popsáno kolem 50 případů celosvětově [19]. Vzhledem k tomu, že jsme dosud nenašli zmínku o tomto onemocnění v naší literatuře, uvádíme případ naší nemocné s touto vzácnou dermatózou.
POPIS PŘÍPADU
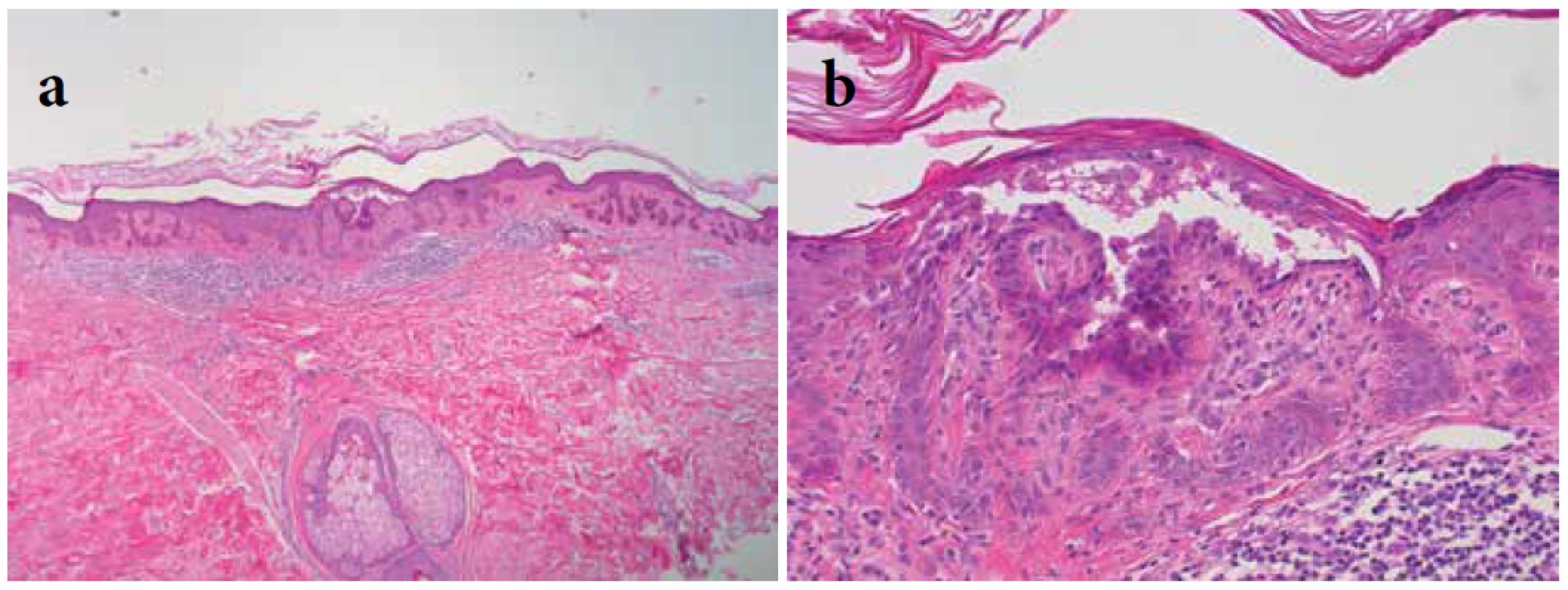
Pacientkou byla 60letá žena osm let dispenzarizovaná pro superficiálně se šířící melanom na zádech, stadium IA (pT1a N0 M0). Dlouhodobě byla léčena pro arteriální hypertenzi (metoprololem, amlodipinem, kombinovaným preparátem perindoprilu a indapamidu), jinak osobní anamnéza byla bez pozoruhodností. Při vyšetření byly patrné četné, bezpříznakové, makulózní, místy splývající pigmentace na trupu, krku a na končetinách, které byly přítomné roky bez výrazné progrese, hodnocené v minulosti klinicky jako lentigo, urticaria pigmentosa a porokeratosis superficialis actinica (obr. 1a, b). Podobné projevy, údajně, měla její matka a bratr. Byla provedena biopsie z projevu na zádech. Histologicky byly patrné úseky epidermis s prstovitě vybíhajícími anastomozujícími hyperpigmentovanými čepy, místy obsahujícími rohové okrouhlé útvary, které byly přítomné ojediněle jak v rozšířené ortokeratotické rohové vrstvě, tak i volně v horním koriu v pruhu mononukleárního infiltrátu. Nález, vzhledem k udanému rodinnému výskytu, umožnil zvážit diagnózu morbus Dowling-Degos (retikulární pigmentovanou anomálii flexur) s rohovými cystami (obr. 2a, b). Současně byla revidována biopsie provedená před třemi roky, která byla zaslána s průvodkou udávající diagnózu „pigmentace v blízkosti jizvy po excizi melanomu” s požadavkem vyloučení recidivy nádoru (bez uvedení výskytu monohočetných projevů), jejíž histologický nález byl hodnocen jako suspektní lentigo solaris. Po dalším prokrájení tohoto vzorku byly pozorovány stejné změny s prstovitě protaženými hyperpigmentovanými epidermálními čepy, navíc však byla prokázána suprabazální akantolýza (obr. 3a, b). Tento nález vedl ke stanovení závěrečné diagnózy morbus Galli-Galli.



V centru excize patrná intraepidermální štěrbina s akantolýzou.
Pacientka byla poučena o povaze onemocnění a neprojevila zájem o léčbu, ani o genetické vyšetření.
DISKUSE
Morbus Dowling-Degos (retikulární pigmentovaná anomálie flexur) je vzácná, autosomálně dominantní nemoc postihující stejně obě pohlaví, s výskytem prakticky v každé generaci [5], výjimečný je sporadický výskyt [8, 17].
První zmínky o nemoci jsou z roku 1938, kdy byla popsaná jako retikulární pigmentovaná anomálie flexur Dowlingem a Freudenthalem a následně popsaná Degosem a Ossipowskim v roce 1954 [7, 9]. Onemocnění vzniká jak v časné adolescenci, tak i v dospělosti s postupnou pomalou progresí klinického nálezu. Přesná incidence není známa [5].
U MDD byly prokázané mutace v několika genech. Ztrátová mutace genu keratin 5 (KRT5) byla identifikována jako první v roce 2006. Betz et al. identifikovali u skupiny pacientů s morbus Dowling-Degos mutaci KRT5, a naznačili tak klíčovou roli keratinů při organizaci buněčné adheze, vychytávání melanozomu, transportu organel a jaderného ukotvení [4].
Další jsou mutace POFUT1, který kóduje protein O-fucosyltransferase 1 a mutace POGLUT1, který kóduje protein O-glucosyltransferase 1. Mutace POFUT1 a POGLUT1 jsou spojeny s variantou MDD, která vynechává flexurální oblasti [3, 15]. V roce 2017 byla dále identifikována mutace genu PSENEN u MDD a v případě přítomnosti spouštecích rizikových faktorů, nikotinismu a obezity, byla tato mutace přítomna také u sdruženého výskytu MDD s hidradenitis suppurativa [18]. Tyto mutace narušují signální dráhy NOTCH, které hrají významnou roli v udržování homeostázy regulováním proliferace a diferenciace melanocytů a keratinocytů a jejich vzájemné interakce [3, 18].
Klinický obraz vykazuje hnědé makuly o průměru kolem 3 mm splývající v typických případech v místě flexur v retikulární hyperpigmentace. Zpravidla vznikají počínající projevy v axilách a v tříslech s pozdějším výskytem na krku, trupu, či vnitřní straně končetin. Erytémové a hnědé papuly s variabilní hyperkeratózou mohou být také přítomny. Někteří pacienti udávali svědění postižených oblastí. Léze podobné komedonům na šíji a zádech, jamkovité periorální jizvy, epidermoidní cysty, či hidradenitis suppurativa jsou další projevy přítomné u některých pacientů [5]. Vzácně jsou popsány případy s projevy nepostihujícími flexurální oblasti [3]. V literatuře byl publikován i ojedinělý případ ženy s histologicky verifikovaným MDD, která měla klinicky jen asymptomatické hypopigmentované makuly na trupu, v axilách a v tříslech. Autoři tak upozornili na rozšíření diferenciální diagnózy u pacientů prezentujících se četnými hypopigmentovanými makulami a na rozmanitou manifestaci MDD [17]. Klinický nález je často nespecifický a zahrnuje široké spektrum nemocí, proto je pro diagnózu nutné histologické vyšetření.
Histologicky je přítomné zvýšené množství melaninu v bazální vrstvě epidermis bez zmnožení melanocytů, prstovitě vybíhající protažené čepy a suprapapilární ztenčení epidermis. V horním koriu bývá přítomen perivaskulární lymfohistiocytární infiltrát s melanofágy [5, 16]. Ralser et al. popsali i formu s rohovými pseudocystami a folikulární hyperkeratózou [18].
Diagnóza je stanovena na základě pečlivého klinického vyšetření kožních lézí, osobní a rodinné anamnézy a histologického vyšetření. V některých případech k diagnóze vedou až opakované biopsie, či je doporučováno odebrání několika biopsií z různých lokalizací. Další laboratorní a zobrazovací metody nejsou nutné [16, 19].
Morbus Galli-Galli (MGG) je v literatuře převážně popisována jako histologická varianta morbus Dowling-Degos [6, 8, 12]. Poprvé byla popsána v roce 1982 Bardachem et al. u dvou sourozenců, kteří měli hyperpigmentace v obličeji a na krku, splňovali diagnostické kritéria MDD, ale měli navíc jedinečný histopatologický nález suprabazální akantolýzy, která je jediným rozlišovacím prvkem mezi klinicky nerozpoznatelnými MGG a MDD [1]. U MGG byla popsána mutace KRT5 a POGLUT1 [3, 12, 14]. Většina autorů podle klinického obrazu, histopatologie a genetiky považuje MGG za akantolytickou variantu MDD [6, 12], jiní autoři vzhledem k odlišující akantolýze považují MGG za samostatnou chorobu [16].
Obě nemoci patří do skupiny nemocí s retikulární pigmentací. Do této skupiny patří, mimo jiné, i syndrom Haber, symetrická akropigmentace Dohi (syn. dyschromatosis symmetrica hereditaria) nebo retikulární akropigmentace Kitamura. Klinický vzhled i histologický nález je u těchto chorob obdobný, proto je zvažován koncept, že se jedná o různé formy jedné chorobné jednotky (tab. 1). V literatuře bylo také publikováno několik vzájemně se překrývajících diagnóz u jednoho pacienta nebo u jeho rodinných příslušníků. Morbus Galli-Galli jako jediná z retikulárních pigmentovaných poruch vykazuje histopatologický rys suprabazální akantolýzy [5, 16].

Diferenciální diagnóza, kromě výše zmíněných nemocí zahrnuje lentigo solaris, verruca seborrhoica reticularis, acanthosis nigrans, porokeratosis superficialis actinica, morbus Darier, morbus Grover, morbus Hailey-Hailey, lichen planus pigmentosus inversus, papilomatosis reticularis et confluens (Gougerot a Carteaud syndrom), prurigo pigmentosa aj. Retikulární pigmentace se však může objevit i u dalších dermatóz, jako je „terra firma-forme dermatosis“, „špinavý krk“ u atopické dermatitidy, síťová hyperpigmentace u erythema ab igne či u livedo reticularis aj. [5, 20].
I když MDD má benigní povahu, bylo popsáno několik případů koexistence kožních malignit včetně spinocelulárního karcinomu, keratoakantomu i amelanotického melanomu u pacientů s MDD. Přesný patomechanismus za touto asociací zatím není úplně znám, nicméně autoři vyslovili spekulace o možné spojitosti MDD a těchto malignit s poruchou folikulární proliferace a keratinizace, anomálií pilosebaceózní jednotky a s mutací KRT5 [10, 11, 13, 21].
Terapie ve většině případů nemá žádoucí výsledek. Lokální kortikosteroidy, tretinoin, adapalen, kyselina azealová a hydrochinon byly zkoušeny s různým efektem [5].
Jsou známe případy, kde byla úspěšná léčba s Er: YAG laserem v kombinaci s lokálním kortikosteroidem a kyselinou fusidovou [22, 23]. Kožní postižení může být příčinou psychických obtíží se snížením kvality života vyžadující spolupráci s psychologem či psychiatrem [2, 19].
Případ naší nemocné, i když nevykazoval převažující postižení flexur, vzhledem k rodinnému výskytu, typické histologii, popsaným výskytem rohových pseudocyst i případů bez postižení flexur dostatečně svědčí pro diagnózu morbus Galli-Galli. V rutinní praxi též ukazuje na význam poskytnutí dostatečných klinických údajů na průvodce k histologickému vyšetření, které mohou navést k diagnóze tohoto vzácného onemocnění.
Čestné prohlášení
Autoři v souvislosti s tématem práce v posledních 12 měsících nespolupracovali s žádnou farmaceutickou firmou.
Do redakce došlo dne 13. 11. 2019.
Adresa pro korespondenci:
MUDr. Petra Kňažeková
Dermatovenerologická klinika 1. LF UK a VFN
U Nemocnice 499/2
128 08 Praha 2
e-mail: petra.knazekova@vfn.cz
Zdroje
1. BARDACH, H., GEBHART, W., LUGER, T. Genodermatosis in a pair of brothers: Dowling-Degos, Grover, Darier, Hailey-Hailey or Galli-Galli disease? Hautarzt, 1982, 33(7), p. 378–383.
2. BASAVARAJ, K. H., NAVYA, M. A., RASHMI, R. Relevance of psychiatry in dermatology: Present concepts. Indian J Psychiatry, 2010, 52(3), p. 270–275.
3. BASMANAV, F. B., OPRISOREANU, A. M., PASTERNACK, S. M. et al. Mutations in POGLUT1, encoding protein O-glucosyltransferase 1, cause autosomal-dominant Dowling-Degos disease. Am J Hum Genet, 2014, 94(1), p. 135–143.
4. BETZ, R. C., PLANKO, L., EIGELSHOVEN, S. et al. Loss-of-function mutations in the keratin 5 gene lead to Dowling-Degos disease. Am J Hum Genet, 2006, 78(3), p. 510–519.
5. BOLOGNIA, J. L., JORIZZO, J. L., SCHAFFER, J. V. Dermatology. 3rd ed., Vol. One, Philadelphia: Elsevier/Saunders, 2012, p. 1064–1074. ISBN 978--0723435716.
6. BRAUN-FALCO, M., VOLGGER, W., BORELLI, S. et al. Galli-Galli disease: An unrecognized entity or an acantholytic variant of Dowling-Degos disease? J Am Acad Dermatol, 2001, 45(5), p. 760–763.
7. DEGOS, R., OSSIPOWSKI, B. Dermatose pigmentaire réticulée des plis. Ann Dermatol Syphiligr, 1954, 81(2), p. 147–151.
8. DESAI, C. A., VIRMANI, N., SAKHIYA, J. et al. An uncommon presentation of Galli–Galli disease. Indian J Dermatol Venereol Leprol, 2016, 82(6), p. 720–723.
9. DOWLING, G. B., FREUDENTHAL, W. Acanthosis nigricans. Proc R Soc Med, 1938, 31(9), p. 1147–1150.
10. FENSKE, N. A., GROOVER, C. E., LOBER, C. W. et al. Dowling-Degos disease, hidradenitis suppurativa, and multiple keratoacanthomas. A disorder that may be caused by a single underlying defect in pilosebaceous epithelial proliferation. J Am Acad Dermatol, 1991, 24(5 pt 2), p. 888–892.
11. GUPTA, V., SAHNI, K., KHUTE, P. et al. Dowling--Degos disease and malignant melanoma: Association or mere coincidence? Indian J Dermatol Venereol Leprol, 2015, 81(6), p. 627–628.
12. HANNEKEN, S., RÜTTEN, A., PASTERNACK, S. M. et al. Systematic mutation screening of KRT5 supports the hypothesis that Galli-Galli disease is a variant of Dowling-Degos disease. Br J Dermatol, 2010, 163(1), p. 197–200.
13. HOHMANN, C. B., KÖCHE, B., BONAMIGO, R. R. et al. Case for diagnosis. Dowling-Degos disease. An Bras Dermatol, 2010, 85(2), p. 241–243.
14. KONO, M., SAWADA, M., NAKAZAWA, Y. et al. A Japanese Case of Galli-Galli Disease due to a Previously Unreported POGLUT1 Mutation. Acta Derm Venereol, 2019, 99(4), p. 458–459.
15. LI, M., CHENG, R., LIANG, J. et al. Mutations in POFUT1, encoding protein O-fucosyltransferase 1, cause generalized Dowling-Degos disease. Am J Hum Genet, 2013, 92(6), p. 895–903.
16. MÜLLER, C. S. L., PFÖHLER, C., TILGEN, W. Changing a concept – controversy on the confusing spectrum of the reticulate pigmented disorders of the skin.J Cutan Pathol, 2009, 36, p. 44–48. doi: 10.1111/j.1600-0560.2008.00995.x.
17. PICKUP, T. L., MUTASIM, D. F., Dowling-Degos disease presenting as hypopigmented macules. J Am Acad Dermatol, 2011, 64(6), p. 1224–1225.
18. RALSER, D. J., BASMANAV, F. B., TAFAZZOLI, A. et al. Mutations in γ-secretase subunit-encoding PSENEN underlie Dowling-Degos disease associated with acne inversa. J Clin Invest, 2017, 127(4), p. 1485–1490.
19. RICE, A. S., COOK, C. Dowling Degos Disease. [online, updated 2019 May 12]. In: StatPearls [internet]. Treasure Island (FL): StatPearls Publishing, 2019 [cit. 25.7.2019]. Dostupné na www: https://www.ncbi.nlm.nih.gov/books/NBK531470/
20. ŠTORK, J., DŮRA, M., PETRÁČKOVÁ, M. et al. Prurigo pigmentosa - popis případu. Čes-slov Derm, 2018, 93(3), p. 102–106.
21. UJIHARA, M., KAMAKURA, T., IKEDA, M. et al. Dowling-Degos disease associated with squamous cell carcinomas on the dappled pigmentation. Br J Dermatol, 2002, 147(3), p. 568–571.
22. WENZEL, J., TAPPE, K., GERDSEN, R. et al. Successful treatment of Dowling-Degos disease with Er:YAG laser. Dermatol Surg, 2002, 28(8), p. 748–750.
23. Yun, J. H., Kim, J. H., Choi, J. S. et al. Treatment of Dowling-Degos disease with fractional Er:YAG laser. J Cosmet Laser Ther, 2013, 15(6), p. 336–339.
Štítky
Dermatologie Dětská dermatologieČlánek vyšel v časopise
Česko-slovenská dermatologie

2019 Číslo 6
- Nástroje k hodnocení závažnosti psoriázy v klinických studiích
- Systémová léčba atopické dermatitidy konečně i u dětí
- Inovace v hojení ran: krytí Zetuvit Plus Silicone Border pro optimální management exsudátu z ran
Nejčtenější v tomto čísle
- Svědění konečníku
- Spitzoidní nádory – diagnosticky obtížné situace. Série případů
- Celková léčba atopické dermatitidy – evropské doporučené postupy a současný stav
- Růžová papula na lýtku
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy