
Periodické horečky a jiná autoinflamatorní onemocnění
Periodic fevers and other autoinflammatory diseases
Autoinflammatory diseases represent a relatively new and rapidly evolving group of rare disorders associated with mutations of genes encoding proteins with a key regulatory role in inflammatory response. Gradual discovery of mechanisms that link genetic disorder with its biochemical and immunological consequences leading to continuous or episodic inflammatory stimulation has enabled introduction of directed immunotherapies. Periodic fever syndromes belong to the so far best-known entities: familial Mediterranean fever, mevalonate kinase deficiency, cryopyrinopathies and TNF-receptor associated periodic syndrome. These inherited disorders usually manifest in childhood with variably long febrile episodes accompanied with the spectrum of other skin and organ inflammatory features and elevation of laboratory markers of inflammation. Uncontrolled disease may lead to secondary amyloidosis. Directed anti-inflammatory therapy can prevent evolution of organ damage. In children benign syndrome of periodic fever with aphtae, pharyngitis and cervical adenitis is the most common self-limited disorder without clear genetic disposition. Following other autoinflammatory disease groups are described – pyogenic syndromes, disorders with skin and bone manifestations, granulomatous diseases, monogenic vasculopathies and diseases associated with proteasome disorder. Diagnosis of autoinflammatory diseases is often delayed due to their extreme rarity. Increasing efficacy and availability of molecular-genetic testing and centralization of diagnostics and clinical care in a specialized center for children as well as adults can in the future improve quality of care for patients with these rare conditions.
Keywords:
autoinflammatory diseases (AID), periodic fever syndromes, FMF, CAPS, MKD, TRAPS, PFAPA, NGS
Autoři:
Šárka Fingerhutová 1; Eva Jančová 2; Markéta Tesařová 3; Lenka Dvořáková 4; Pavla Doležalová 1
Působiště autorů:
Centrum dětské revmatologie a autoinflamatorních onemocnění, Klinika dětského a dorostového lékařství 1. LF UK a VFN v Praze
1; Centrum imunonefrologie, Klinika nefrologie 1. LF UK a VFN v Praze
2; Laboratoř pro studium mitochondriálních poruch, Klinika dětského a dorostového lékařství 1. LF UK a VFN v Praze
3; Laboratoř diagnostiky DNA, Klinika dětského a dorostového lékařství 1. LF UK a VFN v Praze
4
Vyšlo v časopise:
Čas. Lék. čes. 2018; 157: 122-129
Kategorie:
Přehledové články
Souhrn
Autoinflamatorní onemocnění jsou relativně novou a rychle rostoucí skupinou vzácných onemocnění spojených s mutacemi genů kódujících proteiny s klíčovou rolí v regulaci zánětlivé odpovědi. Postupné odhalování mechanismů spojujících genetickou odchylku s jejími biochemickými a imunologickými důsledky vedoucími k trvalé či epizodické stimulaci zánětlivé aktivity umožňuje postupné zavádění cílené imunoterapie. Nejdéle známou skupinou těchto chorob jsou periodické horečky – familiární středomořská horečka, deficit mevalonátkinázy, kryopyrinopatie a periodický syndrom asociovaný s receptorem pro tumor nekrotizující faktor. Tato dědičná onemocnění se obvykle manifestují v dětství různě dlouhými epizodami horečky provázené spektrem dalších zánětlivých projevů kožních i orgánových a zvýšením laboratorní zánětlivé aktivity. Hlavním rizikem je rozvoj sekundární amyloidózy. Cílená protizánětlivá léčba může zabránit vzniku orgánového poškození. U dětí je nejčastější benigní, spontánně ustupující syndrom periodické horečky s afty, faryngitidou a krční adenitidou, jehož genetický podklad není zatím objasněn. Mezi další autoinflamatorní choroby patří tzv. pyogenní syndromy, onemocnění s převažující kožní a kostní manifestací, granulomatózní onemocnění, monogenní vaskulopatie a onemocnění spojená s poruchou proteasomu. Diagnostika autoinflamatorních chorob bývá pro jejich vzácnost opožděná často až do dospělosti. Zvýšení efektivity a dostupnosti molekulárně genetické analýzy a centralizace diagnostiky a péče na specializovaném pracovišti pro děti i dospělé mohou do budoucna zlepšit kvalitu péče o pacienty s těmito vzácnými chorobami.
Klíčová slova:
autoinflamatorní onemocnění (AID), syndromy periodických horeček, FMF, CAPS, MKD, TRAPS, PFAPA, NGS
ÚVOD
Autoinflamatorní onemocnění (AID) jsou nově popsanou skupinou vysoce vzácných, většinou vrozených zánětlivých onemocnění. Pojem autoinflamatorní se objevil v odborné literatuře na konci minulého století s cílem zdůraznit jejich odlišnost od autoimunitních chorob (1). Za hlavní příčinu zánětlivých projevů je považována porucha mechanismů vrozené imunity, často spojená s mutacemi genů kódujících proteiny s klíčovou rolí v regulaci zánětlivé odpovědi (2). Spektrum těchto chorob se v posledních desetiletích trvale rozrůstá. Mezi nejdéle známé a relativně nejčastější jednotky patří tzv. syndromy periodické horečky. Další známější skupiny AID jsou uvedeny v tab. 1 (2–4).

Celosvětově nejčastější monogenní periodickou horečkou je familiární středomořská horečka, která spolu se syndromem deficitu mevalonátkinázy patří mezi autosomálně recesivně dědičná onemocnění. Periodické syndromy asociované s kryopyrinem (kryopyrinopatie) a periodický syndrom asociovaný s receptorem pro tumor nekrotizující faktor jsou děděny autosomálně dominantně (3). Poněkud stranou stojí syndrom periodické horečky s aftózní stomatitidou, faryngitidou a krční lymfadenopatií, u kterého dosud nebyla objevena kauzální mutace (5).
Kromě syndromů periodické horečky se další nově objevená či nově mezi AID zařazená monogenní zánětlivá onemocnění řadí do skupin podle jejich hlavních manifestací. Přítomnost sterilního hnisavého zánětu charakterizuje tzv. pyogenní syndromy. Představiteli skupiny AID s převažující kožní a kostní manifestací jsou syndromy DITRA (deficiency of the IL‐36 receptor antagonist) a SAPHO (synovitis, acne, pustulosis, hyperostosis, osteitis). Mezi granulomatózní onemocnění se řadí syndrom juvenilní sarkoidózy. K nověji popsaným stavům patří onemocnění spojená s poruchou proteasomu a onemocnění asociovaná s vaskulopatiemi (tab. 1) (6).
ETIOLOGIE AID
Na základě zdokonalování molekulárně genetických metod i možností konfirmace patogenních mutací funkčními, především imunologickými studiemi, jsme v současné době svědky progresivního nárůstu počtu AID, u kterých se začínají rozkrývat jejich etiopatogenetické mechanismy. Důsledkem toho se mění klasifikace již známých AID, ale i rychle přibývajících nových syndromů v této oblasti. Dříve přesně stanovené hranice mezi autoinflamatorními a autoimunitními onemocněními se začínají stírat (7).
S nástupem inovativních technologií charakteru sekvenování nové generace(NGS – next generation sequencing) se výrazně zvýšila účinnost detekce kauzálních mutací na genové úrovni, a dochází tak ke komplikování zprvu jasně daných vzorců dědičnosti dle Mendela. Interpretace výsledků genetických analýz nebývá vždy zcela jednoznačná. Jedná se například o problematiku mutací s nízkou penetrancí (8, 9). Korelace genotyp-fenotyp je současným celosvětovým problémem, který bude potřeba na základě funkčních studií postupně objasnit. Možnosti výskytu mozaicismu, již několikrát popsaného u jednotlivých AID, komplikují situaci u klinicky suspektních pacientů s negativním výsledkem dříve provedeného Sangerova sekvenování (10, 11). Negativita, ale i pozitivita výsledku genetické analýzy tedy ještě zdaleka nemusí znamenat nalezení příčiny nebo vyloučení suspektního onemocnění. K interpretaci výsledků je zapotřebí hlubší spolupráce mezi klinikem, imunologem, genetikem a specialistou na informační technologie (12).
Využití léčebné strategie na základě znalosti etiopatogeneze ilustrujeme příkladem kryopyrinopatií. Všechny fenotypy tohoto onemocnění jsou způsobeny mutacemi v genu NLRP3 kódujícího kryopyrin, označovaný také jako NALP3. NALP3 patří do velké skupiny proteinů, které mají klíčovou roli ve správném fungování vrozené imunity. Jejich aktivací je spouštěna kaskáda buněčné odpovědi, která ústí v aktivaci kaspázy 1 s následným zvýšeným uvolňováním především interleukinu 1 (IL-1) a 18 (IL-18) s jejich prozánětlivými účinky. Objev funkčního dopadu mutace NLRP3 in vitro vedl k následnému terapeutickému testu blokádou IL-1, což se u pacienta setkalo s takřka zázračným efektem (13). Tento úspěch translačního výzkumu se stal základem vývoje cílené terapeutické strategie u řady AID, u nichž byla prokázána nadprodukce prozánětlivých mediátorů, zejména IL-1.
SYNDROMY PERIODICKÉ HOREČKY
Hlavní charakteristikou syndromů periodické horečky jsou opakující se epizody horeček doprovázené systémovým zánětem, variabilním výskytem jiných orgánových příznaků a možným rozvojem závažných komplikací, zejména reaktivní (sekundární) amyloidózy (3). Míra pochopení etiopatogenetického procesu určuje současné terapeutické možnosti (6). Pro řadu těchto stavů je k dispozici vysoce efektivní imunoterapie. Cílem léčby je kontrola aktivity onemocnění, prevence orgánového poškození a zvýšení kvality života pacientů (12).
K manifestaci onemocnění dochází obvykle již v časném dětství. Nízká prevalence, velká variabilita projevů a nedostatečná informovanost lékařů o těchto nově popsaných chorobách jsou hlavními příčinami časté diagnostické prodlevy dosahující až řádu let (14). Časné stanovení diagnózy a následně i nastavení cílené léčby je pro prevenci vniku nevratného poškození zásadní (12). Na základě rozvoje možností genetického vyšetření dochází v posledních letech k identifikaci vyššího počtu pacientů s AID. Tyto choroby jsou rozpoznávány nejen u dětí, ale i mezi dospělými, kteří byli často řadu let neúspěšně léčeni pro blíže nespecifikované horečnaté stavy. Především u těchto pacientů dochází po stanovení správné diagnózy a zahájení cílené léčby k významnému zlepšení kvality života.
Periodické syndromy asociované s kryopyrinem
Periodické syndromy asociované s kryopyrinem (CAPS – cryopyrin-associated periodic syndromes) neboli kryopyrinopatie jsou skupinou zánětlivých onemocnění různého spektra závažnosti. Všechny fenotypy CAPS jsou způsobeny autosomálně dědičně přenesenou mutací, případně mutací de novo, v genu NLRP3 (viz výše). Dle tíže onemocnění rozlišujeme tři hlavní fenotypy, dále pak řadu překryvných či ne zcela vyhraněných syndromů. Jedná se o familiární chladovou kopřivku (FCAS – familial cold autoinflammatory syndrome), Muckleův-Wellsův syndrom (MWS) a chronický infantilní neurologický, kožní a kloubní syndrom (CINCA – chronic infantile neurologic cutaneous articular neboli NOMID – neonatal onset multisystem inflammatory disease) (13).
FCAS čili familiární chladová urtika je nejméně závažným fenotypem onemocnění. Klinické příznaky se objevují v návaznosti na expozici chladu (po 1–2 hodinách) a obvykle vymizí do 24 hodin (15). Onemocnění se typicky manifestuje v průběhu prvního roku života, kdy se objevuje především vyrážka doprovázená teplotami. Mezi další projevy patří zimnice, konjunktivitida a artralgie.
Nejzávažnější průběh je popisován u syndromu CINCA. Systémový zánět se projevuje obvykle již v novorozeneckém věku horečkami s exantémem (obr. 1a) a vysokou laboratorní zánětlivou aktivitou (16). Nejzávažnějším postižením jsou neurologické manifestace ve smyslu chronické aseptické meningitidy vedoucí k syndromu nitrolební hypertenze, jejímž důsledkem je mozková atrofie a psychomotorická retardace. Zánět v oblasti vnitřního ucha vede k senzorineurální ztrátě sluchu (16). Až u poloviny pacientů se v průběhu času rozvine hypertrofická artropatie (16).
Muckleův-Wellsův syndrom se závažností svých projevů řadí mezi výše zmíněné jednotky. Epizody horečky mohou být vyvolané chladem či stresem, ale mohou se objevovat i bez jasné příčiny. Bývají provázeny podobně jako u FCAS exantémem (obr. 1b), konjunktivitidou, bolestmi hlavy, artritidou či artralgiemi. Na rozdíl od FCAS je častou komplikací MWS rozvoj senzorineurální hluchoty. Hlavním nebezpečím neléčeného onemocnění je vysoké riziko rozvoje amyloidózy (25 % pacientů) (16).
K léčbě kryopyrinopatií jsou využívány preparáty blokátorů IL-1 (12). Účinnost anakinry, kankinumabu i rilonaceptu byla ověřena studiemi s dětskými i dospělými pacienty všech fenotypů a vedla k jejich schválení regulačními úřady v této indikaci (12). V České republice je dosud hrazen pouze kanakinumab (přípravek Ilaris), anakinra (přípravek Kineret) je k dispozici zatím pouze v režimu off-label.
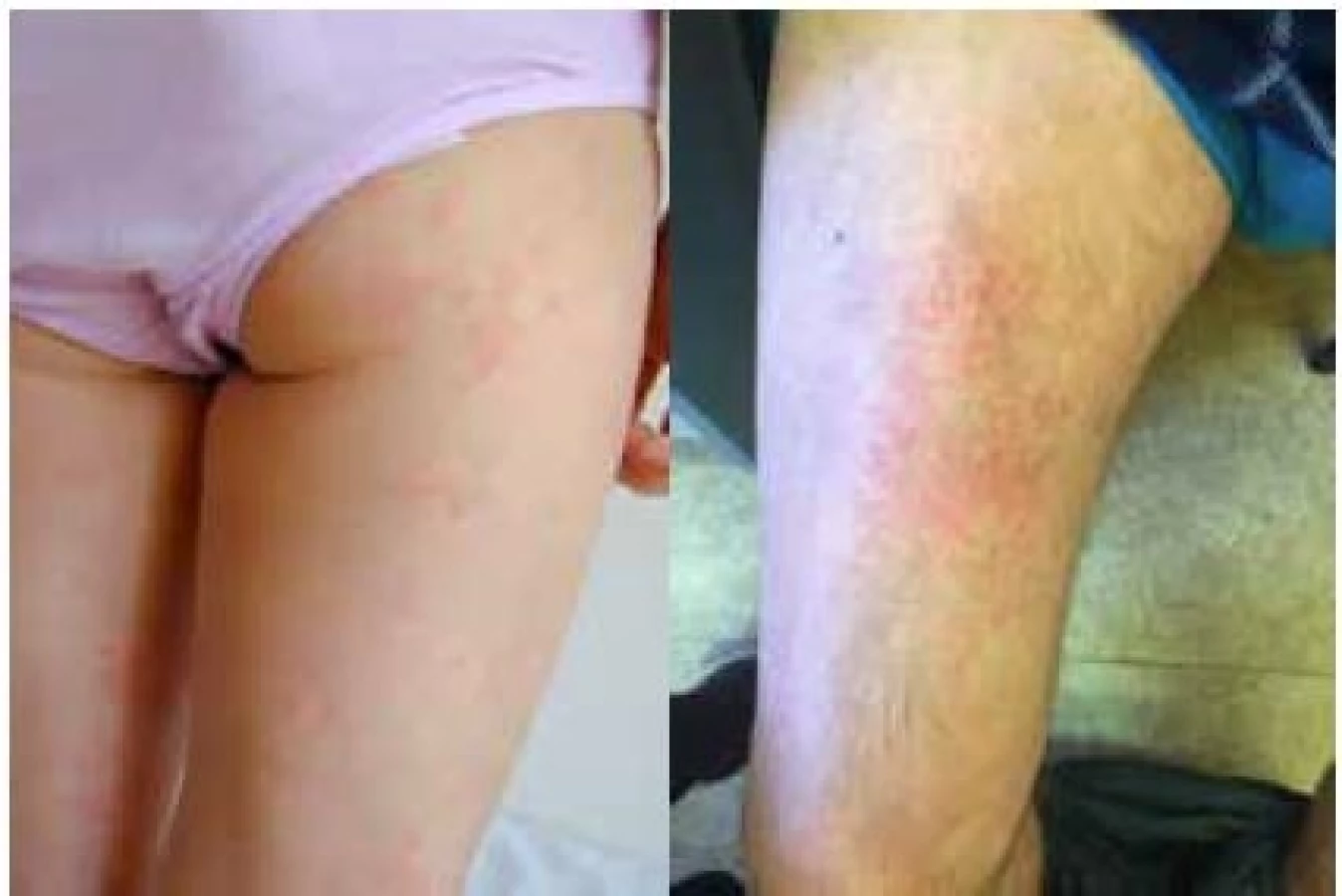
Familiární středomořská horečka
Familiární středomořská horečka (FMF – familial Mediterranean fever) je celosvětově nejčastější monogenní horečkou. Nízké zastoupení v naší populaci způsobuje její vazba na etnický původ. Nejvyšší manifestace dosahuje v oblastech kolem Středozemního moře, populačně je to pak u aškenázských a sefardských Židů, Arabů, Turků či v arménské populaci (17). V roce 1997 byla jako jedna z prvních mutací způsobující autoinflamatorní onemocnění popsána právě mutace v genu MEFV (Mediterranean fever) (18). Tento gen lokalizovaný na krátkém raménku šestnáctého chromosomu (16p13.3) kóduje protein pyrin (marenostrin) (19). Syndromologicky je toto onemocnění popisováno již od roku 1908 (19). Jeho patofyziologickým mechanismem je nedostatečná transkripce inhibičních faktorů v regulaci inflamasomu NLRP3, která působí nadměrné uvolňování IL-1 (19).
Klinickým korelátem jsou krátkodobé epizody horeček (několik hodin až 3 dny) s těžkými projevy serozitidy. Nejčastější manifestací jsou kruté bolesti břicha při akutní peritonitidě (až u 90 % pacientů) či bolesti na hrudi z důvodu jednostranné pleuritidy (až u 40 % pacientů). Více než 50 % pacientů postihuje akutní přechodná artritida velkých kloubů (3). Někdy se objevuje vyrážka, která vzhledem i lokalitou připomíná erysipel (19). K manifestaci onemocnění dochází v dětském či mladém dospělém věku, obvykle do 20 let. Nejzávažnější dlouhodobou komplikací je sekundární renální amyloidóza (20).
Před zavedením kolchicinu do léčebné praxe byla amyloidóza hlavním příčinou smrti těchto pacientů (až v 70 % případů). V kolchicin-rezistentních případech se uplatňuje léčba blokátory IL-1 (20).
Deficit mevalonátkinázy: periodický syndrom asociovaný s mevalonátkinázou
Periodický syndrom asociovaný s mevalonátkinázou (MAPS – mevalonate kinase associated periodic syndrome) označovaný také pojmy deficit mevalonátkinázy (MKD – mevalonate kinase deficiency) či syndrom hyper IgD (HIDS – hyperimmunoglobulinemia D syndrome) je způsoben mutací v genu pro mevalonátkinázu (12q24). Tento enzym se fyziologicky účastní biosyntézy cholesterolu. V případě MKD dochází k poruše tvorby isoprenoidových produktů této cesty a současně k nadprodukci IL-1 beta (3). Zbytková aktivita mevalonátkinázy určuje fenotyp onemocnění. U zcela nepatrné aktivity enzymu (0,1 %) hovoříme o metabolickém onemocnění – mevalonové acidurii.
Jeho diagnostika nebývá vzhledem k závažným projevům (mentální retardace, ataxie, neprospívání, hypotonie, myopatie, katarakta, febrilie) příliš opožděna. Enzymová aktivita v rozmezí 1–8 % normy navozuje klinické projevy MAPS/HIDS (3). Jedná se o recidivující febrilie (délka trvání 4–7 dní) provázené bolestí břicha (někdy s nauzeou, zvracením, průjmem), faryngitidou, krční lymfadenopatií, artralgiemi, artritidou. K manifestaci onemocnění dochází v časném dětském věku s možností přechodu k mírnějšímu fenotypu v dospělosti. Epizody recidivujících zánětlivých stavů s popsanými projevy mohou být spouštěny očkováním, stresem nebo běžným infektem (3).
V laboratorním vyšetření nacházíme nespecificky zvýšené hladiny imunoglobulinu D, u více než 80 % pacientů rovněž i elevaci hladiny imunoglobulinu A (3). Do moči se v průběhu ataky teplot vylučuje substrát hromaděný před enzymatickou blokádou – mevalonát (3). Jeho detekce v moči odebrané v epizodě horečky je považována za vhodné screeningové vyšetření.
Epizodické podávání kortikosteroidů v době ataky může vést k částečnému zmírnění projevů. U závažněji probíhajícího onemocnění se uplatňuje biologická terapie (12). Charakter obtíží, frekvence epizod či nedostatečné potlačení zánětlivé aktivity si u některých dosud popsaných pacientů vyžádalo transplantaci hematopoetických kmenových buněk (HSCT) (12).
Periodický syndrom asociovaný s receptorem pro tumor nekrotizující faktor
Příčinou periodického syndromu asociovaného s receptorem pro tumor nekrotizující faktor (TRAPS – TNF-receptor-associated periodic syndrome) je mutace genu pro receptor TNF alfa (TNFR). Za fyziologických okolností vazba solubilního TNF působí konformační změnu jeho receptoru, která vede k aktivaci další intracelulární kaskády signálních molekul a rozvoji zánětlivé odpovědi. Mutací podjednotky A1 TNFR nedochází k ukončení signálu pro další prozánětlivou kaskádu a k odštěpení receptoru, který má ve své solubilní formě fyziologickou protizánětlivou regulační funkci. V organismu tedy nadále přetrvává prozánětlivý stav (21).
Onemocnění je charakterizováno protrahovanými epizodami horeček, často vyšetřovanými jako horečka neznámého původu. Teploty trvají v rozmezí řady dní až týdnů. Klinický obraz dokreslují myalgie, artralgie, bolesti břicha a oční projevy ve smyslu konjunktivitidy a periorbitálního edému. Na kůži se objevuje proximodistálně migrující palpačně citlivý makulární raš (především na končetinách). Bolestivost postižených míst bývá způsobena fasciitidou (19).
Léčebně se uplatňuje epizodická kortikoterapie v průběhu teplot. Efektivita této terapie však časem klesá. Podávání solubilního receptoru pro TNF etanerceptu vykazuje dle studií podobný trend. Dle současných doporučení je u závažně probíhajícího onemocnění indikována léčba blokátory IL-1 (12).
Periodická horečka s afty, faryngitidou a krční adenitidou
Periodická horečka s afty, faryngitidou a krční adenitidou (PFAPA – periodic fever, aphtae, pharyngitis, cervical adenitis) je nejčastějším syndromem periodických teplot dětského věku (22). Vzhledem k časté pozitivní rodinné anamnéze stran recidivujících teplot, opakovaných angín v předškolním věku či tonzilektomie u rodinných příslušníků se spekuluje o genetickém pozadí, které se zatím nepodařilo definovat (23). Akronym PFAPA vystihuje hlavní projevy onemocnění. Další příznaky bývají nespecifické (únava, nechutenství, bolesti břicha, podrážděnost, artralgie, myalgie) (24). Epizody teplot trvají zpravidla 3–6 dní a recidivují s frekvencí 3–8 týdnů, v mezidobí je dítě zcela asymptomatické (22). K manifestaci projevů dochází obvykle do pěti let věku (22), ke spontánní remisi pak do puberty (25).
Objektivní nález tonzilitidy/tonzilofaryngitidy provázející syndrom PFAPA nelze bez mikrobiologického vyšetření odlišit od streptokokového onemocnění. Lymfadenopatie bývá zpravidla oboustranná a nepřesahuje velikost 5 cm (26). Při laboratorním vyšetření nacházíme v období epizody zvýšené parametry zánětu, které se zcela normalizují po ústupu teplot (27). Vysoké hodnoty CRP a nález tonzilitidy vede lékaře často ke zbytečné preskripci antibiotické terapie, která na průběh epizod PFAPA nemá žádný vliv, ba naopak přispívá ke zvyšování bakteriální rezistence (28).
Standardem léčby je epizodické podávání prednisonu v dávce 1–2 mg/kg jednorázově v začátku febrilní epizody (22). Další léčebnou variantou je podávání kolchicinu (29). Nejúčinnější je tonzilektomie, která vede u více než 90 % dětí k trvalému ústupu onemocnění (30).
PYOGENNÍ SYNDROMY
Syndrom pyogenní artritidy, pyoderma gangrenosum a akné
Příčinou autosomálně dominantního syndromu pyogenní artritidy, pyoderma gangrenosum a akné(PAPA – pyogenic arthritis, pyoderma gangrenosum and acne) je mutace genu pro protein prolin/serin/threonin fosfatázu (CD2 antigen vazebný protein). Tento cytoskeletální protein je exprimován hematopoetickými buňkami, které modulují aktivaci T buněk, cytoskelet a uvolňování IL-1. Mutace pak způsobuje zvýšení afinity tohoto proteinu k pyrinu, následkem čehož dochází k nadprodukci IL-1 stejnou cestou jako v případě FMF (31).
Mezi hlavní klinické projevy patří sterilní hnisavé artritidy, které mohou navazovat na minimální traumata či vznikají spontánně a vedou ke kloubní destrukci. Dalšími typickými projevy jsou pyoderma gangrenosum a akné. Může se objevit recidivující otitida, faryngitida či lymfadenopatie se splenomegalií. Kloubní projevy se v průběhu dospívání zlepšují, kožní se naopak zhoršují (16).
Léčebně se uplatňují blokátory TNF (16).
Deficit antagonisty receptoru pro IL-1
Deficit antagonisty receptoru pro IL-1 (DIRA – deficiency of the interleukin-1 receptor antagonist) se manifestuje velmi časně neutrofilní pustulární dermatózou, periostitidou a aseptickou multifokální osteomyelitidou, které jsou doprovázeny vysoce zánětlivou laboratoří. Přibližně u 2/3 pacientů se kožní projevy objevují již v průběhu novorozeneckého období. Jedná se o autosomálně recesivní onemocnění, které je způsobeno mutací genu kódujícího protein antagonisty receptoru pro IL-1 (IL-1RA) (32, 33). Následkem toho dochází k ztrátě antagonistické aktivity proteinu, a IL-1 tak kontinuálně aktivuje buněčnou zánětlivou odpověď (32).
Tato zánětlivá kaskáda může být vyvolána různými spouštěči, jako například mechanickou stimulací kůže, zavedením katetru/kanyly, intubací (33). Kožní postižení může vyústit v difuzní deskvamaci. Syndrom je často doprovázen anonychií a dolíčkováním nehtů, na sliznicích se tvoří afty. K ostatním příznakům řadíme rozšíření předního konce žeber či klíčků, fúzi krčních obratlů, otoky kloubů (33).
Blokáda IL-1 dramaticky zlepšuje projevy onemocnění a může navodit i jeho dlouhodobou remisi (16).
ONEMOCNĚNÍ S KOŽNÍ A KOSTNÍ MANIFESTACÍ
Deficit antagonisty receptoru pro interleukin 36
Příčinou deficitu antagonisty receptoru pro interleukin 36 (DITRA – deficiency of the interleukin thirty-six receptor antagonist) je ztrátová mutace genu kódujícího antagonistu receptoru pro IL-36. Jedná se o autosomálně recesivní onemocnění. Majoritní exprese IL-36 v kůži předurčuje projevy onemocnění. Důsledkem mutace nedochází k blokádě IL-36 a v těle vzniká nekontrolovaný prozánětlivý stav (6, 32).
Klinicky se jedná o recidivující stavy generalizované pustulární psoriázy doprovázené horečkou, únavou a tělesnou slabostí. U onemocnění nedochází k dalšímu orgánovému postižení.
Terapeuticky se používá blokáda IL-1 (6, 32).
Syndrom synovitidy, akné, pustulózy, hyperostózy a osteitidy
Etiopatogeneze syndromu synovitidy, akné, pustulózy, hyperostózy a osteitidy (SAPHO – synovitis, acne, pustulosis, hyperostosis, osteitis) není dosud plně objasněna. Předpokládá se souhra genetické dispozice v kombinaci s epigenetickými faktory a vlivy prostředí.
Postižení pohybového aparátu zahrnuje sterilní osteitidu, hyperostózu, synovitidu, artropatii a entezopatii. Jedná se o neerozivní artritidu, často s postižením páteře, sternokostálních a sternoklavikulárních kloubů (34). Typické kožní léze zahrnují palmoplantární pustulózu a těžké akné. Interval mezi kožním a osteoartikulárním postižením bývá menší než dva roky.
V léčbě se uplatňují nesteroidní antirevmatika, kortikosteroidy, bisfosfonáty, případně i blokáda cytokinů (IL-1 nebo TNF-α) (34).
GRANULOMATÓZNÍ ONEMOCNĚNÍ
Dětská sarkoidóza: Blauův syndrom
Dětská sarkoidóza, označovaná také jako Blauův syndrom či sarkoidóza s časným začátkem, je familiární či sporadicky se vyskytující onemocnění s autosomálně dominantním způsobem přenosu charakterizované triádou polyartikulární synovitidy, granulomatózní uveitidy a typické vyrážky. Na rozdíl od sarkoidózy začínající v dospělém věku nebo v adolescenci je Blauův syndrom spojen s mutací genu NOD2 (nucleotide-binding oligomerization domain 2). Jedná se o intracelulární receptor pro mikrobiální struktury. Důsledkem mutace je nadměrná aktivace NF-kB (nuclear factor kappa-light-chain-engancer of activated B-cells) i v nepřítomnosti bakterií, což vede k vyvolání prozánětlivého stavu (35).
Patologicko-anatomickým podkladem klinických projevů je obrovskobuněčný granulomatózní zánět. Oproti uveitidě při juvenilní idiopatické artritidě zde bývá postižen i zadní segment oka (35). Dermatitida je charakterizována chronickým nebo intermitentním drobným tmavě červeným až lividním makulopapulózním exantémem, často s lichenoidními prvky (16).
Další systémové a orgánové projevy jsou častější u geneticky negativní sarkoidózy starších dětí, ale mohou provázet i Blauův syndrom. Jedná se o granulomatózní infiltraci lymfatických uzlin a slinných žláz, granulomatózní či intersticiální nefritidu, jaterní granulomy, intersticiální pneumonitidu, kraniální neuropatii, vaskulitidu (16).
Onemocnění obvykle vyžaduje systémovou imunosupresi, ke které se kromě kortikosteroidů používají jak syntetické, tak biologické chorobu modifikující léky (methotrexát, azathioprin, mykofenolát mofetil, blokátory TNF-α a IL-1) (35).
MONOGENNÍ VASKULOPATIE
Infantilní vaskulopatie asociovaná se stimulátorem interferonových genů
Infantilní vaskulopatie asociovaná se stimulátorem interferonových genů (SAVI – STING-associated vasculopathy with onset in infancy) je způsobena gain-of-function mutací v genu TMEM173, který kóduje stimulátor interferonových genů (STING – stimulator of interferon genes). Díky tomu dochází k vazbě ligandu cyklického guanosinmonofosfátu na STING, čímž je aktivována fosforylace kináz a faktoru regulujícího interferon beta a dochází k nadměrné stimulaci transkripce genů odpovědných za interferonovou odpověď (36).
Systémové příznaky se objevují obvykle během několika týdnů po narození. Vaskulopatie postihuje malé a střední cévy. Nejmarkantnější projevy jsou způsobené zúžením a uzávěry kožních cév (teleangiektazie, puchýřnaté až ulcerující projevy na tváři, nose, prstech, chodidlech). Cévní okluze působí resorpci chrupavky ušního boltce, perforaci nosního septa, dystrofické změny nehtů, gangrény prstů. U většiny těchto pacientů byl popsán rozvoj intersticiální plicní nemoci.
Kauzální terapie není zatím k dispozici, slibně se jeví léčba inhibitory Janusovy kinázy (36).
Vaskulopatie typu nodózní polyarteritidy, deficit adenosindeaminázy 2
Onemocnění deficitem adenosindeaminázy 2 (DADA2 – deficiency of adenosine deaminase 2) bylo poprvé popsáno teprve v roce 2014 (13). Jedná se o autosomálně recesivní onemocnění s mutací v genu CECR1 (cat eye syndrome chromosome region 1, 22q11.1) kódujícím enzym adenosindeaminázy 2 (ADA2). Ten je na rozdíl od adenosindeaminázy 1 (ADA1) lokalizován extracelulárně a jeho aktivita tkví v deaminaci adenosinu na inosin. K vzestupu zánětlivé aktivity dochází na základě jejího účinku v regulaci proliferace monocytů a diferenciace makrofágů. Mutace způsobuje prevalenci M1 prozánětlivých buněk (37).
Klinicky se onemocnění projevuje širokým spektrem vaskulopatií. Nejčastějším kožním projevem je livedo reticularis (obr. 2), dále pak ischemie nebo přímo nekrózy prstů. V biopsiích prokazujeme nekrotizující, případně leukocytoklastovou vaskulitidu či panikulitidu. Ostatní projevy onemocnění rovněž souvisí s ischemií tkání při uzávěrech/zúženích malých a středních tepen. Klinické odlišení od vaskulitidy charakteru dětské polyarteritis nodosa (PAN) je nemožné. DADA2 dále typicky postihuje periferní a zejména centrální nervový sytém. Může se manifestovat v jakémkoli věku jinak nevysvětlenou tranzitorní ischemickou epizodou, ischemickou či hemoragickou mozkovou příhodou (typicky lakunární lokalizace), tranzientní mononeuritidou či polyneuropatií (37).
V případě rodinného výskytu časných iktů a zejména při postižení rodiče dítěte s vaskulitidou charakteru PAN může správná diagnóza a adekvátní terapie výrazně zlepšit prognózu mozkové ischemie (37). Laboratorním biomarkerem DADA2 je významné snížení enzymatické aktivity ADA2 zjistitelné v séru pacientů. V terapii se uplatňuje použití blokátorů TNF-α. (37).

ONEMOCNĚNÍ ASOCIOVANÁ S PORUCHOU PROTEASOMU
Chronická atypická neutrofilní dermatóza s lipodystrofií a zvýšenou teplotou
Syndrom chronické atypické neutrofilní dermatózy s lipodystrofií a zvýšenou teplotou (CANDLE – chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature) byl popsán v roce 2010. Je způsoben mutací v genu kódujícím beta 8 podjednotku proteasomu (gen PSMB8). Díky této mutaci nemůže docházet v buňce ke správné proteolýze intracelulárních proteinů a jejich prezentaci imunitnímu systému, následkem je hromadění intermediárních substancí v buňce a akumulace proteinů v tkáních pacientů. Tento stav vede k zvyšování buněčného stresu, aktivaci Janusovy kinázy, produkci interferonu (IFN) a kruhově opět aktivaci genové exprese PSMB8 (16, 38).
Ataky teplot, lipodystrofie a další zánětlivé projevy se začínají objevovat již během prvního roku života ve spojení s neprospíváním. Kožní příznaky zahrnují recidivující anulární edematózní ložiska především na trupu, která se hojí spontánně a zanechávají hyperpigmentace. Častý je nafialovělý otok očních víček. Mezi další projevy patří nosní a ušní chondritida, konjunktivitida, nodulární episkleritida, epididymitida, nefritida, otitida, parotitida, aseptická meningitida nebo lymfadenopatie.
V léčbě se uplatňují nesteroidní antirevmatika nebo kortikosteroidy, zkouší se blokáda Janusovy kinázy (16, 38).
DIAGNOSTIKA A PÉČE O PACIENTY S AID V ČESKÉ REPUBLICE
Evropská doporučení pro diagnostiku a léčbu AID stanovují základní pravidla péče o tyto pacienty. Mezi obecné principy patří multidisciplinární péče ve specializovaném centru s expertízou v dané problematice (12). Komplexní péče vyžaduje účast dětského revmatologa, pediatra/internisty, imunologa, fyzioterapeuta a dalších specialistů podle typu orgánového postižení, např. z oborů ORL, oftalmologie, nefrologie, neurologie, metabolických onemocnění. Interpretace výsledků genetického vyšetření a genetické poradenství v rodinách pacientů vyžadují vysokou úroveň genetické erudice (39). Vedle výše jmenovaných klinických specialistů jsou nedílnou součástí týmu specialisté a odborný personál laboratoří (metabolické, genetické, imunologické). Vzhledem k celoživotnímu charakteru většiny AID má být péče zaměřená na problematiku celé rodiny v kontextu významného ovlivnění kvality života, s možností psychosociální podpory. Hlavním cílem léčby je pak kontrola aktivity onemocnění a prevence vzniku orgánových komplikací (12).
Péče o pacienty s AID celého věkového spektra je v České republice zaštítěna pracovišti VFN v Praze. Centrum dětské revmatologie a autoinflamatorních onemocnění poskytuje péči dětem a ve spolupráci s Centrem imunonefrologie dospělým pacientům. VFN je evropským expertním centrem v rámci Evropské referenční sítě (ERN, viz https://ec.europa.eu/health/ern/overview_en) pro vzácné choroby v oblasti imunodeficitů, autoimunních a autoinflamatorních onemocnění (ERN RITA, viz http://rita.ern-net.eu). Základní informace o AID pro pacienty jsou k dispozici v různých světových jazycích, včetně českého jazyka, na webových stránkách www.pediatric-rheumatology.printo.it. Instrukce k případnému objednání vyšetření v ambulanci periodických horeček jsou uvedeny na webu Kliniky dětského a dorostového lékařství (KDDL) VFN pod sekcí Centrum dětské revmatologie a autoinflamatorních onemocnění (viz www.vfn.cz/pracoviste/kliniky-a-oddeleni/klinika-detskeho-a-dorostoveho-lekarstvi/specializovana-centra/centrum-detske-revmatologie/klinicka-pece).
V laboratořích KDDL VFN byla od roku 2008 postupně zavedena molekulárně genetická diagnostika monogenních periodických horeček a byly analyzovány vzorky DNA u 248 pacientů. V období před zavedením genetické diagnostiky u nás byla analýza prováděna v zahraničních laboratořích v rámci evropské spolupráce na projektu Eurofever (No2007332). Dosud byla geneticky potvrzena diagnóza CAPS u 10 pacientů, TRAPS u 3, MKD u 15 a FMF u 8 pacientů ve věku od 2 do 56 let. Počty vyšetření s pozitivními výsledky jsou vzhledem k vzácnosti onemocnění očekávaně nízké a odpovídají záchytu uváděnému v literatuře (40). Přestože jsou v literatuře u jednotlivých syndromů popsány typické manifestace, v praxi se často setkáváme s jejich neúplným klinickým vyjádřením. Ne zřídka se tedy stává, že při vyslovení suspekce na monogenní příčinu obtíží je vhodné vyšetřit více genů najednou. Z těchto důvodů se celosvětově začalo uplatňovat vyšetření souboru genů v podobě panelu metodou NGS (41). Negativní nález genetické analýzy však nevylučuje přítomnost onemocnění na základě zatím nepopsané mutace (15). Interpretace výsledku analýzy je dále komplikována možností nálezu variant či polymorfizmů s nejasným klinickým významem, mutací s nízkou penetrancí či různou mírou genetického mozaicismu (8–11). Jako u všech stavů splňujících definici vzácných onemocnění by genetické vyšetření AID měl indikovat a interpretovat odborník v dané oblasti (6).
Genetická laboratoř KDDL VFN zavádí vyšetření panelu 89 genů především z oblasti autoinflamatorních onemocnění a některých dalších hereditárních stavů (monogenní vaskulopatie a střevní záněty, hemofagocytující lymfohistiocytózy, autoimunitní lymfoproliferativní syndromy, vrozené poruchy komplementu). Z dalších vyšetření biomarkerů AID nabízejí laboratoře KDDL VFN vyšetření mevalonátu v moči a plánují zavedení analýzy aktivity ADA2.
ZÁVĚR
Metody molekulárně genetických vyšetření přinášejí do klinické praxe nové a účinnější možnosti diagnostiky těchto onemocnění. Současným největším problémem je jejich interpretace. Rozrůstající se skupina AID a růst počtu pacientů s těmito syndromy vyžadují široké spektrum klinické i laboratorní expertízy. Pacienti s podezřením na AID by měli být centralizováni nejen vzhledem k vzácnosti těchto stavů, ale také kvůli rostoucím možnostem cílené léčby mimo schválené či hrazené indikace, která by měla být do budoucna lépe dostupná právě ve specializovaných centrech pro vzácná onemocnění.
Poděkování
Článek byl vytvořen za podpory projektu RVO VFN 64165 a SVV 260373.
Čestné prohlášení
Autorka práce prohlašuje, že v souvislosti s tématem, vznikem a publikací tohoto článku není ve střetu zájmů a vznik ani publikace článku nebyly podpořeny žádnou farmaceutickou firmou. Toto prohlášení se týká i všech spoluautorek.
Seznam použitých zkratek
AID - autoinflamatorní onemocnění
CANDLE - chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature
CAPS - cryopyrin-associated periodic syndromes
CECR1 - cat cye syndrome chromosome region 1
CINCA - chronic infantile neurologic cutaneous articular
DADA2 - deficiency of adenosine deaminase 2
DIRA - deficiency of the interleukin 1 receptor antagonist
DITRA - deficiency of the IL‐36 receptor antagonist
DNA - deoxyribonukleová kyselina
ERN - European Reference Networks
FCAS - familial cold autoinflammatory syndrome
FMF - familial Mediterranean fever
HIDS - hyperimmunoglobulinemia D syndrome
HSCT - hematopoietic stem cell transplantation
IFN - interferon
IgA - imunoglobulin A
IgD - imunoglobulin D
IL - interleukin
MAPS - mevalonate kinase-associated periodic syndrome
MEFV - Mediterranean fever
MKD - mevalonate kinase deficiency
MWS - Muckleův-Wellsův syndrom
NALRP3 - NACHT, LRR and PYD domains-containing protein 3
NF-kB - nukleární factor kappa B
NGS - next generation sequencing
NLRP3 - NLR family pyrin domain containing 3
NOD2 - nucleotide-binding oligomerization domain 2
NOMID - neonatal onset multisystem inflammatory disease
PAN - polyarteritis nodosa
PAPA - pyogenic arthritis, pyoderma gangrenosum and acne
PFAPA - periodic fever, aphtae, pharyngitis, cervical adenitis
PSMB8 - proteasome subunit beta type-8
RITA - rare immunodeficiency, autoinflammatory and autoimmune diseases
SAPHO - synovitis, acne, pustulosis, hyperostosis, osteitis
SAVI - STING-associated vasculopathy with onset in infancy
STING - stimulator of interferon genes
TMEM - transmembránový protein
TNF - tumor nekrotizující faktor
TNFR - receptor TNF
TRAPS - TNF-receptor associated periodic syndrome
Adresa pro korespondenci:
MUDr. Šárka Fingerhutová
Klinika dětského a dorostového lékařství 1. LF UK a VFN v Praze
Ke Karlovu 2, 121 00 Praha 2
Tel.: 736 224 818
e-mail: sarka.fingerhutova@lf1.cuni.cz
Zdroje
1. McDermott MF, Aksentijevich I, Galon J et al. Germline mutations in the extracellular domains of the 55kDa TNF receptor, TNFR1 define a family of dominantly inherited autoinflammatory syndromes. Cell 1999; 97, 133–144.
2. Meiorin S, Espada G, Rosé C. Autoinflammatory diseases in pediatrics. Arch Argent Pediatr 2013; 111(3): 237–234.
3. Federichi S, Caorsi S, Gattorno M. The autoinflammatory diseases. Swiss Med Wkly 2012 Jun 19; 142: w13602.
4. Cetkovská P, Benáková N. Autoinflamatorní syndromy s kožními projevy. Československá dermatologie 2015; 90(4): 144–155.
5. Doležalová P, Król P. Recidivující horečky u dětí. Vox pediatrie. 2010, roč. 10, č.3, 16–20.
6. Fingerhutová Š, Doležalová P. Kožní projevy autoinflamatorních onemocnění. Dermatologie pro praxi 2017; 11(4): 192–198.
7. Milner JD. PLAID: a syndrome of complex patterns of disease and unique phenotypes. J Clin Immunol 2015; 35 : 527–530.
8. Lachman HJ, Papa R, Gerhold K et al. The phenotype of TNF receptor-associated autoinflammatory syndrome (TRAPS) at presentation: a series of 158 cases from the Eurofever/EUROTRAPS international registry. Ann Rheum Dis 2014; 73 (12): 2160–7.
9. Montealegre Sanchez GA, Hashkes PJ. Neurological manifestations of the Mendelian-inherited autoinflammatory syndromes. Dev Med Child Neurol 2009; 51(6): 420–428.
10. Rowczenio DM, Trojer H, Omoyeinmi E et al. Brief report: association of tumor necrosis factor receptor-associated periodic syndrome with gonosomal mosaicism of a novel 24-nucleotide TNFRSF1A deletion. Arthritis Rheumatol. 2016; 68(8): 2044–2049.
11. Rowczenio DM, Gomes SM, Aróstegui JI et al. Late-onset cryopyrin-associated periodic syndromes caused by somatic NLRP3 mosaicism-UK single center experience. Front Immunol 2017; 8 : 1410.
12. Ter Haar NM, Oswald M, Jeyaratnam J et al. Recommendations for the management of autoinflammatory diseases. Ann Rheum Dis 2015; 74(9): 1636–1644.
13. Neven B, Prieur AM, Quartier dit Maire P. Cryopyrinopathies: update on pathogenesis and treatment. Nat Clin Pract Rheumatol 2008; 4(9): 481–489.
14. Toplak N, Grenkel J, Ozen S et al. An international registry on autoinflammatory diseases: the Eurofever experience. Ann Rheum Dis 2012; 71 : 1177–1182.
15. Król P, Doležalová P. Horečka jako hlavní projev nemoci. Pediatrie pro praxi 2011; 12(2): 111–114.
16. Dávila-Seijo P, Hernández-Martín A, Torrelo A. Autoinflammatory syndromes for the dermatologist. Clin Dermatol. 2014; 32 (4): 488–501.
17. Lidar M, Livneh A. Familial Mediterranean fever: clinical, molecular and management advancements. Neth J Med 2007; 65(9): 318–324.
18. The international FMF Consortium. Ancient missense mutations in a new member of the RoRet gene family are likely to cause familial Mediterranean fever. Cell 1997; 90 : 797–807.
19. Braun-Falco M, Ruzicka T. Skin manifestations in autoinflammatory syndromes. J Dtsch Dermato Ges 2011; 9(3): 232–246.
20. Gattorno M, Federici S, Pelagatti MA et al. Diagnosis and management of autoinflammatory diseases in childhood. J Clin Immunol 2008; 28(Suppl. I): 73–83.
21. Lachmann HJ. Clinical immunology review series: an approach to the patient with a periodic fever syndrome. Clin Exp Immunol 2011; 165(3): 301–309.
22. Vanoni F, Theodoropoulou K, Hofer M. PFAPA syndrome: A review on treatment and outcome. Pediatr Rheumatol Online J 2016; 14(1): 38.
23. Cochard M, Clet J, Le L et al. PFAPA syndrome is not a sporadic disease. Rheumatology (Oxford) 2010; 49(10) 1984–1987.
24. Król P, Bohm M, Sula V et al. PFAPA syndrome: clinical characteristics and treatment outcomes in a large single-centre cohort. Clin Exp Rheumatol 2013; 31(6): 980–987.
25. Perko D, Debeljak M, Toplak N et al. Clinical features and genetic background of the periodic fever syndrome with aphthous stomatitis, pharyngitis, and adenitis: a single center longitudinal study of 81 patients. Mediators Inflamm 2015; 2015 : 293417.
26. Long SS. Syndrome of periodic fever, aphthous stomatitis, pharyngitis, and adenitis (PFAPA): What it isn’t. What is it? J Pediatr 1999; 135 (1): 1–5.
27. Stojanov S, Hoffmann F, Kéry A et al. Cytokine profile in PFAPA syndrome suggests continuous inflammation and reduced anti-inflammatory response. Eur Cytokine Netw 2006; 17(2): 90–97.
28. Licameli G, Lawton M, Kenna M et al. Long-term surgical outcomes of adenotonsillectomy for PFAPA syndrome. Arch Otolaryngol Head Neck Surg 2012; 138 (10): 902–906.
29. Padeh S, Stoffman N, Berkun Y. Periodic fever accompanied by aphthous stomatitis, pharyngitis and cervical adenitis syndrome (PFAPA syndrome) in adults. Isr Med Assoc J 2008; 10(5): 358–360.
30. Lantto U, Koivunen P, Tapiainen T et al. Long-term outcome of classic and incomplete PFAPA (Periodic fever, aphtous stomatitis, pharyngitis, and adenitis) syndrome after tonsillectomy. J Pediatr 2016; 179 : 172–177.
31. Smith EJ, Allantaz F, Bennett L et al. Clinical, molecular, and genetic characteristics of PAPA syndrome: a review. Curr Genomics 2010; 11(7): 519–527.
32. Touitou I, Galeotti C, Rossi-Semerano L et al. The expanding spectrum of rare monogenic autoinflammatory diseases. Orphanet J Rare Dis 2013; 8 : 162.
33. Schnellbacher C, Ciocca G, Menendez R et al. Deficiency of interleukin-1 receptor antagonist responsive to Anakinra. Pediatr Dermatol 2013; 30(6): 758–760.
34. Rukavina I. SAPHO syndrome: a review. J Child Orthop 2015; 9(1): 19–27.
35. Caso F, Galozzi P, Costa L et al. Autoinflammatory granulomatous diseases: from Blau syndrome and early onset sarcoidosis to NOD2-mediated disease and Crohn´s disease. RMD Open 2015; 1 (1): e000097.
36. Liu Y, Jesus AA, Marrero B et al. Activated STING in a vascular and pulmonary syndrome. N Engl J Med 2014; 371 (6): 507–518.
37. Caorsi R, Penco F, Shena F et al. Monogenic polyarteritis: the lesson of ADA deficiency. Pediatr Rheumatol 2016, 14 : 51.
38. Kluk J, Rustin M, Brogan PA et al. Chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature syndrome: a report of a novel mutation and review of the literature. Br J Dermatol 2014; 170 (1): 215–217.
39. Shinar Y, Obici L, Aksentijevich I et al. Guidelines for the genetic diagnosis of hereditary recurrent fevers. Ann Rheum Dis 2012; 71 : 1599–1605.
40. Marcuzzi A, Piscianz E, Kleiner G et al. Clinical genetic testing of periodic fever syndromes. Biomed Res Int 2013; 2013 : 501305.
41. Metzker ML. Sequencing technologies – the next generation. Nat Rev Genet 2010; 11 (1): 31–46.
Štítky
Adiktologie Alergologie a imunologie Angiologie Audiologie a foniatrie Biochemie Dermatologie Dětská gastroenterologie Dětská chirurgie Dětská kardiologie Dětská neurologie Dětská otorinolaryngologie Dětská psychiatrie Dětská revmatologie Diabetologie Farmacie Chirurgie cévní Pediatrie Revmatologie Algeziologie Dentální hygienistkaČlánek vyšel v časopise
Časopis lékařů českých

- Diosmin a hesperidin: Co ukazuje farmakokinetika?
- Doc. Jitka Fricová: V USA nasazovali fentanyl poměrně nekriticky, v Česku je situace jiná
- Riziko roztroušené sklerózy u pacientů s psoriázou
- Neuropatie výrazně zpomaluje proces hojení u povrchových ran
Nejčtenější v tomto čísle
- Periodické horečky a jiná autoinflamatorní onemocnění
- Uzlinový syndrom u nemoci z kočičího škrábnutí dětí a dospělých
- Celiakie dětí a dospívajících
- Horizontální přenos genetické informace a jeho význam pro vznik antibiotické rezistence
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy