
Recruitment of the Major Vault Protein by InlK: A Strategy to Avoid Autophagy
L.
monocytogenes is a facultative intracellular bacterium responsible for listeriosis. It is able to invade, survive and replicate in phagocytic and non-phagocytic cells. The infectious process at the cellular level has been extensively studied and many virulence factors have been identified. Yet, the role of InlK, a member of the internalin family specific to L. monocytogenes, remains unknown. Here, we first show using deletion analysis and in vivo infection, that InlK is a bona fide virulence factor, poorly expressed in vitro and well expressed in vivo, and that it is anchored to the bacterial surface by sortase A. We then demonstrate by a yeast two hybrid screen using InlK as a bait, validated by pulldown experiments and immunofluorescence analysis that intracytosolic bacteria via an interaction with the protein InlK interact with the Major Vault Protein (MVP), the main component of cytoplasmic ribonucleoproteic particules named vaults. Although vaults have been implicated in several cellular processes, their role has remained elusive. Our analysis demonstrates that MVP recruitment disguises intracytosolic bacteria from autophagic recognition, leading to an increased survival rate of InlK over-expressing bacteria compared to InlK− bacteria. Together these results reveal that MVP is hijacked by L. monocytogenes in order to counteract the autophagy process, a finding that could have major implications in deciphering the cellular role of vault particles.
Published in the journal:
. PLoS Pathog 7(8): e32767. doi:10.1371/journal.ppat.1002168
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1002168
Summary
L.
monocytogenes is a facultative intracellular bacterium responsible for listeriosis. It is able to invade, survive and replicate in phagocytic and non-phagocytic cells. The infectious process at the cellular level has been extensively studied and many virulence factors have been identified. Yet, the role of InlK, a member of the internalin family specific to L. monocytogenes, remains unknown. Here, we first show using deletion analysis and in vivo infection, that InlK is a bona fide virulence factor, poorly expressed in vitro and well expressed in vivo, and that it is anchored to the bacterial surface by sortase A. We then demonstrate by a yeast two hybrid screen using InlK as a bait, validated by pulldown experiments and immunofluorescence analysis that intracytosolic bacteria via an interaction with the protein InlK interact with the Major Vault Protein (MVP), the main component of cytoplasmic ribonucleoproteic particules named vaults. Although vaults have been implicated in several cellular processes, their role has remained elusive. Our analysis demonstrates that MVP recruitment disguises intracytosolic bacteria from autophagic recognition, leading to an increased survival rate of InlK over-expressing bacteria compared to InlK− bacteria. Together these results reveal that MVP is hijacked by L. monocytogenes in order to counteract the autophagy process, a finding that could have major implications in deciphering the cellular role of vault particles.
Introduction
Listeria monocytogenes is a Gram-positive bacterium responsible for listeriosis, a severe food-borne human infection with an overall mortality rate of 30% [1]. L. monocytogenes has evolved efficient strategies to survive in the intestine and cross the intestinal, blood-brain and placental barriers [2], [3] leading to clinical features of the disease that include gastroenteritis, septicemia, central nervous system infections, and mother-to-child infections [4]. Inside the host, this facultative intracellular bacterium is able to invade phagocytic and non-phagocytic cells, replicate intracellularly, and spread directly from cell-to-cell, thereby escaping the immune response [3]. L. monocytogenes has thus emerged as a paradigm to study host-pathogen interactions and fundamental processes in cell biology [5]. For instance, the study of actin rearrangements upon entry and intracellular movements [6]–[9] is an example of how understanding a bacterial-induced process can yield insight into basic cellular processes. Namely, the listerial virulence factor ActA triggers the recruitment of Arp2/3 complex and Ena/VASP to mediate actin polymerization and propel the bacterium from one infected cell to another without exposure to the extra-cellular milieu [8], [10]. Interestingly, as shown recently ActA also disguises the bacteria from autophagic recognition within the cytosol as ActA - bacteria becomes rapidly ubiquitinated and targeted to autophagy [9], [11]. It is currently viewed that ubiquitin-associated bacteria recognized by the autophagy machinery are trapped by autophagosomal membrane for delivery into the lytic compartment where they undergo degradation by autolysosomes [11], [12]. Interestingly, a variety of studies had noticed that autophagic markers can accumulate around intracytosolic L. monocytogenes, unless bacteria were forming actin tails [13], [14]. Consequently, it has been hypothesized and shown that L. monocytogenes avoids ubiquitination and autophagic recognition by expressing ActA, and ActA mutants are efficiently targeted by autophagy [11]. While the role of ActA in autophagy is now established, the role that many other surface proteins play during Listeria infection remains fragmentary [15].
The vault particle is the largest cytoplasmic ribonucleoprotein complex known to date [16]. Originally identified as contaminants of clathrin-coated vesicles preparation, these complexes were named vault particles because of their barrel shaped morphology resembling the ceiling of cathedrals [17]. Mammalian vaults are composed of the highly conserved major vault protein (MVP) constituting more than 70% of the mass of the particle [16], [18], [19] which spontaneously forms vault particles without the need of other vault components [20]. The two other vault components are the telomerase associated protein (TEP-1) [21] and the vault poly(ADP)ribose polymerase (vPARP) [22]–[24]. Vault preparations have additionally been shown to contain several small untranslated RNAs [25], [26]. Vaults exist in thousands of copies per cell and are widely expressed in all eukaryotic organisms, from Dictyostelium discoideum to mammals, except plants, Saccharomyces cerevisiae, Caenorhabditis elegans and Drosophila melanogaster [27]. Diverse roles have been proposed for MVP and/or vaults [27], including roles in drug resistance [28], cellular differentiation [29], innate immunity [30], virus infections [31], signaling cascades [28], [32]–[35] and cell survival [33], [36]. However, the precise cellular function(s) of MVP and vaults remains poorly understood. In addition, the MVP−/− mice are viable, healthy and show no obvious abnormalities [37], [38].
The genome sequence of L. monocytogenes EGD-e has revealed the presence of 25 genes encoding proteins of the internalin family [39]. Proteins of this family, which are characterized by the presence of leucine-rich-repeats (LRRs), are mostly surface proteins [40]. Their binding to the bacterial surface is mediated by different anchoring domains, in particular the LPXTG motif which allows a sortase A mediated covalent attachment to the peptidoglycan [41]. The invasion protein, Internalin, is one such protein [42]. Comparative post-genomic studies have established that several members of the L. monocytogenes internalin family are absent in L. innocua, a closely related non-pathogenic species [40]. Lmo1290 is an internalin gene absent in L. innocua, herein referred to as inlK, which is expressed at very low levels in brain-heart-infusion medium [43], [44] and induced during infection [43].
In this study we investigated the role of InlK in the infectious process. We first explored the expression of InlK and the virulence phenotype of the inlK deletion mutant. We then searched for potential host partners of InlK and identified MVP. We demonstrated that the InlK/MVP interaction occurs in the cytosol of infected cells at the bacterial surface. Moreover, our results reveal that MVP recruitment protects L. monocytogenes from autophagic recognition, leading to an increase in bacterial survival in infected cells.
Results
L. monocytogenes inlK encodes a virulence factor
The gene lmo1290 ( = inlK) is 1797 bp long. It is located 331 bp downstream from gene lmo1289 which is followed by a transcriptional terminator. Lmo1290 is also followed by a transcriptional terminator upstream from the divergently transcribed oatA gene which encodes a peptidoglycan O–acetyltransferase (Figure 1A) [45]. The inlK gene is present in all 22 L. monocytogenes genomes sequenced to date and absent from the genomes of L. ivanovii and all non-pathogenic Listeria strains including L. innocua (Figure 1A), L. seeligeri, L. welschimeri and L. grayi, suggesting that InlK could be involved in Listeria virulence.
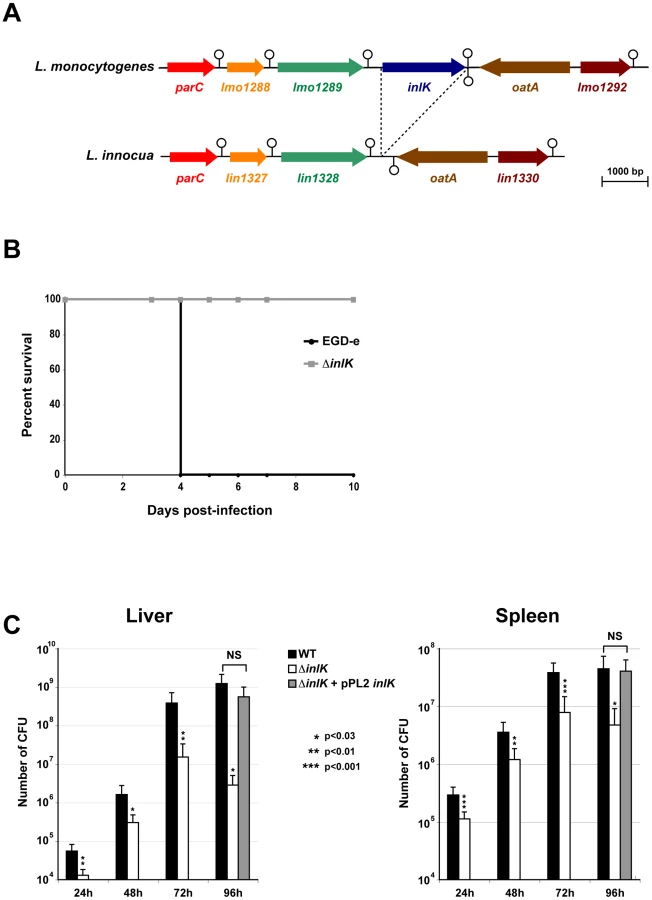
To assess the role of InlK in virulence, we generated an inlK-deletion mutant (ΔinlK) in the strain EGD-e. The ΔinlK mutant grew as rapidly as the wild-type (WT) in broth medium and infected cells (macrophages and epithelial HeLa cells) (data not shown). The LD50 of the ΔinlK mutant after intravenous (i.v.) injection in BALB/c mice was 2.2×104 CFU, compared with 1.7×103 CFU for the WT strain. Inactivation of inlK resulted in complete survival of animals infected intravenously with 104 bacteria (Figure 1B). In contrast, infection with the same number of WT bacteria led to 100% mortality. Moreover, the number of CFU recovered from spleens and livers of i.v. infected BALB/c mice after 24 h, 48 h, 72 h and 96 h of infection was significantly lower (∼1 Log10) for the mutant compared to the WT (Figure 1C), and virulence of the mutant was fully restored by complementation (Figure 1C). Together, these results establish a role for InlK in the virulence of L. monocytogenes.
InlK is expressed in vivo
InlK is a 598 amino acid LPXTG surface protein predicted to be anchored to the peptidoglycan by sortase A (Figure 2A). To address whether L. monocytogenes produces InlK in vitro, we first generated an antibody against a purified recombinant InlK protein (Figure S1A) and used it to detect the protein at the bacterial surface by immunofluorescence. In agreement with previous whole genome transcriptomic results that demonstrated a low expression level of inlK in vitro [44], bacteria grown in brain-heart infusion (BHI) medium were not stained by the InlK antibody (Figure 2B), suggesting that InlK protein was poorly expressed on the surface or not produced. We then showed that InlK was not detected in bacterial total extracts (Figure 2C), also in agreement with previous data indicating that InlK is not present in the cell wall proteome of L. monocytogenes EGD-e grown in BHI medium [46]. Moreover, consistent with the fact that the two major regulators of virulence genes, PrfA and sigmaB, were not required for basal inlK transcription [44], [47], the InlK protein level was also not detectable when bacteria were grown in charcoal supplemented medium or at low pH (data not shown).

To verify that the inlK open reading frame encoded a surface protein, inlK was expressed under the control of two constitutive promoters active in Listeria. We used either the promoter of the protease gene from Lactococcus lactis subsp. cremoris on the multicopy plasmid pPRT-inlK or the promoter PHyper after integration on the chromosome of the plasmid pADc-inlK [48], [49]. InlK antibodies efficiently labeled InlK on the surface of bacteria that constitutively expressed inlK (Figure 2B) and also detected the protein in bacterial total extracts (Figure 2C). This labeling was specific, as the InlK antibody did not label WT or inlK mutant bacteria grown in same conditions. Interestingly, when InlK was over-expressed by Listeria under the control of constitutive promoters, a polypeptide with a lower mass than expected was also detected by Western-blot (Figure 2C) indicating that the protein may be processed. Moreover, InlK was not detected by immunofluorescence at the surface of a ΔsrtA sortase mutant over-expressing inlK (Figure 2B), but was then detected in the supernatant of the culture medium (Figure 2D). Taken together these results established that, when inlK is expressed, the protein is anchored at the bacterial surface in a sortase A-dependent manner.
Recently, a whole genome transcriptomic analysis of L. monocytogenes during infection revealed that the gene inlK was better expressed in vivo compared to growth in BHI [43]. We thus investigated whether the InlK protein was indeed produced in vivo by testing for the presence of anti-InlK antibodies. Purified InlK was submitted to migration on polyacrylamide gel (Figure S1A) and blotted with two different rabbit anti-Listeria sera. As shown in Figure 2E, a rabbit anti-L. monocytogenes serum obtained after immunization with killed bacteria was not able to detect the purified InlK whereas the serum directed against live bacteria detected InlK. This signal was specific to InlK as the antibodies did not label bovine serum albumine (BSA) used at the same concentration.
To confirm in vivo inlK expression, we constructed an expression reporter vector in which the expression of the bioluminescent operon luxABCDE was under the control of inlK promoter (pPL2-PinlK-luxABCDE). This construct was integrated in the chromosome of WT L. monocytogenes EGD-e, and the resulting strain was used to infect cell lines or BALB/c mice [50]. As shown in Figure S1B, inlK was neither expressed in BHI growth medium (right panel) nor in cells infected with bacteria previously grown in BHI (left panel). Conversely, inlK was expressed in vivo in i.v. infected mice, 24 h post-infection (Figure S1C). This signal was specific to inlK expression as it did not superimpose on those obtained with the control strain of L. monocytogenes that contains a bioluminescent reporter of LLO promoter (pPL2-Phly-luxABCDE). Together, these results confirm that InlK is expressed in vivo.
InlK interacts with the Major Vault Protein
To identify InlK interaction partners in the eukaryotic cell, we used InlK as a bait in a large-scale yeast two-hybrid screen and identified the Major Vault Protein (MVP) as a prey with a very high interaction score (Table S1). To confirm this interaction we performed a bacterial pull down assay and showed that GST-MVP purified protein bound to InlK over-expressing bacteria, but not to WT bacteria (Figure 3A). This interaction was specific as (i) the WT strain (which expresses InlA, InlB and InlH) did not bind MVP (data not shown), (ii) the overexpression of InlJ (i.e. another internalin not expressed in BHI [49]) was not able to mediate bacterial binding to MVP, and (iii) InlK over-expressing bacteria were not able to bind another GST fusion protein, GST-ScarA. Finally, bacterial incubation with MVP-GFP transfected cells lysates confirmed the interaction between InlK and MVP (Figure 3B). This interaction occurred when InlK was either expressed on a multicopy plasmid, or integrated in the chromosome (Figure S2A).
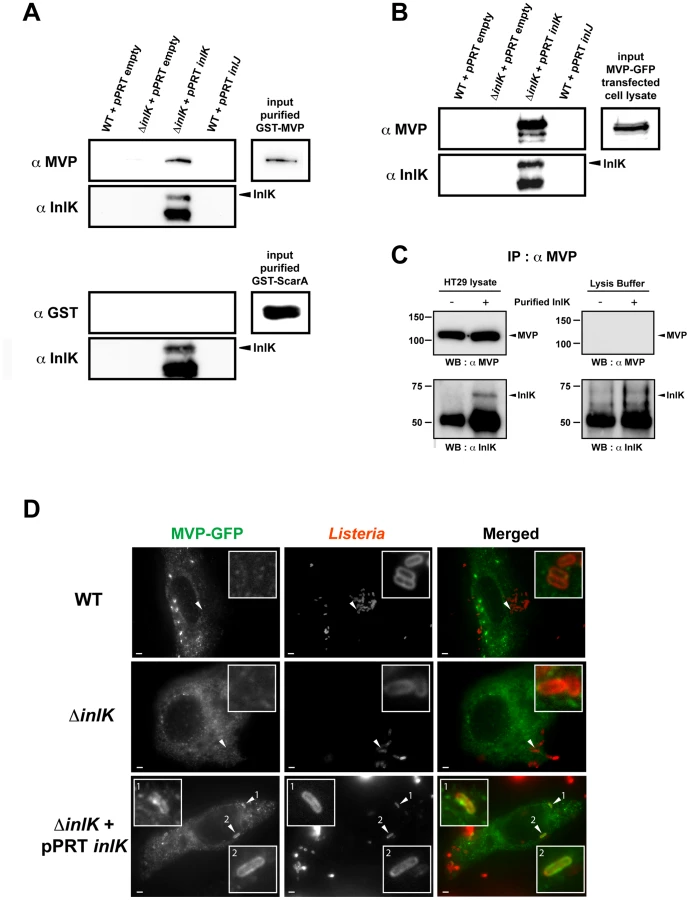
This interaction between purified InlK and endogenous MVP was confirmed by co-immunoprecipitation assays (Figure 3C). Indeed, when purified InlK was incubated with HT29 cell lysate, it interacted with endogenous MVP and the two partners co-immunoprecipitated, as shown using an anti-MVP antibody (Figure 3C). Similar results were obtained with stable HEK293-HTP-InlK cells that were engineered to express InlK in their cytosol, under the control of a tetracyclin inducible promoter (Figure S2B).
The InlK/MVP interaction occurs in the cytosol at the bacterial surface and does not depend on actin polymerization
In agreement with a specific interaction between InlK and MVP, we observed that InlK over-expressing bacteria co-localized with MVP in MVP-GFP transfected HeLa cells whereas the inlK mutant or wild type bacteria that do not express InlK in vitro did not co-localize with MVP (Figure 3D). As MVP has been mainly described as a cytoplasmic protein [51], [52] and InlK is targeted and anchored to the bacterial surface (Figure 2D), we hypothesized that the InlK/MVP interaction should occur in the cytosol of infected cells after lysis of the internalization vacuole. To test this hypothesis, we analyzed the localization of MVP recruiting bacteria. A differential immuno-staining protocol allowing extra - and intracellular Listeria to be distinguished showed that MVP was recruited to intracellular bacteria (Figure 4A). Irrespective of the time post-infection, ∼20% of InlK over-expressing bacteria were observed to recruit MVP [24.3%±3.0; 16.8±2.5; 18.2±1.6 and 18.5%±4.8 (mean ± SEM from n = 3 experiments) at 1 h, 2 h, 4 h and 8 h post-infection respectively] (Figure 4A, right panel). To determine whether MVP was recruited to intracellular bacteria before or after lysis of the internalization vacuole, we used a marker of early times points after vacuole escape, YFP-CBD, a YFP fused phage protein known to bind L. monocytogenes peptidoglycan as soon as the vacuole membrane begins to lyse (Figure S3A) [53]. Cells were co-transfected with MVP-Tomato and YFP-CBD, fixed 4 h post-infection and immuno-stained for actin. As expected, bacteria that polymerized actin were efficiently labeled with YFP-CBD (Figure 4B, Figure S3B inset 2), confirming that YFP-CBD efficiently labels intracytosolic Listeria. Moreover, all MVP-positive bacteria were also labeled with YFP-CBD (Figure 4B, Figure S3B) revealing that MVP was recruited by intracytosolic bacteria after lysis of the internalization vacuole.
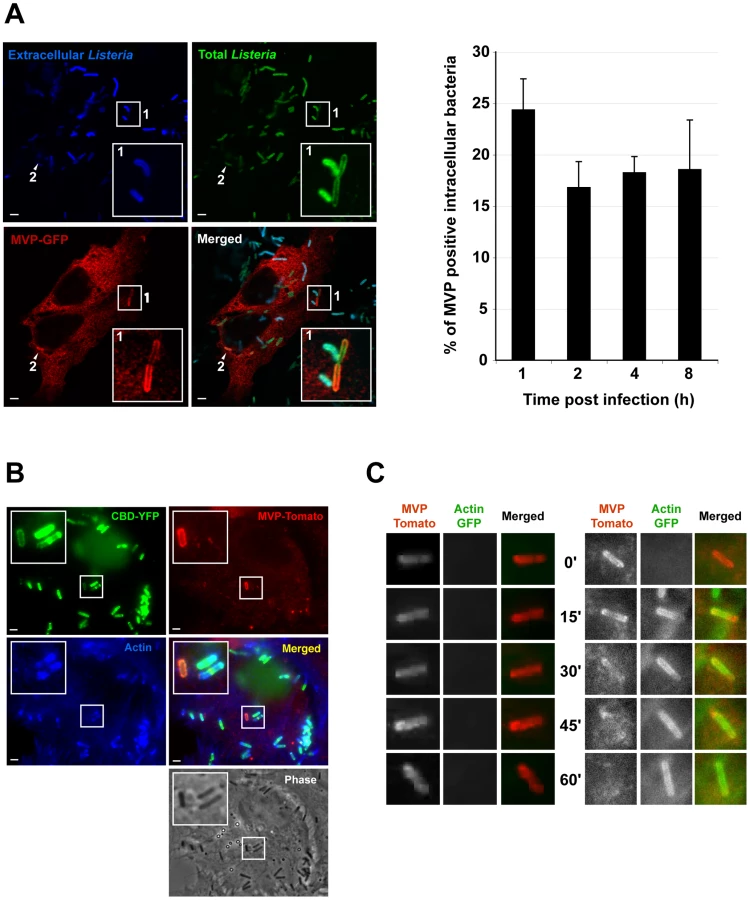
Interestingly, we did not observe the co-recruitment of MVP-GFP and endogenous actin to intracellular bacteria (Figure 4B, Figure S3B). Co-recruitment was also not observed in infected cells previously co-transfected with MVP-GFP and actin-CFP (Figure S3C). We therefore analysed the kinetics of MVP and actin recruitment by performing live-cell imaging. Cells were co-transfected with MVP-Tomato and actin-GFP, and infected with InlK expressing L. monocytogenes. Strikingly, MVP was recruited rapidly by InlK over-expressing bacteria and could then be replaced by actin (Figure 4C and Video S1), showing that MVP recruitment occurs before actin polymerization. We then verified that MVP recruitment occurred independently of actin polymerization using a actA mutant. Intracytosolic ΔactA over-expressing InlK were efficiently labeled with MVP (Figure S3D). This MVP recruitment was more efficient than for wild type bacteria. Indeed, the percentage of intracytosolic ΔactA over-expressing InlK having recruited MVP at 4 h post-infection was 88.3±12.7% (mean ± SEM from n = 3 experiments), compared to the 16.8±2.5% (mean ± SEM from n = 3 experiments) observed when using the InlK over-expressing strains that are able to polymerize actin via ActA. Together, these results suggested that ActA at least partially impairs MVP recruitment. As ActA protects bacteria from autophagy [11], these data also suggested that both InlK and ActA may protect bacteria from autophagy.
MVP recruitment protects Listeria from autophagy
To test if MVP recruitment could lead to autophagic escape, we used two well-established markers of autophagy, p62 (SQSTM1) and LC3 (Atg8) [54]. p62 has emerged as the prototypic adaptor involved in directing cytoplasmic substrates towards autophagic degradation [55]. p62 interacts with ubiquitinated subtrates via its ubiquitin-binding domain, and links them to the autophagosomal structural protein LC3. We infected MVP-transfected HeLa cells with InlK over-expressing bacteria for 4 h and after fixation, immuno-stained for endogenous p62 and actin. No co-localization could be observed between MVP and p62 (Figure 5A, Figure S4A) or MVP and LC3 (Figure 5B, Figure S4B). Interestingly, the vast majority of MVP-positive bacteria were completely devoid of anti-p62 labeling (95.1±2.0%; mean ± SEM from n = 3 experiments) but 4.9±2.0% (mean ± SEM from n = 3 experiments) were stained at one pole with MVP and at the other pole with p62. Similar results were obtained using GFP-LC3 (Figure 5, Figure S4B). As previously demonstrated [11], bacteria that had started to recruit actin were not labeled by p62 (Figure 5A) or GFP-LC3 (Figure 5B). Strikingly, when the MVP-positive bacteria that exhibited a recruitment of LC3 at one pole were examined by live-cell-imaging (Figure 5C), the membrane elongation leading to the autophagosome formation failed to occur (Video S2 and S3). Together these results indicate that bacteria which either recruit MVP or have started to polymerize actin evade autophagic recognition.

We thus studied autophagy marker recruitment by the actA mutant over-expressing InlK. In agreement with our previous observations (Figure S3D), InlK over-expressing ΔactA bacteria efficiently recruited MVP [88.6±12.8% (mean ± SEM from n = 3 experiments)] (Figure 5D and S4C). These MVP positive bacteria were neither surrounded by ubiquitinated proteins nor recognized by LC3 (Figure 5D and S4C). Furthermore, the level of LC3-II, the active form of LC3 that correlates with active autophagy [54], was significantly lower in cells infected with ΔactA+InlK as compared with ΔactA (1.82±0.14 fold) (Figure 5E). Together, these results show that in the absence of ActA, Listeria is able to evade autophagic recognition via MVP recruitment.
MVP-dependent escape from autophagy leads to increased Listeria survival
Autophagy is recognized as a cell-autonomous innate defense mechanism that may control growth of intracellular microbes [56]. We thus tested if MVP-mediated autophagy escape leads to increased bacterial survival. As macrophages are among the cells which express the highest levels of MVP [29], [57], [58], the intracellular survival of WT, WT+InlK, ΔactA and ΔactA+InlK was analysed in RAW 264.7 macrophages (Figure 6A). These four strains (WT, WT+InlK, ΔactA and ΔactA+InlK) were first verified to grow identically in culture medium (data not shown). As previously described by Yoshikawa et al. [11], the intracellular survival rate of ΔactA bacteria at 4 h post-infection was significantly lower than that of WT bacteria (Figure 6B). Strikingly, the expression of InlK by the ΔactA strain restored the intracellular survival rate to the level of WT bacteria (Figure 6B), indicating that InlK could functionally replace ActA in its role in autophagy escape. Infection of MVP-transfected epithelial cells with ΔactA and ΔactA+inlK led to similar results (Figure 6C and 6D).

The intracellular survival of ΔactA and ΔactA+InlK was then analysed in RAW 264.7 macrophages treated with control or MVP siRNA (Figure 6E). As previously observed (Figure 6B), the ΔactA+InlK strain replicated better than the ΔactA strain in control cells (Figure 6E). Strikingly, in MVP-depleted cells, the ΔactA+InlK strain did not replicate faster than the ΔactA strain (Figure 6F), confirming the role of InlK/MVP interaction in survival rate. Taken together, these data show that the specific recruitment of MVP to the bacterial surface via InlK leads to a better survival of L. monocytogenes.
Discussion
L. monocytogenes has emerged as a paradigm to study host-pathogen interactions and fundamental processes in cell biology [5], [59]. However the role of the many proteins expressed on the bacterial surface during Listeria infection remains fragmentary [15]. In this study we report that InlK, a L. monocytogenes surface protein of the internalin family, plays a critical role in Listeria virulence. We show that InlK is anchored to the listerial surface through its LPXTG peptidoglycan anchoring signal by sortase A and is produced during in vivo infection, whereas it cannot be detected on bacteria grown in BHI medium [44] or within the cytosol of tissue-cultured cells. This in vivo specific expression profile was previously described for other virulence factors of L. monocytogenes, e.g. the internalin InlJ, that behaves as an adhesin [49] and recently LntA, a secreted bacterial protein involved in chromatin remodeling and type III interferon response [60]. Furthermore, our results confirm and extend our recently published transcriptomic analysis of L. monocytogenes [43] which identified inlK as a gene highly activated during in vivo infection and that may play a role in the infectious process. Together, our results demonstrate that InlK is a so far undescribed virulence factor of L. monocytogenes.
To enter, survive and spread from cell-to-cell, L. monocytogenes has been shown to interact with several host partners. We revealed here that MVP is a specific cellular interactor of InlK. The highly conserved MVP protein constitutes more than 70% of the mass of the largest cytoplasmic ribonucleoprotein (RNP) complex known, i.e. vault particles [16], [18], [19]. Since its first description in 1986 [61], several putative functions have been attributed to this RNP complex. Data that link the vault complex to various functions have suggested roles in multidrug resistance [28], [62], transport [63], signaling [28], [32]–[35], apoptosis resistance [33], [36] or innate immunity [30]. However, no compelling evidence for a cellular role was reported unequivocally and MVP was mainly considered as a scaffold protein. Nevertheless, vaults were previously found to be implicated in g-herpesvirus (Epstein-Barr and Kaposi's sarcoma virus) [31], [64] and Pseudomonas aeruginosa infectious processes [30]. During Epstein-Barr or Kaposi's sarcoma virus infection, the expression of vault RNAs (vRNAs) was shown to be specifically up-regulated in human lymphocytes [31], [64]. However, the function of this overexpression was not assessed. In addition, not only vRNA but also MVP was reported to be upregulated during viral infection by human T-cell lymphotropic virus type I (HTLV-I) infection [65]. In the case of bacteria, MVP was implicated in host resistance to P. aeruginosa lung infection [30]. Indeed, a rapid recruitment of MVP to lipid rafts was observed when human lung epithelial cells were infected with P. aeruginosa. This recruitment was dependent on bacterial binding to the cystic fibrosis transmembrane conductance regulator CFTR. However, no evidence of direct binding between MVP and bacteria was observed. Our results provide the first report of a direct interaction between a microbial protein and a component of the vault particles. Indeed, we demonstrated that InlK over-expressing L. monocytogenes were able to directly bind MVP. In agreement with previous observations that MVP/vaults are predominantly (>90%) localized in the cytoplasm [28], [51], [52], we established that the InlK/MVP interaction occurs in the cytosol of infected cells, after the disruption of the internalization vacuole, and independently of actin polymerization.
As with a variety of intracellular microbes, intracytosolic L. monocytogenes are recognized by autophagy, a cell-autonomous effector mechanism of innate immunity that protects the cytosol against bacterial invasion [66]. Perrin et al. first demonstrated that cytosolic L. monocytogenes occasionally colocalized with ubiquitin in infected cells, and this association was more frequent in case of the ΔactA strain [13], [14]. More recently, Yoshikawa et al. demonstrated that the recruitment of VASP, Arp2/3 complex and actin via ActA protect bacteria from ubiquitination and autophagic recognition [11]. Here we reveal that L. monocytogenes has a second strategy to escape autophagy in the absence of ActA (Figure 7). Indeed, no significant difference could be observed between the intracellular survival rate of WT and WT+InlK bacteria in infected RAW 267.4 macrophages (Figure 6B), suggesting that when ActA is expressed it is sufficient for Listeria to escape from autophagy. In contrast, in absence of ActA, InlK protects against autophagy. Together, our results show that the bacteria are able, via InlK, to decorate their surface with MVP in order to escape from autophagy (Figure 7). It will be thus of the highest importance to decipher in which cells InlK is expressed in vivo and when the InlK/MVP interaction takes place during infection. These data will be critical to unravel the role of InlK in the pathophysiology of Listeria infection. It will also be of great interest to further study the link between actin polymerization, MVP, autophagy, and pathogen dissemination.

Materials and Methods
Bacterial strains, growth conditions and reagents
Listeria strains (Table S2) were grown in brain-heart infusion (BHI) medium (Difco; BD) and Escherichia coli were grown in Luria-Bertani Medium (LB) medium (Difco; BD). When required, chloramphenicol and erythromycin were used at final concentrations of 7 µg/ml and 5 µg/ml respectively for L. monocytogenes and kanamycin, erythromycin and chloramphenicol were used at final concentration of 50 µg/ml, 150 µg/ml and 35 µg/ml, respectively for E. coli.
Generation of EGD-e ΔinlK mutant strain and inlK over-expressing strains
Generation of ΔinlK mutant strain
Two ∼700 pb fragments flanking inlK gene were PCR amplified from EGD-e chromosomal DNA. The primers used for the inlK 5′ flanking fragment were A (5′-TTG GAT CCG CTG TAG ATT TCA CAA AAG-3′) and B (5′-TAA CAC GCG TAA GTC ATT ATC CTC TCC ACT C-3′), and the primers used for the 3′ fragment were C (5′-GAA AAC GCG TAA AAA ACT ATC CGC CCA C-3′) and D (5′-TTG GTC CAT GGT TAA GCA TTG CTG GTG-3′). After restriction of the amplified 5′ and 3′ fragments with BamHI and MluI, and MluI and NcoI respectively, 5′ and 3′ fragments were coligated in the thermosensitive plasmid pMAD [67] digested by BamHI and NcoI, yielding the pMAD-ΔinlK plasmid. The sequence was verified by sequencing. This plasmid was electroporated into L. monocytogenes EGD-e. Independent colonies were used for allelic exchange in L. monocytogenes wild-type EGD-e, which was performed as previously described [49], generating a ΔinlK isogenic deletion mutant (Table S2). Deletion of the entire inlK gene was confirmed by PCR amplification and sequencing.
Generation of InlK over-expressing strains
To express InlK in L. monocytogenes the pPRT - and pADc - derivative plasmids were constructed as described below. In the pPRT-inlK plasmid, inlK was expressed under the control of the promoter region of the protease gene from Lactococcus lactis subsp. cremoris, which is active in Listeria [49]. This is a multicopy plasmid which expresses an erythromycin resistance gene used for cloning selection.
The pADc-inlK plasmid generated as previously described by Balestrino et al [48] was derived from the integrative pPL2 plasmid, which inserts in the Listeria chromosome at the tRNAArg-attBB site, thereby avoiding the requirement for antibiotic pressure to maintain the plasmid and preventing heterogeneity of InlK expression due to variation in the plasmid copy number.
Cell lines and infection
HeLa cells (human epithelial cervix carcinoma; ATCC CCL2), Jeg3 cells (human placenta choriocarcinoma, ATCC HTB-36) and RAW 267.4 murine macrophages (ATCC TIB-71) were grown as recommended by ATCC (Manassas, VA). Cells were infected with exponentially growing Listeria strains such that the multiplicity of infection was 50 bacteria per cell (MOI50) for epithelial cell lines and MOI10 for RAW 267.4 macrophages. After 1 h of infection for epithelial cell lines and 15 min for RAW 267.4 macrophages, cells were washed and treated with 25 µg/ml of gentamicin. Incubation times were as indicated. All experiments were performed in serum-free medium. Then, cells were washed three times with PBS 1X (Difco, BD) and lysed by adding 500 µl of 0.1% Triton X-100. The number of viable bacteria released from cells was assessed by plating serial dilutions of bacteria on agar plates.
siRNA experiments
2.5×105 RAW 267.4 macrophages per well were plated in 12 wells plates and incubated at 37°C in 10% CO2. 24 h after plating, cells were treated with 80 nM of either a pool of anti-mouse MVP siRNA (ON-TARGETplus SMART pool L-049201-01-005 Mouse MVP, Dharmacon) or control siRNA (ON-TARGETplus Non-targeting siRNA:1, Dharmacon), using Lipofectamine 2000 (Invitrogen) as recommended by the manufacturer. The following day, the medium was changed and the cells were incubated in complete medium for another 24 hours. Infections were performed as above-mentioned and the efficiency of siRNA knock-down was assessed by performing Western-blot on total cell lysates in each experiment (Figure 6E).
InlK purification
The inlK coding sequence (aa 27–568) was amplified using primers lmo1290-Fw: 5′ - GAG TCG GAT CCG GTA TTT GCT GCA GAG CAAC C-3′ and lmo1290-Rev: 5′ - GAG TCG TCG ACA GCC TCT TTA CTT GGT TCT G-3′. The PCR product was purified and ligated with pET28b (Novagen) plasmid after double digestion with BamHI and SalI enzymes. The ligation product was electroporated in E.coli XL-1 Blue and positive clones were selected on 50 µg/ml supplemented BHI and sequenced (BUG 2812). For purification, E.coli BL21(DE3) (Invitrogen) were chemically transformed with the purified His6-InlK-His6 expressing pET28b plasmid. Bacteria were grown in 50 µg/ml supplemented LB until OD600 0.6 and IPTG was added at the final concentration 1 mM for 4 additional hours. Bacteria were lysed using a French press and the supernatant was recovered. His6-InlK-His6 purification was performed using TALON His-Tag Purification Resins (Clontech). Increased concentration of imidazole (0–200 mM) in Tris-HCl 20 mM, NaCl 0.5 M (pH = 8) were used for purification and elution of InlK. The purified protein was dialysed against Tris-HCl 20 mM, NaCl 0.5 M (pH = 8) buffer and concentrated using AmiconUltra centrifugal filter (Millipore).
Antibodies and reagents
The primary antibodies used in this study were anti-actin mouse monoclonal (mAb) (AC-15; Sigma-Aldrich), anti-LRP mAb (MVP was also named LRP for Lung resistance protein) (Ref:610512; BD Biosciences), anti-p62 mAb (Ref:610832, BD Biosciences), anti-ubiquitin mAb (FK-2, Affiniti), anti-Atg8 (LC3) rabbit polyclonal (pAb) (Novus Biologicals, Ref:NB100-2331), anti-killed L. monocytogenes pAb (R11), anti-live L. monocytogenes pAb (R177). Monoclonal antipeptide antibody that recognizes ActA (A16) was obtained as previously described [68]. An anti-InlK polyclonal rabbit antibody (R190) was generated against His6-InlK-His6 recombinant protein (aa 27–568) deleted from its signal peptide and peptidoglycan-anchoring sequence and affinity-purified on a ECH Sepharose 4B column (GE Healthcare) coupled with 2.5 mg His6-InlK-His6 recombinant protein expressed from pET28b-InlK plasmid as described above. The polyclonal pre-immune serum of R190 (pre-immune R190) was recovered from rabbits before they were s.c injected with purified InlK. The secondary antibodies were Alexa Fluor 488 - and 546-conjugated goat anti-mouse and anti-rabbit, respectively (Molecular Probes) and HRP-conjugated goat anti-mouse and goat anti-rabbit (AbCys). Alexa fluor 647-conjugated phalloidin was purchased from Molecular Probes; DAPI from Roche Applied Sciences; and the Amersham ECL Plus kit from Ge Healthcare.
The GST-tagged purified recombinant MVP protein was purchased from Abnova (Ref:H00009961-P01).
Immunofluorescence microscopy
Cells were fixed in 4% paraformaldehyde (PFA) in 1X PBS for 20 minutes at room temperature. Cells were then rinsed in 1X PBS before incubation in blocking solution (0.5% BSA, 50 mM NH4Cl in PBS, pH 7.4) containing 0.05% saponin. Cells were then incubated with the primary antibodies diluted in the blocking solution for 30 min at room temperature, rinsed 5 times in 1X PBS and further incubated for 30 minutes with the secondary antibodies diluted in blocking solution. Where needed, fluorescent phalloidin was added with the secondary antibodies to label actin. Cells were then rinsed 5 times in 1X PBS and mounted on glass coverslip using Fluoromount mounting medium (EMS, PA). The differential immuno-staining between extra - and intracellular Listeria was previously described [69]. Samples were analysed either with a Zeiss Axiovert 135 epifluorescence microscope connected to a CCD camera or with a Zeiss LSM510 confocal microscope (Carl Zeiss, Germany). Images were acquired with a 100X oil immersion objective and images were processed using MetaMorph (Universal Imaging Corp.).
Plasmids
The MVP-GFP plasmid that encodes EGFP fused to MVP C-terminus has been previously described [70]. To construct MVP-CFP (BUG 2908) and MVP-Tomato (BUG 2909), the MVP coding sequence was isolated from the MVP-GFP (BUG 2907) plasmid by double enzyme digestion (HindIII and BamHI) and ligated in pECFP-N1 and ptdTomato-N1. Briefly, ptdTomato-N1 was constructed by replacing EGFP, from pEGFP-N1 vector (Invitrogen), by tdTomato, from ptdTomato-LCa vector (BUG 2420) [69]. Plasmid encoding CBD-YFP (BUG 2305) [53], actin-GFP (BUG 2421), actin-CFP (BUG 2155) and GFP-LC3 (BUG 3046) [71] were described elsewhere. Cells transfections were performed with FuGENE HD (Roche) as recommended by the manufacturer.
Bacterial pull down assay
To test the binding of bacteria to GST-MVP, L. monocytogenes strains were grown in BHI to an OD600 of 1.0, and 1 ml of each culture was taken for each reaction. Bacterial cells were washed twice in buffer with 20 mM HEPES pH 7.5, 150 mM NaCl, resuspended in 1 ml of binding buffer (20 mM HEPES pH 7.5, 150 mM NaCl, 1 mM CaCl2, 1 mM MgCl2, and 0.2% BSA), and incubated at room temperature on a rotating wheel for 30 min. GST-MVP recombinant protein was added to a final concentration of 1 µg/ml and samples were incubated with rotation for an additional hour. Samples were centrifuged and pellets were washed three times in 20 mM HEPES pH 7.5, 300 mM NaCl, 0.05% Tween 20 and three times in buffer lacking Tween 20. The final bacterial pellets were resuspended in 20 microliters of 2X sample buffer, boiled for 10 min, and stored at −20°C before migration on 8% SDS/PAGE gels and Western blotting.
To analyse the binding of bacteria to transfected MVP-GFP HeLa cell lysates, HeLa cells were grown on 75 cm2 flask, then transfected with MVP-GFP plasmid 24 h prior to the experiment. Cells were lysed at 4°C for 10 min in a 1 ml of lysis buffer (Tris-HCl 50 mM, pH 7.5, NaCl 150 mM, EDTA 2 mM, NP40 1%, AEBSF 1 mM, Na3VO4 3 mM). Cells were scraped and the lysates were incubated with rotation for additional 15 min. Lysates were cleared by 15 min centrifugation at 13 000 g at 4°C. L. monocytogenes strains were grown in BHI to an OD600 of 1.0, and 1 ml of each culture was taken for each reaction. Bacterial cells were washed twice in lysis buffer and were resuspended in cell lysate supernatants, mainly corresponding to cytoplasmic fraction, and incubated at room temperature on a rotating wheel for 1 hour. Samples were centrifuged to pellet bacteria, and washed five times in washing buffer (Tris-HCl 50 mM, pH 7.5, NaCl 150 mM, EDTA 2 mM, NP40 0.2%, AEBSF 1 mM, Na3VO4 3 mM). The final bacterial pellets were treated as described above.
In each experiments bacterial inoculi were counted to ensure that an equal number of bacteria were used for pull-down assay.
Co-immunoprecipitation
HT29 (colon epithelial cells, ATCC HTB-38) cells were cultured to confluence in 75 cm3 flask. Cells were lysed for 10 min at 4°C in 1 ml of lysis buffer (Tris-HCl 50 mM, pH 7.5, NaCl 150 mM, EDTA 2 mM, NP40 1%, AEBSF 1 mM, Na3VO4 3 mM). Cells were scraped and the lysates were incubated with rotation for additional 15 min. Lysates were cleared by 15 min centrifugation at 13 000 g at 4°C and were incubated overnight at 4°C with 20 µg of purified InlK. Then, co-immunoprecipitation was performed using 1 µg of anti-MVP antibody. After five washes in lysis buffer, samples were resuspended in 20 microliters of 2X sample buffer, boiled for 10 min, and stored at −20°C before migration on 8% SDS/PAGE gels and Western blotting.
Stable HEK293-HTP Blue and HEK293-HTP InlK were constructed as previously described [60]. When necessary cells were treated with doxycyclin 24 h prior the assay. Co-immunoprecipitations were performed using anti-MVP antibody as described above.
Yeast two-hybrid screening
The InlK encoding sequence (aa 27–568) was amplified by PCR from EGD-e and cloned into pB29 (N-bait-LexA-C fusion) plasmid. Randomly primed cDNA from human placenta poly(A) were constructed into a prey plasmid derived from pBTM116. The two-hybrid screen was performed by Hybrigenics (www.hybrigenics.com). The DNA inserts of the positive clones were sequenced to identify the corresponding gene in GenBank database using a fully automated procedure. Results of the yeast two-hybrid screening are recapitulated in Table S1.
Immunoblotting
Cells were seeded into 6 well plates and incubated 24 h before infection. Infections were done as described above. At the indicated times cells were lysed and lysates were analysed by Western blot. The immunoblots shown are representative of three independent experiments. Analysis of immunoblots was performed using G:Box Gel documentation system (Syngene).
Murine infection experiments
All experiments were performed according to Institut Pasteur guidelines for laboratory animal husbandry. For determination of LD50, groups of 8-week-old BALB/c female mice (Charles River Laboratory) were injected i.v with increasing concentrations of L. monocytogenes WT strain or DinlK mutant. LD50 were determined by the probit method at 10 days after inoculation.
Bacterial growth in mice was determined by injecting 6 - to 8-week-old female BALB/c mice intravenously with a sublethal bacterial inoculum, 104 CFU. After 24, 48 72 and 96 h of infection, liver and spleen were dissected in sterile conditions and the numbers of CFU were determined by plating serial dilutions of organ (liver and spleen) homogenates on BHI agar medium.
Ethics statement
All animals were handled in strict accordance with good animal practice as defined by the relevant national and local animal welfare bodies, and all animal work was approved by the Institut Pasteur animal experimentation committee which comply with European regulations (directive 2010/63/EU of the European parliament and of the council of 22 September 2010 on the protection of animals used for scientific purposes).
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. DortetLVeiga-ChaconECossartP 2009 Listeria monocytogenes. SchaechterM Encyclopedia of Microbiology 3rd ed. Oxford Elsevier 182 198
2. DissonOGrayoSHuilletENikitasGLanga-VivesF 2008 Conjugated action of two species-specific invasion proteins for fetoplacental listeriosis. Nature 455 1114 1118
3. HamonMBierneHCossartP 2006 Listeria monocytogenes: a multifaceted model. Nat Rev Microbiol 4 423 434
4. LecuitM 2007 Human listeriosis and animal models. Microbes Infect 9 1216 1225
5. CossartPSansonettiPJ 2004 Bacterial invasion: the paradigms of enteroinvasive pathogens. Science 304 242 248
6. BierneHGouinERouxPCaroniPYinHL 2001 A role for cofilin and LIM kinase in Listeria-induced phagocytosis. J Cell Biol 155 101 112
7. BonazziMVeigaEPizarro-CerdaJCossartP 2008 Successive post-translational modifications of E-cadherin are required for InlA-mediated internalization of Listeria monocytogenes. Cell Microbiol 10 2208 2222
8. KocksCGouinETabouretMBerchePOhayonH 1992 L. monocytogenes-induced actin assembly requires the actA gene product, a surface protein. Cell 68 521 531
9. MostowySCossartP 2011 Autophagy and the cytoskeleton: New links revealed by intracellular pathogens. Autophagy 7 780 782
10. CossartP 2000 Actin-based motility of pathogens: the Arp2/3 complex is a central player. Cell Microbiol 2 195 205
11. YoshikawaYOgawaMHainTYoshidaMFukumatsuM 2009 Listeria monocytogenes ActA-mediated escape from autophagic recognition. Nat Cell Biol 11 1233 1240
12. ThurstonTLRyzhakovGBloorSvon MuhlinenNRandowF 2009 The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat Immunol 10 1215 1221
13. BirminghamCLCanadienVGouinETroyEBYoshimoriT 2007 Listeria monocytogenes evades killing by autophagy during colonization of host cells. Autophagy 3 442 451
14. PerrinAJJiangXBirminghamCLSoNSBrumellJH 2004 Recognition of bacteria in the cytosol of Mammalian cells by the ubiquitin system. Curr Biol 14 806 811
15. BierneHCossartP 2007 Listeria monocytogenes surface proteins: from genome predictions to function. Microbiol Mol Biol Rev 71 377 397
16. TanakaHKatoKYamashitaESumizawaTZhouY 2009 The structure of rat liver vault at 3.5 angstrom resolution. Science 323 384 388
17. RomeLKedershaNChuganiD 1991 Unlocking vaults: organelles in search of a function. Trends Cell Biol 1 47 50
18. AndersonDHKickhoeferVASieversSARomeLHEisenbergD 2007 Draft crystal structure of the vault shell at 9-A resolution. PLoS Biol 5 e318
19. MikyasYMakabiMRaval-FernandesSHarringtonLKickhoeferVA 2004 Cryoelectron microscopy imaging of recombinant and tissue derived vaults: localization of the MVP N termini and VPARP. J Mol Biol 344 91 105
20. StephenAGRaval-FernandesSHuynhTTorresMKickhoeferVA 2001 Assembly of vault-like particles in insect cells expressing only the major vault protein. J Biol Chem 276 23217 23220
21. KickhoeferVALiuYKongLBSnowBEStewartPL 2001 The Telomerase/vault-associated protein TEP1 is required for vault RNA stability and its association with the vault particle. J Cell Biol 152 157 164
22. KickhoeferVASivaACKedershaNLInmanEMRulandC 1999 The 193-kD vault protein, VPARP, is a novel poly(ADP-ribose) polymerase. J Cell Biol 146 917 928
23. LiuYSnowBEKickhoeferVAErdmannNZhouW 2004 Vault poly(ADP-ribose) polymerase is associated with mammalian telomerase and is dispensable for telomerase function and vault structure in vivo. Mol Cell Biol 24 5314 5323
24. Raval-FernandesSKickhoeferVAKitchenCRomeLH 2005 Increased susceptibility of vault poly(ADP-ribose) polymerase-deficient mice to carcinogen-induced tumorigenesis. Cancer Res 65 8846 8852
25. StadlerPFChenJJHackermullerJHoffmannSHornF 2009 Evolution of vault RNAs. Mol Biol Evol 26 1975 1991
26. van ZonAMossinkMHSchoesterMSchefferGLScheperRJ 2001 Multiple human vault RNAs. Expression and association with the vault complex. J Biol Chem 276 37715 37721
27. BergerWSteinerEGruschMElblingLMickscheM 2009 Vaults and the major vault protein: novel roles in signal pathway regulation and immunity. Cell Mol Life Sci 66 43 61
28. SteinerEHolzmannKElblingLMickscheMBergerW 2006 Cellular functions of vaults and their involvement in multidrug resistance. Curr Drug Targets 7 923 934
29. SchroeijersABReursAWSchefferGLStamAGde JongMC 2002 Up-regulation of drug resistance-related vaults during dendritic cell development. J Immunol 168 1572 1578
30. KowalskiMPDubouix-BourandyABajmocziMGolanDEZaidiT 2007 Host resistance to lung infection mediated by major vault protein in epithelial cells. Science 317 130 132
31. MrazekJKreutmayerSBGrasserFAPolacekNHuttenhoferA 2007 Subtractive hybridization identifies novel differentially expressed ncRNA species in EBV-infected human B cells. Nucleic Acids Res 35 e73
32. KimELeeSMianMFYunSUSongM 2006 Crosstalk between Src and major vault protein in epidermal growth factor-dependent cell signalling. FEBS J 273 793 804
33. KolliSZitoCIMossinkMHWiemerEABennettAM 2004 The major vault protein is a novel substrate for the tyrosine phosphatase SHP-2 and scaffold protein in epidermal growth factor signaling. J Biol Chem 279 29374 29385
34. LiangPWanYYanYWangYLuoN 2010 MVP interacts with YPEL4 and inhibits YPEL4-mediated activities of the ERK signal pathway. Biochem Cell Biol 88 445 450
35. YuZFotouhi-ArdakaniNWuLMaouiMWangS 2002 PTEN associates with the vault particles in HeLa cells. J Biol Chem 277 40247 40252
36. RyuSJParkSC 2009 Targeting major vault protein in senescence-associated apoptosis resistance. Expert Opin Ther Targets 13 479 484
37. MossinkMHde GrootJvan ZonAFranzel-LuitenESchoesterM 2003 Unimpaired dendritic cell functions in MVP/LRP knockout mice. Immunology 110 58 65
38. MossinkMHvan ZonAFranzel-LuitenESchoesterMKickhoeferVA 2002 Disruption of the murine major vault protein (MVP/LRP) gene does not induce hypersensitivity to cytostatics. Cancer Res 62 7298 7304
39. GlaserPFrangeulLBuchrieserCRusniokCAmendA 2001 Comparative genomics of Listeria species. Science 294 849 852
40. BierneHSabetCPersonnicNCossartP 2007 Internalins: a complex family of leucine-rich repeat-containing proteins in Listeria monocytogenes. Microbes Infect 9 1156 1166
41. BierneHMazmanianSKTrostMPucciarelliMGLiuG 2002 Inactivation of the srtA gene in Listeria monocytogenes inhibits anchoring of surface proteins and affects virulence. Mol Microbiol 43 869 881
42. BonazziMCossartP 2006 Bacterial entry into cells: a role for the endocytic machinery. FEBS Lett 580 2962 2967
43. CamejoABuchrieserCCouveECarvalhoFReisO 2009 In vivo transcriptional profiling of Listeria monocytogenes and mutagenesis identify new virulence factors involved in infection. PLoS Pathog 5 e1000449
44. Toledo-AranaADussurgetONikitasGSestoNGuet-RevilletH 2009 The Listeria transcriptional landscape from saprophytism to virulence. Nature 459 950 956
45. AubryCGoulardCNahoriMADecalfJBonecaIG 2011 OatA, a peptidoglycan O-acetyltransferase involved in Listeria monocytogenes immune escape and critical for virulence. J Infect Dis Accepted
46. PucciarelliMGCalvoESabetCBierneHCossartP 2005 Identification of substrates of the Listeria monocytogenes sortases A and B by a non-gel proteomic analysis. Proteomics 5 4808 4817
47. McGannPRaengpradubSIvanekRWiedmannMBoorKJ 2008 Differential regulation of Listeria monocytogenes internalin and internalin-like genes by sigmaB and PrfA as revealed by subgenomic microarray analyses. Foodborne Pathog Dis 5 417 435
48. BalestrinoDHamonMADortetLNahoriMAPizarro-CerdaJ 2010 Single-cell techniques using chromosomally tagged fluorescent bacteria to study Listeria monocytogenes infection processes. Appl Environ Microbiol 76 3625 3636
49. SabetCToledo-AranaAPersonnicNLecuitMDubracS 2008 The Listeria monocytogenes virulence factor InlJ is specifically expressed in vivo and behaves as an adhesin. Infect Immun 76 1368 1378
50. BronPAMonkIRCorrSCHillCGahanCG 2006 Novel luciferase reporter system for in vitro and organ-specific monitoring of differential gene expression in Listeria monocytogenes. Appl Environ Microbiol 72 2876 2884
51. KickhoeferVAVasuSKRomeLH 1996 Vaults are the answer, what is the question? Trends Cell Biol 6 174 178
52. van ZonAMossinkMHScheperRJSonneveldPWiemerEA 2003 The vault complex. Cell Mol Life Sci 60 1828 1837
53. HenryRShaughnessyLLoessnerMJAlberti-SeguiCHigginsDE 2006 Cytolysin-dependent delay of vacuole maturation in macrophages infected with Listeria monocytogenes. Cell Microbiol 8 107 119
54. MizushimaNYoshimoriTLevineB 2010 Methods in mammalian autophagy research. Cell 140 313 326
55. PankivSClausenTHLamarkTBrechABruunJA 2007 p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 282 24131 24145
56. LevineBMizushimaNVirginHW 2011 Autophagy in immunity and inflammation. Nature 469 323 335
57. IzquierdoMASchefferGLFlensMJGiacconeGBroxtermanHJ 1996 Broad distribution of the multidrug resistance-related vault lung resistance protein in normal human tissues and tumors. Am J Pathol 148 877 887
58. SunnaramBLGandemerVSebillotMGrandgirardNAmiotL 2003 LRP overexpression in monocytic lineage. Leuk Res 27 755 759
59. CossartPToledo-AranaA 2008 Listeria monocytogenes, a unique model in infection biology: an overview. Microbes Infect 10 1041 1050
60. LebretonALakisicGJobVFritschLThamTN 2011 A Bacterial Protein Targets the BAHD1 Chromatin Complex to Stimulate Type III Interferon Response. Science 331 1319 1321
61. KedershaNLRomeLH 1986 Isolation and characterization of a novel ribonucleoprotein particle: large structures contain a single species of small RNA. J Cell Biol 103 699 709
62. PerssonHKvistAVallon-ChristerssonJMedstrandPBorgA 2009 The non-coding RNA of the multidrug resistance-linked vault particle encodes multiple regulatory small RNAs. Nat Cell Biol 11 1268 1271
63. SuprenantKA 2002 Vault ribonucleoprotein particles: sarcophagi, gondolas, or safety deposit boxes? Biochemistry 41 14447 14454
64. NandyCMrazekJStoiberHGrasserFAHuttenhoferA 2009 Epstein-barr virus-induced expression of a novel human vault RNA. J Mol Biol 388 776 784
65. SakakiYTerashiKYamaguchiAKawamataNTokitoY 2002 Human T-cell lymphotropic virus type I Tax activates lung resistance-related protein expression in leukemic clones established from an adult T-cell leukemia patient. Exp Hematol 30 340 345
66. OgawaMYoshikawaYMimuroHHainTChakrabortyT 2011 Autophagy targeting of Listeria monocytogenes and the bacterial countermeasure. Autophagy 7 310 314
67. ArnaudMChastanetADebarbouilleM 2004 New vector for efficient allelic replacement in naturally nontransformable, low-GC-content, gram-positive bacteria. Appl Environ Microbiol 70 6887 6891
68. Boujemaa-PaterskiRGouinEHansenGSamarinSLe ClaincheC 2001 Listeria protein ActA mimics WASp family proteins: it activates filament barbed end branching by Arp2/3 complex. Biochemistry 40 11390 11404
69. VeigaEGuttmanJABonazziMBoucrotEToledo-AranaA 2007 Invasive and adherent bacterial pathogens co-Opt host clathrin for infection. Cell Host Microbe 2 340 351
70. van ZonAMossinkMHSchoesterMHoutsmullerABSchefferGL 2003 The formation of vault-tubes: a dynamic interaction between vaults and vault PARP. J Cell Sci 116 4391 4400
71. MostowySBonazziMHamonMAThamTNMalletA 2010 Entrapment of intracytosolic bacteria by septin cage-like structures. Cell Host Microbe 8 433 444
72. GouinEMengaudJCossartP 1994 The virulence gene cluster of Listeria monocytogenes is also present in Listeria ivanovii, an animal pathogen, and Listeria seeligeri, a nonpathogenic species. Infect Immun 62 3550 3553
73. LevraudJPDissonOKissaKBonneICossartP 2009 Real-time observation of Listeria monocytogenes-phagocyte interactions in living zebrafish larvae. Infect Immun 77 3651 3660
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2011 Číslo 8
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Tumor Cell Marker PVRL4 (Nectin 4) Is an Epithelial Cell Receptor for Measles Virus
- Two Group A Streptococcal Peptide Pheromones Act through Opposing Rgg Regulators to Control Biofilm Development
- Differential Contribution of PB1-F2 to the Virulence of Highly Pathogenic H5N1 Influenza A Virus in Mammalian and Avian Species
- Recruitment of the Major Vault Protein by InlK: A Strategy to Avoid Autophagy
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy