
Svalová biopsie u myotonické dystrofie v éře molekulární genetiky
Muscular biopsy in myotonic dystrophy in the era of molecular genetics
The histopatological features of both so far defined types of myotonic dystrophy (DM1 and DM2) are very similar, the affected muscles show a typical pattern of changes. The examination of muscle biopsies in situ may also bring about revealing of expansions of CUG or CCUG repetitons in transcripts of the mutated DNA, the genetic basis of the diseases, which may be demonstreated by FISH (fluorescence in situ hybridization) as intranuclear focal accumulation of the transcript. In order to evaluate the importance of muscle biopsy including FISH at the time, when the diagnosis of DM is based on DNA analysis from blood lymphocytes, we analysed the results of histopathological examination of 34 patients, where the biopsy was indicated by clinical suspicion of myotonic dystrophy. The diagnosis was verified by mutation analysis of DNA for both DM1 and DM2. The examination was supplemented by FISH in 27 patients. The diagnosis of DM1 was confirmed in 13 patients, DM2 in 10 patients, the diagnosis of DM1 or DM2 was not confirmed in 11 patients. The biopsy brought about confirmation or precision-specification of the clinical diagnosis or suggested the diagnosis of DM in the majority of the patients. The bioptic findings in some patients with DM were atypical: in two cases there was inflammatory infiltration, in one case a picture resembling congenital myopathy. On the other hand, histopathological features of myotonic dystrophy were present in several biopsies where the diagnosis of DM was not confirmed. Focal accumulation of CUG/CCUG repeats in myonuclei was demonstrated by fluorescence in situ hybridization – in accordance with the DNA examination – in 23 patients, 13 with DM1 and 10 with DM2. Mutation analysis of the DNA isolated from blood leukocytes represent the method of choice if the diagnosis of DM is suspected. On the other hand, muscle biopsy may reveal histopathological features that may indicate subsequent molecular genetic examination of the patient if the results of the clinical examination are equivocal or atypical. FISH in the biopsy may reveal the intranuclear focal accumulation of the RNA transcripts in cells and tissues of the affected patiens and, in this way, to suggest a possible participation of them in the pathogenesis of the disease.
Key words:
myotonic dystrophy – biopsy – molecular genetics – diagnosis
Autoři:
Z. Lukáš 1; I. Kroupová 1; J. Bednařík 2; M. Falk 3; L. Fajkusová 4; J. Sedláčková 4; I. Valášková 5; S. Voháňka 2
Působiště autorů:
Ústav patologie LF MU a FN Brno
1; Neurologická klinika LF MU a FN Brno
2; Biofyzikální ústav AV ČR, Brno
3; Centrum molekulární biologie
a genové terapie hematoonkologické
kliniky LF MU a FN Brno
4; Oddělení klinické genetiky FN Brno
5
Vyšlo v časopise:
Cesk Slov Neurol N 2007; 70/103(4): 395-401
Kategorie:
Krátké sdělení
Podpořeno grantem IGA MZ ČR NR 8052-3
Souhrn
Histopatologické rysy obou v současnosti definovaných forem myotonické dystrofie (MD1 i MD2) jsou velmi podobné, vykazují charakteristický komplex změn. Vyšetření svalových biopsií in situ může kromě toho přinést i zjištění přítomnosti expanzí repetic CUG resp. CCUG v transkriptu mutované DNA, která tvoří genetický základ onemocnění. Expandované úseky lze znázornit ve tkáňových řezech metodou fluorescenční in situ hybridizace (FISH) jako fokální intranukleární inkluze. Pro posouzení významu svalové biopsie v době, kdy je diagnostika MD1 a MD2 založena na DNA analýze lymfocytů z periferní krve, jsme analyzovali výsledky histopatologického vyšetření souboru 34 nemocných, u kterých byla biopsie provedena na základě klinického podezření na myotonickou dystrofii. Diagnóza MD byla potvrzena analýzou DNA pro oba typy onemocnění. FISH byla provedena u 27 z nich. Mutační analýzou DNA byla potvrzena MD1 v 13 případech, MD2 v 10 případech, u 11 pacientů nebyla diagnóza MD1 či MD2 potvrzena. Ve většině případů byl bioptický histopatologický nález u prokázané MD hodnocen buď jako typický pro MD1 či MD2, anebo nález kompatibilní s touto diagnózou. Bioptické nálezy u některých pacientů s MD byly atypické: ve 2 případech byla přítomna zánětlivá infiltrace, v jednom případě obraz připomínající kongenitální myopatii. Naproti tomu histopatologické rysy MD byly zjištěny v některých biopsiích, u nichž diagnóza MD1 ani MD2 nebyla potvrzena. Fokální intranukleární inkluze prokazované FISH byly prokázány u 23 pacientů – ve shodě s výsledky analýzy DNA – u 13 s MD1 a u 10 s MD2. Metodou volby při podezření na myotonickou dystrofii je mutační analýza DNA z periferní krve. V případě provedené svalové biopsie z důvodu atypického klinického obrazu, který nevedl k podezření na MD, však může zhodnocení biopsie vést k dodatečnému molekulárně-genetickému vyšetření pacienta. Metoda FISH v biopsii je zajímavá z teoretického pohledu, neboť umožňuje zkoumat retinovanou RNA v jádrech jednotlivých buněk a tkání postižených pacientů, a tak může napovědět o jejich možné účasti v patogenezi onemocnění.
Klíčová slova:
myotonická dystrofie – biopsie – molekulární genetika – diagnóza
Úvod
Myotonická dystrofie (MD) je autosomálně dominantní neuromuskulární onemocnění s incidencí asi 1/8000. Je to nejčastější forma svalové dystrofie postihující dospělou populaci [1].
Příčinou „klasické“ myotonické dystrofie (DM1) je na molekulárně-genetické úrovni nestabilní trinukleotidová repetitivní sekvence (CTG)n v genu kódujícím protein-kinázu (DMPK - dystrophia myotonica protein-kinase) na chromozomu 19q13.3 [2,3]. Expanze trinukleotidové sekvence má za následek retenci transkriptu genu DMPK v jádře [ 4], která generuje fenotyp MD [5]. Molekulárně genetická diagnostika CTG expanzí na úrovni DNA je možná Southern blot analýzou [2,6] nebo pomocí PCR [7,8]. Retinovanou RNA v jádrech lze prokázat i ve tkáňových řezech hybridizací in situ, fluorescenční (FISH) či chromogenní [5,9,10]. FISH se zakládá na afinitě fluorochromem označené próby ke komplementárním sekvencím DNA či RNA ve tkáních (buňkách, kulturách).
V 90. letech byla studována a postupně rozpoznávána varianta myotonické dystrofie postihující zejména proximální svalové skupiny (MD druhého typu, MD2, PROMM), u níž nebyla prokázána expanze na 19q13.3 [11,12], avšak byla zjištěna repetice (CCTG)n nukleotidů v genu ZNF9 na chromozomu 3q21 [13]. Také tuto repetitivní sekvenci lze prokázat DNA analýzou, a stejně jako u prvního typu byla vypracována i varianta detekce retinované RNA intranukleárně in situ [14,15].
Bylo prokázáno, že je to RNA obsahující zmíněné tri - či tetranukleotidové expanze, která odpovídá za klinické rysy obou forem onemocnění. Expanze jsou inkorporované do ribonukleárních inkluzí, přitom dojde k dysregulaci RNA-vazebných proteinů: ke snížené regulaci rodiny muscleblind proteinů (MBNL) a ke zvýšené regulaci CUG-vazebných proteinů (CUG-BP). Obě tyto dysregulované skupiny proteinů pozměňují či poškozují sestřih transkriptů, které se uplatní v patogenezi MD, zejména transkript chloridového kanálu, receptoru pro inzulin a další [16].
Klinický obraz MD je dobře vymezen, vyznačuje se však mimořádnou variabilitou. I když je MD pokládána za onemocnění kosterního svalstva zahrnující distální myotonii, progresivní slabost a dystrofii, postihuje i další systémy. Postižení často trpí alterací srdečního a hladkého svalstva, dále bývají přítomny katarakta, hypersomnie, abnormální odezva na aplikaci glukózy a u mužů navíc předčasná plešatost a testikulární atrofie. Poruchy srdeční vodivosti mohou mít za následek fatální arytmii, a tak ovlivnit prognózu onemocnění. Nápadným rysem je variabilita fenotypu jak mezi, tak uvnitř postižených rodin [16]. Mnozí pacienti s MD mají příznaky onemocnění již od dětství, zatímco v jiných případech mohou být časné symptomy zjištěny dříve u pacientových dětí než u pacienta samotného. Velmi charakteristická forma onemocnění počínající in utero je pozoruhodná a málo obvyklá [17]. Zatím však byla popsána jen u prvního typu – MD1.
Elektromyografický nález (EMG) není pro diagnostiku rozhodující, jsou-li přítomny jasné klinické symptomy případně rodinná anamnéza. EMG vyšetření je však nezbytné pokud myotonie není zřetelná nebo chybí. V klinicky vyvinutých případech jsou nejčastější několik sekund trvající serie výbojů se snižující se frekvencí a amplitud, provokované inzercí či pohybem jehlové elektrody – myotonické výboje. Volní EMG vykazuje myopatický vzorec [17].
Charakteristické histopatologické rysy onemocnění u MD1 i MD2 jsou velmi podobné. Zahrnují směs neurogenních i myogenních změn. Neexistuje však jediný znak, který by byl typický a charakteristický pro MD, avšak jen seskupení určitých znaků může přinést podporu klinickému podezření na toto onemocnění. Mezi tyto znaky patří zejména zvýšená frekvence a zřetězení vniřních i sarkolematických jader. Častá je i přítomnost atrofických angulárních vláken nebo jaderných shluků s minimem sarkoplazmy, dále prstenčitá vlákna (ring fibres, Ringbinden), přítomnost sarkoplazmatických mas a velikostně-typová dysproporce s nižšími hodnotami průměrů vláken 1 a endo-perimyziální fibróza. V některých případech byla popsána přítomnost degenerativních strukturálních změn až nekróz a známek regenerace [18]. Tyto znaky jsou patrně společné pro MD1 i MD2, i když některé sestavy v recentních sděleních ukazují na rozdíly v typu atrofických vláken (viz níže).
Na naších pracovištích jsme v 90. letech a dříve neměli k dispozici molekulárně genetické diagnostické metody rozlišující oba typy MD a diagnostka spočívala na elektrofyziologických a klinických nálezech většinou doplněných svalovou biopsií. Později byly u nás vypracovány metody mutační analýzy DNA pacientů postižených MD1 i MD2 [19,20] a v minulých letech jsme postupně zaváděli do diagnostické praxe fluorescenční in situ hybridizaci. Vyšetření svalových biopsií in situ (vyšetření in situ znamená vyšetření přítomnosti určitých tkání a orgánů v topografické návaznosti na struktury okolní (situs viscerum), na úrovni tkáně nebo i buňky je to přítomnost a poloha jádra, organel, či sloučenin (glykogen, železo, enzym, protein), a to v kontextu přítomných struktur, na rozdíl od vyšetření extra situm, třeba v homogenátech nebo v extraktech. Patří sem tedy jak histopatologické vyšetření, tak i molekulárně genetické vyšetření: může kromě histopatologické analýzy přinést i zjištění přítomnosti expanzí repetic CUG resp. CCUG v transkriptu mutované DNA, která tvoří genetický základ onemocnění. Expandované úseky RNA lze znázornit ve tkáňových řezech metodou fluorescenční in situ hybridizace (FISH) jako fokální intranukleární inkluze.
V tomto sdělení uvedeme výsledky diagnostiky obou forem MD ze svalových excizí pacientů na základě klinické indikace. Výsledky histopatologické analýzy biopsie a výsledky RNA hybridizace in situ jsou srovnány s výsledky vyšetření DNA metodou PCR.
Materiál a metodika
Analyzovali jsme soubor 34 pacientů (17 mužů od 15 do 64 let a 17 žen od 16 měsíců do 75 let), u kterých byla provedena svalová biopsie na základě klinického podezření z diagnózy myotonické dystrofie. Diagnóza MD1 nebo MD2 byla potvrzena či vyloučena pomocí DNA analýzy u 34 pacientů (tab. 1). V tabulce není zařazeno 13 předchozích bioptických vyšetření z doby, kdy vyšetření MD pomocí molekulárně genetických metod ještě nebylo zavedeno.
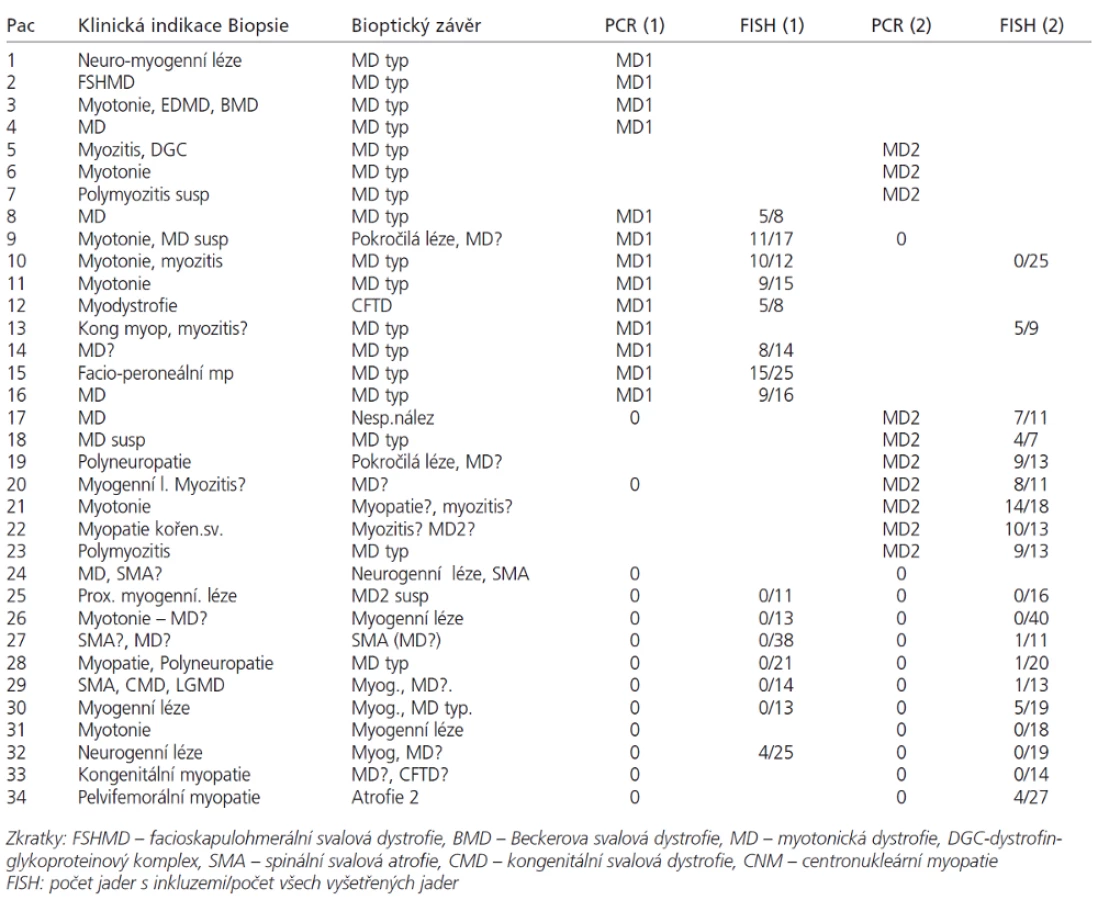

Svalové biopsie byly hluboce zmrazeny a kryostatové řezy zpracovány konvenčními histologickými a histochemickými metodami [21,22] doplněnými u 27 nemocných o RNA FISH. V každém vzorku byly hodnoceny tyto parametry: přítomnost vnitřních jader (normálně do 3 %, u myogenních lézí více než 5 %, u MD a centronukleární myopatie dosahuje frekvence až 65–100 %). Dále je to přítomnost atrofických vláken (v normě jen ojedinělá, jako dolní konec Gaussovy křivky), přítomnost prstenčitých vláken a sarkoplazmatických mas (v normě nepřítomna), velikostně-typová dysproporce s nižšími hodnotami průměrů vláken 1, a endo-perimyziální fibróza (v normě se též nevyskytují). Na základě těchto parametrů byla míra pravděpodobnosti dg. MD odstupňována takto: typický nález (v tabulce MD typ) obsahuje nad 65 % vnitřních jader, atrofie, případně ještě další znaky; nález kompatibilní s dg. MD (v tabulce MD?) znamená nález mírně zvýšeného počtu (nad 5 %) vnitřních jader a přítomnost atrofií.
In situ hybridizace vycházela z citovaných prací [8,9], upravená podle Thorntona (osobní sdělení). Řezy (6mm) byly napjaty na skla (SuperFrost Plus slides Menzel-Glaser), vysušeny při pokojové teplotě 30 minut, fixovány v modifikované Carnoyově tekutině (73% etanol; 25% kyselina octová; 2% formalin) 30 minut při 4 °C a promyty v PBS. Následovala inkubace v prehybridizačním pufru (30% formamid a 2x SSC) a hybridizace s probou (1 ng/ml) 2 hodiny při 37 °C v hybridizačním pufru (30% formamid, 2x SSC, 0,02% BSA, 66 mg/ml yeast tRNA, 2 mM vanadyl komplex). Sekvence próby pro MD1: (5’-CAG CAG CAG CAG CAG CAG CAG-3’2’ –O-Me-RNA5’Fl), sekvence próby pro MD2 (5’-CAG GCA GGC AGG CAG GCA GG-3’2’ –O-Me-RNA5’Fl, ). Po hybridizaci byly řezy oprány ve 30% formamidu a 2x SSC po 30 minut při 45 °C, a 40 min při pokojové teplotě. Nakonec zamontovány ve směsi s DAPI (Vectashield Mountaing Medium with DAPI, Vector Laboratories, USA) a hodnoceny ve fluorescenčním mikroskopu Leica DMRXA2.
Hodnocení výsledků RNA-FISH: jediným parametrem je přítomnost tečkovitých signálů retinované RNA v subsarkolematických i vnitřních jádrech svalových vláken. V pozitivních jádrech jsou většinou přítomny 1-4 jednotlivé signály. Z technických důvodů (tloušťka řezu) nemusejí být v každém nákroji jádra přítomné RNA signály zastiženy, proto hodnotíme biopsie jako MD-pozitivní, je-li přítomno aslepoň 30 % pozitivních jader. Dalším problémem je možná nespecifická pozitivita, vysvětlitelná interferencí autofluoreskujícího lipopigmentu, který emituje široké spektrum vlnových délek [9]. Tato autofluorescence je někdy obtížně odlišitelná od specifické fluorescence užitého fluorochromu.
Pro hodnocení validity klinických i histopatologických nálezů a FISH byly výsledky molekulárně genetického vyšetření pacientů brány jako kritérium správné diagnózy.
Výsledky
Histopatologické parametry
Jednotlivé parametry a výsledky histopatologického nálezu, RNA-FISH, DNA analýzy na základě klinické indikace biopsie jsou shrnuty v tab. 1. Z tabelárních údajů vyplývá, že vysoká či zvýšená frekvence vnitřních jader (obr. 1) byla v naší sestavě zjištěna s jedinou výjimkou ve všech pozitivních biopsiích obou skupin pacientů. Přítomnost atrofií (obr. 1) byla zjištěna ve všech biopsiích obou skupin. Atrofie vláken 1 převažovala v biopsiích pacientů s MD1, zatímco u MD2 nebyla zjištěna ani v jediném případě. U MD2 byla přítomna jen atrofie typu 2 nebo obou typů vláken. Velikostně typová dysproporce základních typů s vyššími hodnotami vláken 1 (obr. 2) byla nalezena u 30-60 % případů MD1, MD2 i MD nerozlišené. Prstenčitá vlákna a sarkoplazmatické masy (obr. 3 a 4) byly v našich pozitivních biopsiích zjištěny celkem v 18 % všech pacientů obou skupin, ve 20 % u pacientů s MD1 a 30 % u pacientů s MD2. Fibróza byla zjištěna asi v polovině vyšetřených případů, 2krát dokonce i zánětlivá infitrace (jednou s pozitivní reakcí MAC v cévní stěně a HLA1 v sarkolemě).




V několika případech byly histopatologické nálezy netypické. U pacienta 12 byla DNA analýzou prokázána MD 1 a bioptická diagnóza zněla kongenitální typová disproporce svalových vláken (CFTD). U pacienta 19 byla zjištěna tak pokročilá léze, že histopatologická diagnóza nebyla možná, zatím co DNA analýza prokázala repetici CTG svědčící pro MD 1. Pacient 24 s prokázanou SMA měl v biopsii obraz neodlišitelný od MD. U pacientů 21 a 22 byla přítomna již zmíněná zánětlivá infiltrace, u MD velmi vzácná.
RNA fluorescenční in situ hybridizace
V pozitivních jádrech byly většinou přítomny 1-4 jednotlivé signály (obr. 5 a 6 pro DM1 a DM2). Pozitivní signály byly zjištěny i v některých jádrech intramuskulárních cév. V těchto jádrech však byly většinou jen 1-2 signály. Kromě toho, několik pozitivních signálů bylo nalezeno i v jádrech cévních endotelií (nedemonstrováno). Biopsie, v nichž bylo nalezeno více než 30 % pozitivních jader svalových vláken, byly hodnoceny jako pozitivní pro zkoumaný typ MD. Ve většině případů, kdy byla provedena FISH (34 ze 36), odpovídaly její výsledky nálezům DNA analýzy z periferní krve. V jádrech u pacientů 13 a 30, kde diagnóza MD2 nebyla mutační analýzou DNA potvrzena, byly pozitivní signály přítomny. Nespecifické pozitivní signály snad mohou být způsobeny interferencí autofluoreskujícího lipopigmentu, která při hodnocení výsledků FISH často působí komplikace [9]. Lipofuscin excituje a emituje široké spektrum vlnových délek, takže generuje signál prakticky v každém z užívaných kanálů, zatímco specifický signál fluoresceinu – FITC (495/520) resp Texaské červeně (596/620) je vidět pouze s filtrem odpovídajících vlnových délek. Při nejasnostech v topografii vysílaného signálu pomůže vyšetření laserovým konfokálním mikroskopem.

Diskuse a závěry
Klinická indikace biopsie zahrnuje samozřejmě jen část případů MD diagnostikovaných v našem regionu, protože u většiny pacientů byla a je diagnostikována na základě klinického a elektrofyziologického nálezu doplněného DNA analýzou [20].
Porovnáme-li jednotlivé parametry histopatologického vyšetření vedoucího k diagnóze MD (tab. 1), zjišťujeme, že nejčastějším a patrně nejspecifičtějším histopatologickým parametrem je zvýšená frekvence vnitřních jader [18,21,22]. Přítomnost atrofií, ať ve formě jaderných shluků (nuclear clumps) s minimem sarkoplazmy nebo jako malá angulární vlákna, je dalším znakem přítomným prakticky ve všech našich pozitivních biopsiích. U MD1 je udávána atrofie vláken 1 [17,18], avšak u MD2 nacházejí Schoser et al [23] a Vihola et al [24] převážně atrofii typu 2. Naproti tomu autoři Day et al [25] popisují u MD2 atrofii vláken bez udání typů. Tento parametr však má cenu jen ve spojení s dalšími, protože atrofie vláken typu 2 je znakem inaktivity, vyskytujícím se u celého spektra neuro - i myopatií včetně myozitid a dalších onemocnění pohybového aparátu. Atrofie svalových vláken je však i základním znakem neurogenních (i některých dalších myogenních) lézí, takže její specificita je tím pro histopatologickou diagnostiku MD limitovaná. Nicméně, naše výsledky podporují údaje citovaných autorů o převaze atrofie vláken typu 1 u MD1 a atrofie vláken typu 2 u MD2.
Velikostně-typová dysproporce základních typů vláken [26] je však odlišným znakem, který nemusí nutně znamenat atrofii některého typu, pouze vyšší průměrné hodnoty velikosti (průměrů) vláken typu 2 než má typ 1. Prstenčitá vlákna [27] jsou dosti specifickým nálezem u MD, ale jejich frekvence v našich pozitivních biopsiích je nevysoká, stejně jako v publikovaných větších sestavách [17]. Oba jmenované parametry jsou přítomny jen v části biopsií s průkazem obou typů MD.
Naše nálezy ukazují, že histopatologický obraz u MD1 i 2 je poměrně charakteristický a před érou molekulárně-genetické diagnostiky podpořil diagnózu myotonické dystrofie.V několika případech histopatologické vyšetření nevedlo ke správnému diagnostickému závěru: u 2 pacientů s DNA analýzou potvrzenou diagnózou DM skýtal bioptický nález obraz myozitidy, u dalšího pacienta bioptická diagnóza zněla kongenitální typová disproporce svalových vláken (CFTD). Nález zánětlivé infiltrace je u DM vskutku ojedinělý, její přítomnost u svalových dystrofií jako DMD, či FSHMD, nebo i u neurogenních atrofií však byla popsána opakovaně [21]. Velikostní typová dysproporce je ovšem znak nespecifický a bývá přítomen nejen u CFTD, ale i u myotonické dystrofie, u dalších kongenitálních myopatií (nemaline myopathy), ale i u spinální svalové atrofie. V naší sestavě byl tento případ ojedinělým bioptickým nálezem MD bez přítomnosti vnitřních jader.
V éře molekulárně-genetické diagnostiky MD1 a MD2 však diagnostika myotonické dystrofie pomocí histopatologických změn pozbývá významu a metodou volby při typickém klinickém a elektrofyziologickém nálezu (který se u obou geneticky definovaných chorob poněkud liší [20] je jednoznačně DNA diagnostika z periferní krve. Pozitivita FISH nálezů však těsně koreluje s nálezy DNA analýzy. Znalost histopatologického obrazu charakteristického pro MD však může být přínosná v případě, že svalová biopsie byla provedena z výše uvedených důvodů: histologický nález známek typických pro MD může vést k dodatečnému vyšetření DNA z lymfocytů periferní krve, nebo i ze zbytku bioptického materiálu. DNA analýza z lymfocytů je samozřejmě jednodušší, protože obsahuje daleko víc jaderné DNA na hmotnostní či objemovou jednotku než sval. A konečně, na zbytku biopsie lze pomocí FISH odhalit i přítomnost intranukeárních fokusů s expandovanou RNA (viz výše).
Metoda FISH v biopsii je zajímavá z teoretického pohledu, neboť umožňuje zkoumat retinovanou RNA v jádrech jednotlivých buněk a tkání u postižených pacientů, a tím i mechanizmus rozvoje klinické manifestace. Je nutné dořešit i metodické aspekty jako prezervace či fixace tkání, upřesnit vyhodnocení pozitivních nálezů a vztah k výsledkům DNA analýzy. Nejsou dosud známy rozdíly v expanzi DNA v jednotlivých tkáních a orgánech postižených pacientů; zřejmě však takové variace existují [28]. Také zmapování RNA inkluzí v tkáních pacientů s MD a jejich případný vztah k velikosti expanze nejsou v současné době dostatečně prozkoumány.
Přijetí k recenzi: 25. 1. 2007
Přijetí do tisku: 8. 2. 2007
prof. MUDr. Zdeněk Lukáš, CSc.
Ústav patologie FN Brno,
Jihlavská 20, 625 00 Brno
e-mail: zlukas @fnbrno.cz
Zdroje
1. Harper PS. Myotonic dystrophy. London: WB Saunders 2001.
2. Brook JD, McCurah ME, Harley HG, Buckler AJ, Church D, Aburatani H et al. Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3´ end of a transcript encoding a protein kinasae family member. Cell 1992; 68 : 799-808.
3. Fu YH, Pizzuti A, Fenwick Jr RG, King J, Rajnarayan S, Dunne PW et al. An unstable triplet repeat in a gene related to myotonic dystrophy. Science 1992; 255 : 1256-1258.
4. Davis BM, McCurach ME, Taneja K, Singer RH, Housman DE. Expansion of a CUG trinucleotide repeat in the 3´ untranslated region of myotonic dystrophy protein kinase transcripts results in nuclear retention of transcripts. Proc Natl Acad Sci 1997; 94 : 7388-7393.
5. Mankodi A, Logigian E, Callahan L, McClain C, White R, Henderson D et al. Myotonic dystrophy in transgenic mice expressing an expanded CUG repeat. Science 2000; 289 : 1769-1772.
6. Buxton J, Shelbourne P, Davies J, Jones C, Van Tongeren T, Aslanidis C et al. Detection of an unstable fragment of DNA specific to individuals with myotonic dystrophy. Nature 1992; 355 : 547-548.
7. Cheng S, Barcelo JM, Korneluk RG. Characterization of large CTG repeat expansions in myotonic dystrophy alleles using PCR. Hum Mut 1996; 7 : 304-310.
8. Schalling M, Hudson TJ, Buetow KH, Housman DE. Direct detection of novel trinucleotide repeats in the human genome. Nature genetics 1993; 4 : 135-139.
9. Taneja KL. Localization of trinucleotide repeat sequences in myotonic dystrophy cells using a single fluorochrome –labeled PNA probe. Biotechniques 1998; 24 : 472-476.
10. Jiang H, Mankodi A, Swanson MS, Moxley RT, Thornton CA. Myotonic dystrophy type 1 is associated with nuclear foci of mutant RNA, sequestration of muscleblind proteins and deregulated alternative splicing in neurons. Human molecular genetics 2004; 13 : 3079-3088.
11. Thornton CA, Griggs RC, Moxley RT. Myotonic dystrophy with no trinucleotide repeat expansion. Ann Neurol 1994; 35 : 269-272.
12. Ricker K, Koch MC, Lehmann-Horn F, Pongraz D, Otto M, Heine R et al. Proximal myotonic myopathy: a new dominant disorder with myotonia, muscle weakness and cataracts. Neurology 1994; 44 : 1448-1452.
13. Liquori CL, Ricker K, Moseley ML, Jacobsen JF, Kress W, Naylor SL et al. Myotoni dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science 2001; 293 : 864-867.
14. Sallinen R, Vihola A, Bachinski LL, Huoponen K, Haapasalo H, Hackman P et al. New methods of molecular diagnosis and demonstration of the (CCTG)n mutation in myotonic dystrophy type 2 (MD2). Neuromusc Disord 2004; 14 : 274-283.
15. Cardani R, Mancinelli E, Sansone V, Rotondo G, Meola G. Biomolecular identification of (CCTG)n mutation in myotonic dystrophy type 2 (DM2) by FISH on muscle biopsy. Eur H Histochem 2004; 48 : 437-442.
16. Day JW, Ranum LPW. RNA pathogenesis of the myotonic dystrophies. Neuromusc Disord 2005; 15 : 5-16.
17. Ranum LPW, Day JW. Myotonic dystrophy: RNA pathogenesis comes into focus. Am J Hum Genet 2004; 74 : 793-804.
18. Pongratz D, Schultz D, Koppenwallner C, Hubner G. Wertigkeit der Muskelbiopsie in der diagnostik der dystrophia myotonica (Curschmann-Steinert). Klin Wschr 1979; 57 : 215-224.
19. Falk M, Vojtěšková M, Lukáš Z, Kroupová I, Froster U. Simple procedure for automatic detection of unstable alleles in myotonic dystrophy and Huntington´s disease loci. Genetic testing 2006; 10 : 83-94.
20. Voháňka S, Bednařík J, Fajkusová L, Sedláčková J. Myotonická dystrofie typ 2: vzácné nebo časté onemocnění v České rerpublice? Česk Slov Neurol N 2005; 68/101 : 390-393.
21. Dubowitz V. Muscle biopsy. A practical approach. Bailliére Tindall 1985.
22. Carpenter S, Karpati G. Pathology of skeletal muscle. Oxford University Press 2001.
23. Schoser BG, Schneider-Gold C, Kress W, Goebel HH, Reilich P, Koch MC et al. Muscle pathology in 57 patients with myotonic dystrophy type 2. Muscle Nerve 2004; 29 : 275-281.
24. Vihola A, Bassez G, Meola G, Zhang S, Haapasalo H, Paetau A et al. Histopathological differences of myotonic dystrophy type 1 (DM1) and PROMM/DM2. Neurology 2003; 60 : 1854-1857.
25. Day JW, Ricker K, Jacobsen JF, Rasmussen LJ, Dick KA, Kress W et al. Myotonic dystrophy type 2: molecular diagnosis and clinical spectrum. Neurology 2003; 60 : 657-664.
26. Bundschu HD, Hauger W, Lang HD. Myotonische dystrophie Curschmann-Steinert. Dtsch Med Wschr 1975; 100 : 1337-1341.
27. Hübner G. Ringbinden der quergestreiften Muskulatur. Acta neuropath 1977; 38 : 27-32.
28. Kinoshita M, Takahashi R, Hasegawa T, Komori T, Nagasawa R, Hirose K et al. (CTG)n expansions in various tissues from a myotonic dystrophy patient. Muscle Nerve 1996; 19 : 240-242.
Štítky
Dětská neurologie Neurochirurgie NeurologieČlánek vyšel v časopise
Česká a slovenská neurologie a neurochirurgie

2007 Číslo 4
- Využití tiapridu v léčbě poruch chování u geriatrických pacientů
- Naděje budí časná diagnostika Parkinsonovy choroby založená na pachu kůže
- Kontinuální infuze tiapridu v léčbě hyperaktivního deliria – kazuistika
- Přibývající důkazy o přínosu léčebného konopí u pacientů s chronickou bolestí
- Nízká hladina vitamínu D v prenatálním období může zvýšit riziko roztroušené sklerózy
Nejčtenější v tomto čísle
- Cervikální dystonie
- Hladina D-dimerů u pacientů s akutní ischemickou cévní mozkovu příhodou
- Trombóza esovitého splavu – současný pohled na diagnostiku a léčbu
- Repetitivní transkraniální magnetická stimulace a chronický subjektivní nonvibrační tinnitus
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy