
A Genetic Association Study of Serum Acute-Phase C-Reactive Protein Levels in Rheumatoid Arthritis: Implications for Clinical Interpretation
Background:
The acute-phase increase in serum C-reactive protein (CRP) is used to diagnose and monitor infectious and inflammatory diseases. Little is known about the influence of genetics on acute-phase CRP, particularly in patients with chronic inflammation.
Methods and Findings:
We studied two independent sets of patients with chronic inflammation due to rheumatoid arthritis (total 695 patients). A tagSNP approach captured common variation at the CRP locus and the relationship between genotype and serum CRP was explored by linear modelling. Erythrocyte sedimentation rate (ESR) was incorporated as an independent marker of inflammation to adjust for the varying levels of inflammatory disease activity between patients. Common genetic variants at the CRP locus were associated with acute-phase serum CRP (for the most associated haplotype: p = 0.002, p<0.0005, p<0.0005 in patient sets 1, 2, and the combined sets, respectively), translating into an approximately 3.5-fold change in expected serum CRP concentrations between carriers of two common CRP haplotypes. For example, when ESR = 50 mm/h the expected geometric mean CRP (95% confidence interval) concentration was 43.1 mg/l (32.1–50.0) for haplotype 1 and 14.2 mg/l (9.5–23.2) for haplotype 4.
Conclusions:
Our findings raise questions about the interpretation of acute-phase serum CRP. In particular, failure to take into account the potential for genetic effects may result in the inappropriate reassurance or suboptimal treatment of patients simply because they carry low-CRP–associated genetic variants. CRP is increasingly being incorporated into clinical algorithms to compare disease activity between patients and to predict future clinical events: our findings impact on the use of these algorithms. For example, where access to effective, but expensive, biological therapies in rheumatoid arthritis is rationed on the basis of a DAS28-CRP clinical activity score, then two patients with identical underlying disease severity could be given, or denied, treatment on the basis of CRP genotype alone. The accuracy and utility of these algorithms might be improved by using a genetically adjusted CRP measurement.
: Please see later in the article for the Editors' Summary
Published in the journal:
. PLoS Med 7(9): e32767. doi:10.1371/journal.pmed.1000341
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pmed.1000341
Summary
Background:
The acute-phase increase in serum C-reactive protein (CRP) is used to diagnose and monitor infectious and inflammatory diseases. Little is known about the influence of genetics on acute-phase CRP, particularly in patients with chronic inflammation.
Methods and Findings:
We studied two independent sets of patients with chronic inflammation due to rheumatoid arthritis (total 695 patients). A tagSNP approach captured common variation at the CRP locus and the relationship between genotype and serum CRP was explored by linear modelling. Erythrocyte sedimentation rate (ESR) was incorporated as an independent marker of inflammation to adjust for the varying levels of inflammatory disease activity between patients. Common genetic variants at the CRP locus were associated with acute-phase serum CRP (for the most associated haplotype: p = 0.002, p<0.0005, p<0.0005 in patient sets 1, 2, and the combined sets, respectively), translating into an approximately 3.5-fold change in expected serum CRP concentrations between carriers of two common CRP haplotypes. For example, when ESR = 50 mm/h the expected geometric mean CRP (95% confidence interval) concentration was 43.1 mg/l (32.1–50.0) for haplotype 1 and 14.2 mg/l (9.5–23.2) for haplotype 4.
Conclusions:
Our findings raise questions about the interpretation of acute-phase serum CRP. In particular, failure to take into account the potential for genetic effects may result in the inappropriate reassurance or suboptimal treatment of patients simply because they carry low-CRP–associated genetic variants. CRP is increasingly being incorporated into clinical algorithms to compare disease activity between patients and to predict future clinical events: our findings impact on the use of these algorithms. For example, where access to effective, but expensive, biological therapies in rheumatoid arthritis is rationed on the basis of a DAS28-CRP clinical activity score, then two patients with identical underlying disease severity could be given, or denied, treatment on the basis of CRP genotype alone. The accuracy and utility of these algorithms might be improved by using a genetically adjusted CRP measurement.
: Please see later in the article for the Editors' Summary
Introduction
Genetic variants at the CRP locus influence the low, basal levels of serum C-reactive protein (CRP) in apparently healthy individuals [1]–[5]. The primary motivation for these existing studies has been the observation of a strong correlation between basal CRP and future cardiovascular risk, and the possibility, now looking less likely, that CRP has a causal role in this process [6],[7]. The widespread clinical application of CRP measurement, however, is not related to its low-level variation within the basal range, but to the high levels seen as part of the human acute-phase response. Serum CRP levels greater than 10 mg/l are generally considered indicative of an infectious or inflammatory process [8]. CRP expression increases rapidly following an inflammatory stimulus, leading to serum levels that may exceed 500 times baseline, making CRP an ideal marker for the diagnosis and monitoring of inflammatory processes. Serum CRP is extensively used in all medical specialities for this purpose [8]. Surprisingly, there has been little published data on the role of CRP variants in determining the magnitude of this acute-phase response, and none in the context of chronic active inflammation.
The demonstration of a strong, genetically determined and hence lifelong, influence on the magnitude of acute-phase CRP in any particular patient would have a number of important implications. Firstly, as a diagnostic test, CRP thresholds are used not only as a component of formal clinical algorithms, but also as part of a physician's intuitive decision-making process when diagnosing inflammatory disease and making treatment decisions. As we discuss later, this raises the possibility that false reassurance could be given to a patient with disease, or optimal treatment withheld, because they are genetically predisposed to only a modest rise in acute-phase CRP. Secondly the demonstration of a strong genetic effect on the acute-phase CRP response also raises the possibility that this may have consequences in terms of individual susceptibility to diseases in which the innate immune system plays a protective (or deleterious) role.
The existing literature on the effects of CRP genetics on acute-phase CRP levels consists of five studies, each of which measured the magnitude of CRP rise following a single noxious stimulus [9]–[13]. As we discuss later, this approach may be complicated by the difficulty in quantifying or standardising stimuli between patients or a difficulty in inducing a marked CRP rise. We have therefore taken a different approach, which we believe offers distinct advantages. We have studied genetic determinants of acute-phase CRP in patients with rheumatoid arthritis, a chronic inflammatory disease characterised by a marked acute-phase response.
By studying patients with rheumatoid arthritis we made use of the historical tendency to record the erythrocyte sedimentation rate (ESR) alongside CRP as an alternative and independent marker of inflammation. Although ESR has become less widely used because of its slow response following an inflammatory stimulus, this is not important in chronic inflammation where both CRP and ESR are at a steady state. The factors contributing to ESR are not fully understood, but seem to be related to intrinsic erythrocyte membrane properties, notably the charge, with a lesser influence from the concentration of large plasma proteins such as immunoglobulin or fibrinogen [14],[15]. There is no evidence that serum CRP levels directly influence ESR: indeed the slow fluctuation in ESR following the onset or resolution of inflammation is in stark contrast to the rapid expression and short half-life of CRP [8],[16],[17]. The incorporation of ESR as a covariate in our model fulfils two purposes. First, it provides an independent adjustment for the underlying inflammatory status of each of our patients (a consequence of their synovial joint inflammation). Without this adjustment, the random error associated with dramatically differing levels of inflammation across the cohort (random with respect to either recruitment into the study, or with respect to underlying CRP genotype) may be so great as to make the relatively small effect of CRP genotype on CRP levels difficult to demonstrate with any level of significance. Second, since most physicians have an intuitive appreciation of the relationship between CRP and ESR in a variety of inflammatory settings, the incorporation of ESR into our models allows us to generate a clinically interpretable estimate of the magnitude of genetic effect at all levels of underlying inflammatory drive. Our results are therefore directly relevant to the conditions in which CRP quantification contributes to clinical decision making.
In this paper we study the association between genetic variants at the CRP locus and acute-phase CRP in patients with chronic active inflammation due to rheumatoid arthritis.
Methods
Study Participants
The discovery study was carried out in a cohort of 281 UK rheumatoid arthritis cases (patient set 1). Details on the recruitment of these patients have been published elsewhere [18]. The mean age of participants was 61 y (SD 12), 79% were female. Replication was performed in 414 rheumatoid arthritis patients recruited in New Zealand (Wellington and Christchurch) and Australia (Adelaide) (patient set 2). The mean age was 62 y (SD 13) and 70% were female. All patients were of self-reported white European ancestry and met 1987 ACR revised criteria for the diagnosis of rheumatoid arthritis [19]. Data were available for at least one pair of measurements for CRP and ESR (Westergren method) collected simultaneously as part of routine clinical management. For some of the individuals in patient set 2, data were available for serial CRP/ESR pairs, measured at different times. For these individuals, each CRP/ESR pair was arbitrarily assigned an integer value, and a random number generator used to identify one of these pairs at random to carry forward for analysis.
Ethical approval for the collection of DNA and patient data was obtained locally at each recruitment site (from the Guy's hospital Research Ethics Committee, London UK, the University Hospital Lewisham ethics committee, London, UK, the Upper South B regional ethics committee of New Zealand, the MultiRegion Ethics committee of New Zealand, and the Research and Ethics committee, Repatriation General Hospital, Adelaide Australia). Written informed consent was obtained from each patient. Patient identity was encrypted at the site of collection and all data were analysed anonymously.
SNP Selection and Genotyping
SNPs were selected to tag common variation at CRP. We used previously published genotype data on 22 SNPs densely covering CRP in 799 unrelated, healthy UK individuals as our reference dataset for this population [20]. Using these data in turn captured all of the CRP variation observed in the HapMap CEPH population. Tagging SNPs were selected using the tagger facility of Haploview (employing an aggressive tagging strategy, r2 = 0.9 threshold) [21].
Genotyping on patient set 1 was performed by matrix-assisted laser desorption and ionization-time of flight (MALDI-TOF) mass spectrometry (Sequenom) using the iPLEX assay. Genotyping on patient set 2 was outsourced to Kbioscience Ltd using their variant of competitive allele-specific PCR (KASPar) (www.kbioscience.co.uk/chemistry/chemistry_application_note.html). Duplicate genotyping was performed on 5% of patients to ensure reproducibility (100% concordance observed). Predefined quality control exclusion criteria were: any individual genotyped at <90% of markers, any marker genotyping in <90% individuals, any marker with a minor allele frequency <5%, and any marker out of Hardy-Weinberg equilibrium (p<0.01). Ten individuals were excluded from patient set 2 because of poor genotyping, otherwise genotyping quality control criteria were met and all markers were in Hardy-Weinberg equilibrium. Population haplotype structure was inferred using Haploview, and individual patient haplotypes were inferred using PHASE v. 2.1 [21],[22]. Only individuals for whom haplotypes could be assigned with a posterior probability >0.95 were carried forward for haplotype association analysis. Six individuals were excluded after failing to meet this requirement.
Statistical Analysis
The standard CRP assay becomes nonlinear below 1 mg/l leading to the common practice of reporting values as a range (i.e., <1) or rounded to an integer (i.e., “0”). We assigned all CRP measurements reported as <1 mg/l a value of 0.5 mg/l. An initial review identified four data outliers with very low ESR but increased CRP; three from patient set 1 and one from patient set 2. We postulated that these individuals were in the early stages of an acute inflammatory episode (when the rise in ESR lags behind the rise in CRP) and excluded them from further analysis.
Both CRP and ESR distributions were positively skewed so a log-transformation was applied for analysis. The association between SNP genotype and the quantitative markers of inflammation (CRP or ESR) was evaluated by linear model, with genotype modelled as an additive allelic effect. All parameter estimates given throughout the paper are expressed relative to the major allele. An initial assessment was made of the association between CRP SNPs and the “raw” serum CRP levels, unadjusted for ESR. In addition, the direct association between CRP SNPs and ESR itself was made. In all subsequent models the association between CRP genotype (either at SNP or haplotype level) was assessed with ESR, as an independent marker of background inflammatory status, included as a covariate in the model. The relationship between CRP and ESR was nonlinear, so the final model included a significant quadratic term:
Since patients and data in this study originated from four sites of collection (South London, Wellington, Christchurch, and Adelaide), a random effects parameter was included where relevant to reflect these clusters of data.
In the haplotype analyses, a comparison of haplotype effect was achieved by simultaneously entering each haplotype into the model with the remaining, commonest, haplotype (H1) acting as the reference. Expected CRP values were derived from the regression parameters by back-transforming to the original scale, with the mean of the log-distribution reflecting the median of the untransformed distribution.
Results
In patient set 1 the median serum CRP was 11 mg/l (range 1–195; 5th, 95th centiles 5, 88). 51.8% of CRP measurements were >10 mg/l. The median ESR was 22.5 mm/h (range 2–132; 5th, 95th centiles 4, 83). The six genotyped SNPs (with minor allele frequency) were rs2808632 (0.28), rs3093059 (0.07), rs1800947 (0.08), rs1205 (0.35), rs876538 (0.23), and rs11265257 (0.40). In a preliminary analysis, unadjusted for background inflammatory status (ESR), significant associations between serum CRP and SNPs rs1205, rs11265257, and rs1800947 were observed (Tables S1 and S2). In contrast, ESR was not associated with any CRP SNP (Table S3).
Table 1 shows the association of CRP SNPs with serum CRP level, incorporating ESR as a covariate to adjust for the underlying inflammatory status of that individual. A conservative Bonferroni correction for six tests would require p<0.008 to achieve overall significance, hence both rs1205 and rs1125257 are strongly associated with CRP, with the minor allele associated with lower CRP level. We looked for departure from a simple additive model of allele effect but none was seen.
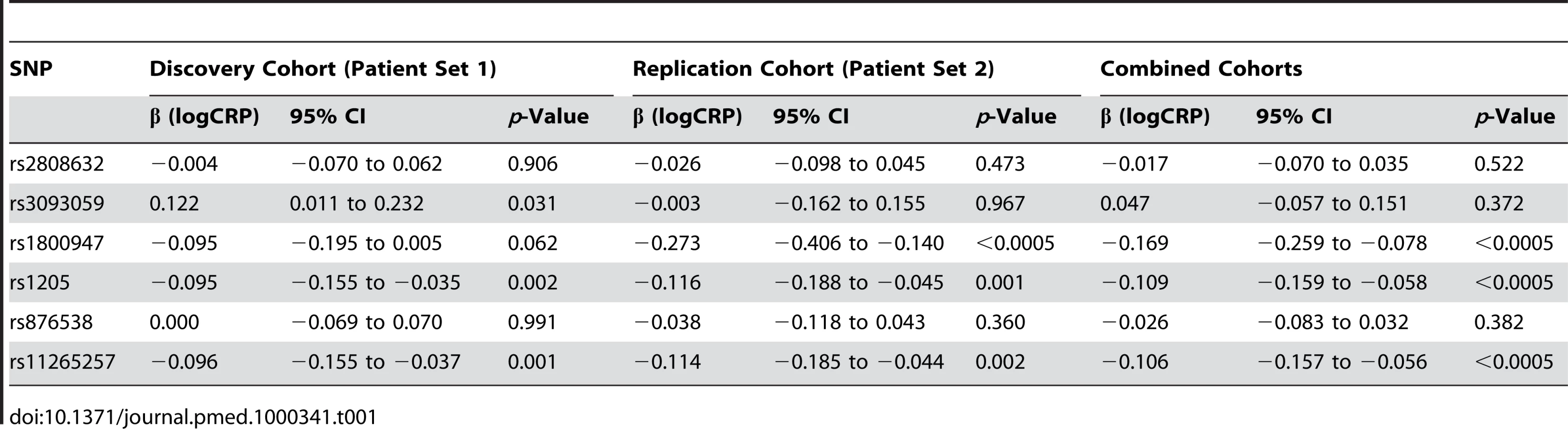
In patient set 2 the median CRP was 5 mg/l (range 0.25–334; 5th, 95th centiles 0.25, 60). 34.9% of CRP measurements were >10 mg/l. The median ESR was 16 mm/h (range 2–106; 5th, 95th centiles 4, 58). Overall, the level of inflammation observed in the patients in set 2 was therefore slightly less than that observed in set 1, suggesting either less severe disease or better disease control; this does not impact on the interpretation of our findings. The SNP minor allele frequencies were rs2808632 (0.34), rs3093059 (0.05), rs1800947 (0.07), rs1205 (0.34), rs876538 (0.24), and rs11265257 (0.41). Initial analysis of patient set 2 confirmed the association of CRP genotype with “raw” serum CRP levels, but not with ESR (Tables S1–S3). Table 1 demonstrates the replication in patient set 2 of the significant association of rs1205 and rs11265257 with ESR-adjusted serum CRP. It also shows strong association for rs1800947, a more rare SNP with only borderline association in patient set 1.
Having replicated the initial genetic associations, we performed a combined analysis using pooled data from both cohorts. In addition to the inclusion of a “cohort” term in our combined model (as outlined in the Methods section), we also looked for potential SNP × cohort interactions that might complicate the interpretation of this analysis. None of the key SNP effects we observed differed significantly from one cohort to another. This combined analysis again emphasised the high level of significance attached to our identified SNPs (Table 1).
While Table 1 shows the highly significant associations between SNPs at CRP and serum CRP level, it does not provide easily interpretable parameter estimates. In Table 2 we have therefore shown the geometric mean and 95% confidence intervals (CIs) modelled for each genotype (with reference to the major allele homozygotes) at the associated SNPs, in our combined dataset. It should be reiterated that the geometric mean of the log-transformed CRP distribution can be considered equivalent to the median of the untransformed CRP distribution. Since the predicted CRP levels depend on the background inflammatory status (as assessed by ESR) in this model, we have illustrated expected geometric mean CRP at two levels of background inflammation (ESR 40 mm/h and ESR 80 mm/h).

Considerable evidence, from the basal CRP literature, supports the hypothesis that there are multiple functional SNPs at the CRP locus [23]–[25]. We tested this hypothesis directly in the combined dataset by performing an analysis conditioned on the most significant SNPs and were able to demonstrate an independent association from three SNPs entered simultaneously into the regression model (Table S4). It is therefore evident that an analysis of the effect of CRP genotype on serum CRP based on individual SNPs alone is likely to markedly underestimate the overall effect size. We therefore performed a haplotype analysis. This approach has the advantage of providing a realistic estimate of the integrated effect of multiple SNPs on acute-phase serum CRP levels, while also only considering those SNP combinations that are actually found in the population and are of direct clinical relevance.
Table 3 demonstrates the inferred haplotype structure and haplotype frequencies in the two patient sets. Table 4 demonstrates the association of CRP haplotypes with serum CRP level, with reference to the commonest haplotype, H1, which is set as the baseline. Once again, individual haplotypes were consistently associated with acute-phase serum CRP with high levels of significance in both patient sets individually and in a combined analysis.


Regression parameters were used to illustrate expected CRP values depending on background inflammatory status (defined this by ESR) and CRP haplotype. In the setting of chronic inflammation we would expect both ESR and CRP to be at steady state and in equilibrium. Figure 1A illustrates the expected geometric mean (median) CRP (95% CI) for individuals homozygous for haplotype H1 compared with the expected median for H4 homozygotes. H1 homozygotes have a median CRP 232% higher than H4 homozygotes at each level of inflammation (p<0.0005).

The percentage of the population carrying any specific haplotype pair is modest (7% for H1 homozygotes and 1% for H4 homozygotes), so we illustrate that the effect on CRP level is not confined solely to rare haplotype pairs but can also be demonstrated between larger groupings in the population. In Figure 1B, we have compared individuals carrying any combination of the H1 or H5 haplotypes (i.e., H1 H1, H1 H5, or H5 H5), with individuals carrying any combination of the remaining haplotypes. The H1/H5 group, constituting 11% of the population have an acute-phase CRP 82% higher than the 45% of individuals carrying two of the remaining haplotypes (p<0.0005). Individuals with one haplotype from each of these two groups will naturally have a CRP level falling somewhere in the middle.
We have observed that the magnitude of CRP haplotypic effect on serum CRP, over and above the variation in CRP expected due to underlying inflammatory status, is therefore large. To exclude the possibility that these observations were being unduly influenced by rare data outliers we performed additional analyses. Firstly we reran the model after excluding the 5% of individuals with the highest or lowest overall CRP levels. No meaningful changes to the observed significance levels or parameter estimates were observed (Table S5). Secondly we assessed the impact of individuals to the model by their Cook's distances. All Cook's distances were <1, and in addition an exclusion of the 5% of individuals with the greatest Cook's distance again resulted in no material change to the overall effect sizes and significance levels (Table S6).
Discussion
Summary and Comparison with Current Literature
We have shown a highly significant association between CRP genotype and acute-phase serum CRP level, and to our knowledge we are the first to demonstrate this in a group of patients that includes a considerable number with chronic active inflammation (40% have serum CRP >10 mg/l). We believe that the size of the genetic effect we have observed is large enough to have a clinically relevant impact on the assessment of inflammatory disease activity, which in turn may influence therapeutic decision making.
A key issue in the study of acute-phase CRP genetics is how to quantify the level of inflammatory stimulus driving the acute-phase response. Without this adjustment, random error introduced by varying levels of inflammation across our study population may be so large as to mask the genetic effects owing to variation at the CRP locus. We explained in the introduction that we believe that the incorporation of ESR into our analysis enables us to quantify the background level of inflammation in the chronic inflammatory setting. We hypothesized that there is no biological plausibility for a causal link to underlie the normal CRP/ESR correlation. The data from this study provide additional support for this assumption, by showing no association between CRP polymorphisms and ESR, and therefore support the hypothesis that CRP and ESR are highly correlated, but truly independent, markers of inflammation.
The use of ESR allowed us to find, with high levels of significance, SNPs and haplotypes associated with high and low acute-phase CRP expression. Existing studies addressing this question have all measured the magnitude of CRP rise following a single noxious stimulus. The largest of these looked at the postmyocardial infarction CRP rise and, like us, found higher levels in carriers of the rare allele of rs3093059 and also the A allele of rs3091244, which is on the same haplotype. Conversely lowest CRP increases were seen in carriers of the rare alleles of rs1800947 and rs1205 and the haplotype (H4), which contains both of these SNPs [12]. Other studies have found an association of rs1800947 with lower serum CRP after coronary artery bypass surgery and after oesophagectomy, and rs1130864 with higher serum CRP postcoronary artery bypass surgery [10],[11],[13]. All these studies are complicated by the inability to reliably compare surgical trauma from one procedure to another. Early studies also showed an association between rs1130864 and CRP levels following periodontal therapy and following strenuous exercise, but these stimuli only elicited a low-grade acute-phase response [9],[13]. The findings reported in this study therefore confirm those of earlier reports, but extend them to demonstrate highly significant genetic effects on CRP levels in the setting of chronic inflammation.
In terms of the magnitude of the genetic effect, there is a great deal of variability in the literature discussed above. For example in the cardiac surgery study of Brull and colleagues individuals homozygous for the common allele of rs1130864 had CRP levels that were only 20% higher than heterozygotes or individuals homozygous for the rare allele (164 versus 194 mg/l) [13]. In contrast Suk et al., who examined the increase in CRP concentration following myocardial infarction saw almost 600% difference between genotypes associated with low and high CRP concentrations (11 versus 77 mg/l) [12]. Much of this difference can undoubtedly be explained by the SNP chosen for the study, and also methodological issues such as the level of control for underlying inflammatory stimulus. It is clear, however, that the large effect of CRP genotypes that we have observed in this study in the setting of chronic inflammation, is consistent with large effects that have already been described following acute stimuli (such as those described by Suk et al. [12].).
There is also consistency between the SNPs we find associated with elevated CRP in chronic inflammation and those documented in basal CRP studies. In particular both rs1205 and rs1800947 have been widely associated with lower basal CRP expression [2]–[5]. Similar to the acute-phase literature, there is also variation in the magnitude of genotype effects in basal CRP studies, ranging from an approximate 50% difference to a difference of more than 500% in CRP concentrations between high and low CRP associated genotypes, from study to study and depending on which SNP or haplotype is examined [1],[2],[5]. It is possible, therefore, that the effect we report in the setting of chronic inflammation is simply an extension of the same effect that is observed with basal CRP. This observation is of interest, because it suggests that the low grade stimulation of CRP expression by “metabolic stress” may share a common pathway with the expression of CRP in response to classical inflammatory stimuli [2],[26].
Clinical Implications of the Data
By incorporating ESR we can use the model parameters to calculate the expected median CRP for an individual carrying any combination of CRP haplotype at any level of background inflammation. This finding showed that the genetic associations translated into marked and clinically relevant effects on acute-phase CRP. Although serum CRP is interpreted as part of a complete clinical picture, we propose that, for a newly symptomatic patient, a CRP of 9 mg/l would be reassuring and might trigger a different clinical decision from a CRP of 30 mg/l; similarly for a patient with known inflammatory disease a CRP of 15 mg/l may be observed, but a CRP of 50 mg/l is likely to trigger a change in treatment; yet our data suggest that these CRP differences could occur simply owing to genetics, independent of any difference in underlying disease activity.
In addition to these intuitive examples, it is worth considering specific clinical algorithms that incorporate CRP. For example, rheumatoid arthritis disease activity is frequently assessed using the DAS-28 score (incorporating a count of tenderness and swelling at 28 specific joints, a global assessment of disease activity by visual analogue scale [VAS], and a biochemical marker of inflammation, either ESR or CRP [http://www.reuma-nijmegen.nl/www.das-score.nl/index.html]). In the United Kingdom a DAS-score threshold is used to determine who receives funding for expensive biological therapies. While the DAS28-ESR is more frequently used at present, it seems inevitable that the DAS28-CRP will increase in popularity, and this is where the effect of genetics becomes important [27]. A patient with a genetically determined CRP of 66 mg/l, six swollen and tender joints, and a VAS of 50 mm will have a DAS score of 5.24. A patient with a genetically determined low acute-phase CRP of 20 mg/l, and an identical VAS would require an additional five tender or ten swollen joints to achieve this DAS score. In other words, a patient with genetically determined low CRP would be required to have considerably worse disease to be considered for the same treatment as a patient with genetically determined high CRP.
A review of the recent literature reveals multiple studies proposing a predictive value for acute-phase CRP; including the prediction of all-cause hospital mortality, death and complications in various critical care scenarios, cancer relapse, and the progression of inflammatory arthritis [28]–[35]. Our data do not contradict these study findings, but they suggest that in some situations the predictive ability of CRP could be significantly improved by the provision of a genetic adjustment. Ironically, while CRP measurement offers many advantages over ESR, we have demonstrated a potential drawback in that common genetic variants strongly influence serum levels. In contrast ESR, which is determined by a number of different physiological processes (as discussed earlier), would seem less likely to be strongly influenced by any individual genetic variant or locus (this, of course, remains to be tested).
Although our study was conducted only in patients of white European ancestry, a comparison with existing studies in different ethnic groups raises the possibility of population differences in the acute-phase CRP response, because of population differences in the haplotype architecture at CRP. Of the trans-ethnic mapping studies we are aware of, the most extreme difference appears to be between African American and Filipino populations [36],[37]. In African Americans haplotypes tagged by the minor allele of rs180097 (H4) are very rare (<1%), but in Filipinos this haplotype is common (10%). Similarly haplotypes with the common allele of rs1800974 but the rare allele of rs1205 (H2) are found on 16% of African American but on 43% of Filipino chromosomes. Both these haplotypes are associated with lower acute-phase CRP, and it is intriguing to question whether this would translate into a global tendency towards lower acute-phase CRP in Filipinos compared with African Americans. This hypothesis clearly requires experimental confirmation.
We would also like to speculate on the wider issue of whether the magnitude of acute-phase CRP rise in response to inflammatory stimuli has any implications for patients in terms of the physiological or pathological response to disease. Producing more CRP may offer benefits, perhaps by enhancing clearance of pathogenic bacteria, or by enhancing the clearance of apoptotic cells and reducing susceptibility to systemic autoimmunity [8]. However excessive CRP production may also have deleterious consequences, by promoting inflammation and tissue damage in unwanted situations. This finding has been demonstrated in a rat model of myocardial infarction, where injection of human CRP is associated with a poor outcome; a situation that can be reversed by synthetic CRP inhibitors [23]. Whether naturally occurring, genetically determined variation in the level of acute-phase response translates into differing outcomes in these varied clinical situations remains to be tested.
Study Limitations
We recognise the limitations to our study. There may be considerable heterogeneity within our study populations, which were recruited over four different sites, with CRP and ESR quantified in four different laboratories. We recognise that although patients self-reported white European ancestry, there may be subtle differences in population ancestry, particularly between patients recruited from the Northern and Southern hemispheres, and that this has not been accounted for by a principle components analysis. In addition we were unable to account for variables such as medication history, which may be important given the reported effect of drugs such as statins on basal CRP levels.
Although our study aim was to assess the influence of genetics on the acute-phase CRP response, we appreciate that 60% of our study participants had a serum CRP concentration of ≤10 mg/l and only 8% had very high serum concentration of CRP (>50 mg/l). The recruitment of patients with uncontrolled inflammation is of course difficult as these patients tend to be aggressively treated.
As this study was conducted in patients with rheumatoid arthritis we need to question whether or not our data can be extrapolated to other chronic inflammatory diseases. CRP itself has never been reported as a susceptibility gene in rheumatoid arthritis and, although we did not perform a disease association study here, the allele frequencies we observe in our patient sets are very close to those widely reported elsewhere for non-rheumatoid arthritis patients. We therefore have no reason to suppose that rheumatoid arthritis is a “special case” in terms of its influence on the acute-phase response. We would therefore hypothesize that our findings can be extrapolated to other chronic inflammatory diseases.
Conclusion
CRP can be measured easily and standard assays offer great accuracy. While the extreme sensitivity and rapid response to changing levels of inflammation make CRP measurement an invaluable clinical tool, our data introduce a note of caution. Acute-phase CRP may be strongly influenced by common genetic variants and CRP concentrations should therefore be interpreted in light of this. If technology advances to the stage of allowing rapid bedside genetic testing, then a personalised, genetically adjusted CRP level may prove to be a useful diagnostic and predictive biomarker.
Supporting Information
Zdroje
1. CarlsonCS
AldredSF
LeePK
TracyRP
SchwartzSM
2005 Polymorphisms within the C-reactive protein (CRP) promoter region are associated with plasma CRP levels. Am J Hum Genet 77 64 77
2. MillerDT
ZeeRY
SukDJ
KozlowskiP
ChasmanDI
2005 Association of common CRP gene variants with CRP levels and cardiovascular events. Ann Hum Genet 69 623 638
3. CrawfordDC
SandersCL
QinX
SmithJD
ShephardC
2006 Genetic variation is associated with C-reactive protein levels in the Third National Health and Nutrition Examination Survey. Circulation 114 2458 2465
4. KathiresanS
LarsonMG
VasanRS
GuoCY
GonaP
2006 Contribution of clinical correlates and 13 C-reactive protein gene polymorphisms to interindividual variability in serum C-reactive protein level. Circulation 113 1415 1423
5. LangeLA
CarlsonCS
HindorffLA
LangeEM
WalstonJ
2006 Association of polymorphisms in the CRP gene with circulating C-reactive protein levels and cardiovascular events. JAMA 296 2703 2711
6. DaneshJ
WheelerJG
HirschfieldGM
EdaS
EiriksdottirG
2004 C-reactive protein and other circulating markers of inflammation in the prediction of coronary heart disease. N Engl J Med 350 1387 1397
7. DaneshJ
PepysMB
2009 C-reactive protein and coronary disease: is there a causal link? Circulation 120 2036 2039
8. PepysMB
HirschfieldGM
2003 C-reactive protein: a critical update. J Clin Invest 111 1805 1812
9. D'AiutoF
CasasJP
ShahT
HumphriesSE
HingoraniAD
2005 C-reactive protein (+1444C>T) polymorphism influences CRP response following a moderate inflammatory stimulus. Atherosclerosis 179 413 417
10. MotoyamaS
MiuraM
HinaiY
MaruyamaK
UsamiS
2009 C-reactive protein 1059G>C genetic polymorphism influences serum C-reactive protein levels after esophagectomy in patients with thoracic esophageal cancer. J Am Coll Surg 209 477 483
11. PerryTE
MuehlschlegelJD
LiuKY
FoxAA
CollardCD
2009 C-Reactive protein gene variants are associated with postoperative C-reactive protein levels after coronary artery bypass surgery. BMC Med Genet 10 38
12. SukDJ
ChasmanDI
CannonCP
MillerDT
ZeeRY
2006 Influence of genetic variation in the C-reactive protein gene on the inflammatory response during and after acute coronary ischemia. Ann Hum Genet 70 705 716
13. BrullDJ
SerranoN
ZitoF
JonesL
MontgomeryHE
2003 Human CRP gene polymorphism influences CRP levels: implications for the prediction and pathogenesis of coronary heart disease. Arterioscler Thromb Vasc Biol 23 2063 2069
14. RopesMW
RossmeislE
BauerW
1939 The relationship between the erythrocyte sedimentation rate and the plasma proteins. J Clin Invest 18 791 798
15. ZlonisM
1993 The mystique of the erythrocyte sedimentation rate. A reappraisal of one of the oldest laboratory tests still in use. Clin Lab Med 13 787 800
16. AnderssonR
MalmvallBE
BengtssonBA
1986 Acute phase reactants in the initial phase of giant cell arteritis. Acta Med Scand 220 365 367
17. Osei-BimpongA
MeekJH
LewisSM
2007 ESR or CRP? A comparison of their clinical utility. Hematology 12 353 357
18. SteerS
LadB
GrumleyJA
KingsleyGH
FisherSA
2005 Association of R602W in a protein tyrosine phosphatase gene with a high risk of rheumatoid arthritis in a British population: evidence for an early onset/disease severity effect. Arthritis Rheum 52 358 360
19. ArnettFC
EdworthySM
BlochDA
McShaneDJ
FriesJF
1988 The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum 31 315 324
20. RhodesB
MeekJ
WhittakerJC
VyseTJ
2008 Quantification of the genetic component of basal C-reactive protein expression in SLE nuclear families. Ann Hum Genet 72 611 620
21. BarrettJC
FryB
MallerJ
DalyMJ
2005 Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21 263 265
22. StephensM
DonnellyP
2003 A comparison of bayesian methods for haplotype reconstruction from population genotype data. Am J Hum Genet 73 1162 1169
23. PepysMB
HirschfieldGM
TennentGA
GallimoreJR
KahanMC
2006 Targeting C-reactive protein for the treatment of cardiovascular disease. Nature 440 1217 1221
24. RidkerPM
PareG
ParkerA
ZeeRY
DanikJS
2008 Loci related to metabolic-syndrome pathways including LEPR, HNF1A, IL6R, and GCKR associate with plasma C-reactive protein: the Women's Genome Health Study. Am J Hum Genet 82 1185 1192
25. VerzilliC
ShahT
CasasJP
ChapmanJ
SandhuM
2008 Bayesian meta-analysis of genetic association studies with different sets of markers. Am J Hum Genet 82 859 872
26. KushnerI
SamolsD
MagreyM
2010 A unifying biologic explanation for “high-sensitivity” C-reactive protein and “low-grade” inflammation. Arthritis Care Res (Hoboken) 62 442 446
27. FransenJ
WelsingPM
de KuijzerRM
van RielPL
2004 Disease activity scores using C-reactive protein: CRP may replace ESR in the assessment of RA disease activity. Ann Rheum Dis 62 Suppl 1 151
28. ArtzAS
WickremaA
DinnerS
GodleyLA
KocherginskyM
2008 Pretreatment C-reactive protein is a predictor for outcomes after reduced-intensity allogeneic hematopoietic cell transplantation. Biol Blood Marrow Transplant 14 1209 1216
29. BajwaEK
KhanUA
JanuzziJL
GongMN
ThompsonBT
2009 Plasma C-reactive protein levels are associated with improved outcome in ARDS. Chest 136 471 480
30. KimDK
OhSY
KwonHC
LeeS
KwonKA
2009 Clinical significances of preoperative serum interleukin-6 and C-reactive protein level in operable gastric cancer. BMC Cancer 9 155
31. KoikeY
MikiC
OkugawaY
YokoeT
ToiyamaY
2008 Preoperative C-reactive protein as a prognostic and therapeutic marker for colorectal cancer. J Surg Oncol 98 540 544
32. KompotiM
DrimisS
PapadakiA
KotsomytisK
PoulopoulouC
2008 Serum C-reactive protein at admission predicts in-hospital mortality in medical patients. Eur J Intern Med 19 261 265
33. MoonJM
ChunBJ
2009 Predicting the complicated neutropenic fever in the emergency department. Emerg Med J 26 802 806
34. PooleCD
ConwayP
ReynoldsA
CurrieCJ
2008 The association between C-reactive protein and the likelihood of progression to joint replacement in people with rheumatoid arthritis: a retrospective observational study. BMC Musculoskelet Disord 9 146
35. van der Helm-van MilA
leCS
vanDH
BreedveldFC
ToesRE
2007 A prediction rule for disease outcome in patients with recent-onset undifferentiated arthritis: how to guide individual treatment decisions. Arthritis Rheum 56 433 440
36. RhodesB
WongA
NavarraSV
VillaminC
VyseTJ
2008 Genetic determinants of basal C-reactive protein expression in Filipino systemic lupus erythematosus families. Genes Immun 9 153 160
37. RhodesB
MorrisDL
SubrahmanyanL
AubinC
de LeonCF
2008 Fine-mapping the genetic basis of CRP regulation in African Americans: a Bayesian approach. Hum Genet 123 633 642
Štítky
Interní lékařstvíČlánek vyšel v časopise
PLOS Medicine
2010 Číslo 9
- Ivabradin zlepšuje kvalitu života starších pacientů se srdečním selháním
- Fixní kombinace kandesartan/amlodipin v terapii arteriální hypertenze
- Inovace v hojení ran: krytí Zetuvit Plus Silicone Border pro optimální management exsudátu z ran
- Rána vizitkou (nejen) chirurga
- Patogeneze vzniku keloidní jizvy
Nejčtenější v tomto čísle
- Seventy-Five Trials and Eleven Systematic Reviews a Day: How Will We Ever Keep Up?
- A Genetic Association Study of Serum Acute-Phase C-Reactive Protein Levels in Rheumatoid Arthritis: Implications for Clinical Interpretation
- Persistence with Statins and Onset of Rheumatoid Arthritis: A Population-Based Cohort Study
- The Haunting of Medical Journals: How Ghostwriting Sold “HRT”
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy