
-related risk of mild cognitive impairment and dementia for prevention trials: An analysis of four cohorts
In an analysis of four longitudinal cohort studies, Deborah Blacker and colleagues estimate the risk of mild cognitive impairment and dementia by APOE-status.
Published in the journal:
. PLoS Med 14(3): e32767. doi:10.1371/journal.pmed.1002254
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pmed.1002254
Summary
In an analysis of four longitudinal cohort studies, Deborah Blacker and colleagues estimate the risk of mild cognitive impairment and dementia by APOE-status.
Introduction
At present, 48 million people worldwide have dementia, and this number is projected to increase to 131 million by 2050 [1]. Consequently, prevention of Alzheimer disease, the most common cause of dementia, has become a major research focus, with several prevention trials now underway [2–7]. The feasibility of these trials will in part depend on the ability to recruit individuals at risk of developing disease during the trial period. One strategy to achieve this focuses on individuals at high genetic risk. The Alzheimer’s Prevention Initiative [8] is embarking on two clinical trials targeting cognitively unimpaired individuals at highest genetic risk for Alzheimer disease, one trial in an extended early-onset Columbian kindred carrying a fully penetrant presenilin 1 mutation, and the Generation Study (NCT02565511), a trial in individuals aged 60–75 y who carry two copies of the Alzheimer disease risk allele apolipoprotein E epsilon 4 (APOE-e4). The Generation Study is a double-blind, randomized, placebo-controlled clinical trial of two different anti-amyloid agents in approximately 1,300 participants. Recruitment is through several sources, notably in the United States through the GeneMatch [9] Alzheimer disease prevention registry (NCT02564692). High-volume recruitment efforts are required because the APOE-e4/e4 genotype occurs in approximately 1%–2% of the general population, so thousands of individuals must be screened to identify eligible participants. An assessment of absolute risk among trial-eligible individuals in a meaningful time frame is essential for the informed consent process in the trial, as well as trial design. However, although numerous studies [10–14] document that APOE-e4 increases the relative risk of Alzheimer disease (compared to no copies of APOE-e4, there is a 2 - to 4-fold increase in risk for one copy of APOE-e4, and an 8-to 15-fold increase for two copies), its effect on absolute risk is less clear.
When this study was begun, available estimates of absolute risk of dementia for APOE-e4 carriers were largely based on models developed from relative risks observed in one population and incidence data from another, often from case–control samples. The Risk Evaluation and Education for Alzheimer’s Disease (REVEAL) study [15,16] developed risk estimates [17] based on observed absolute risks in first-degree relatives versus spouses in a family sample [18], and then applied relative risks by sex, age, and genotype from a large meta-analysis [14]. A more recent effort [19], also reported on the 23andMe website [20], applied relative risks from a recent European genome-wide association study (GWAS) sample [21] to incidence estimates from the Rochester [22] and Personnes Agées QUID (PAQUID) [23] cohorts to compute lifetime risks by APOE genotype. Since that time, estimates from a single convenience cohort have been published, also with high incidence rates [24].
Because the available estimates of the APOE-associated incidence of mild cognitive impairment (MCI) or dementia are primarily based on models of disease onset rather than prospective observations, and because APOE also affects longevity and risk for diseases other than dementia, we developed new estimates in population-based cohorts to better inform both trial designers and potential participants. For potential Generation Study participants, the outreach and recruitment protocol for those who do not know their APOE genotype includes institutional review board (IRB)–approved processes for obtaining their genotype and inviting them to a trial site for an initial disclosure visit. To ensure an appropriate disclosure setting during trial enrollment, some prospective participants without the APOE-e4/e4 genotype are also invited for this initial genetic disclosure visit. Our aims were to use prospective data to determine 5-y and lifetime risk of MCI or dementia by age and APOE-e4 dose among those as similar as possible to eligible trial participants (age 60–75 y, normal cognition) and to identify sources of heterogeneity that may account for variation in risk across populations.
Methods
Ethics statement
The Rotterdam Study was approved by a medical ethics committee according to the Population Study Act Rotterdam Study, executed by the Ministry of Health, Welfare and Sport of the Netherlands; written informed consent was obtained from all participants. The Framingham Heart Study was reviewed by the IRB at Boston University Medical Center, and all participants gave written informed consent. The Sacramento Area Latino Study on Aging (SALSA) was reviewed by the IRBs at the University of Michigan and at the University of California at San Francisco and at Davis, and all participants gave written informed consent. Collection of data for the National Alzheimer’s Coordinating Center (NACC) Uniform Data Set cohort was reviewed by the appropriate local IRB at each participating Alzheimer’s Disease Center, and all participants gave written informed consent; research using the NACC database was approved by the University of Washington IRB. The IRBs at Partners HealthCare in Boston and the University of Massachusetts Amherst provided additional approvals for the secondary data analysis reported here. This study is reported as per STROBE reporting guidelines (S1 Checklist).
Cohort selection
We sought available data from longitudinal population-based cohorts based on the following attributes: recruitment and an initial cognitive evaluation at or before age 60 y (because the Generation Study is recruiting individuals aged 60–75 y, and we wanted our risk assessments to be maximally relevant to those entering the trial), ongoing surveillance for assessment of MCI and dementia, and available APOE genotypes. Many aging-focused cohorts (e.g., the Religious Orders Study [25] and the Cache County Study [26]) did not meet these criteria because of initial ascertainment at older ages. We also sought as broad ethnic representation as possible: we were able to include one Hispanic population with limited sample size, but no African-American cohort was available with the requisite data.
Three population-based cohorts were analyzed: the Framingham Heart Study [27], the Rotterdam Study [28], and the SALSA Study [29,30]. For comparison, we also included the NACC Uniform Data Set longitudinal convenience cohort [31] (from the multi-site Alzheimer’s Disease Center Program funded by the US National Institute on Aging) because we believed that NACC participants might resemble those volunteering for the Generation Study in terms of key demographic variables and level of research interest.
Sample selection for the present analyses
Within each cohort, we selected participants with known APOE genotype who were cognitively unimpaired at the time of their first visit within the 60–75-y age window, and included all available subsequent visit information until diagnosis of MCI or dementia. For the two longer-term studies, the Framingham Heart Study and the Rotterdam Study, individuals could contribute to multiple age strata for the stratified analyses, but they were included only once in our regression analyses (see “Statistical analysis” below). APOE genotype was measured in 94.1% (Rotterdam Study), 68.5% (Framingham Heart Study), 76.1% (NACC), and 92.0% (SALSA) of otherwise eligible (i.e., cognitively normal in the age window of 60–75 y) cohort participants, and only these individuals were included in the current study. On average, individuals without APOE genotype available were slightly older, except in the Framingham Heart Study, where they were slightly younger; in all cases the mean difference between those with and without APOE available was less than 1 y. Those without APOE genotype were more likely to be female in the NACC and Rotterdam cohorts, and more likely to be male in the SALSA and Framingham cohorts, but these differences were also small—within 1%–2%, except for the Rotterdam cohort, where females were 66.1% of those without genotype and 54.8% of those with genotype.
Ascertainment and assessment methods for each cohort
The original Framingham Heart Study cohort was recruited in 1948–1953 based on residence in Framingham, Massachusetts, for a longitudinal study of cardiovascular disease (mean age at enrollment 45 y). A cohort of offspring of the original participants and their spouses was established in 1971–1975 (mean age at enrollment 37 y). Details of study procedures have been published elsewhere [27]. Cognitive status has been monitored in the original cohort since 1975, when a comprehensive neuropsychological battery was administered, followed by neurological assessment of participants with lower cognitive test scores [32]. Since 1981, this cohort has been assessed at each examination with a Mini-Mental State Examination (MMSE), where participants were flagged for further cognitive screening if they scored below predefined cutoffs based on education and prior performance. The offspring cohort has undergone similar monitoring with serial MMSEs since 1991. Participants identified as having possible cognitive impairment based on these screening assessments (or in reports of cognitive concerns by the participant, family, treating physician, or Framingham ancillary study investigators, or through review of outside medical records) are invited to undergo additional annual neurological and neuropsychological examinations. A dementia review panel including a neurologist and a neuropsychologist reviews each case of possible cognitive decline and dementia and categorizes participants based on the best available information (from serial neurological and neuropsychological assessments, telephone interviews with caregivers, medical records, neuroimaging, and, when available, autopsy data) and assigns a diagnosis and onset date for dementia according to DSM-IV criteria and for MCI based on Petersen et al. [33] criteria. Diagnoses made prior to 2001 have been re-reviewed to update diagnostic criteria. Participants who entered the sample for the present analyses at a visit prior to MMSE administration but who were cognitively unimpaired at subsequent study visits had this designation extended back to their earlier visits. For our regression analyses, these individuals were included with the baseline visit as the first visit with MMSE administration within our age window (60–75 y).
For the Rotterdam Study, individuals over 55 y in 1990 residing in a specific district of the City of Rotterdam, the Netherlands, were invited to participate, with additional waves invited in 2000 (age >55 y) and 2005 (age >45 y). Details of study procedures have previously been published [28]. In brief, all participants were interviewed at home and examined at the study center every 4 to 5 y. Participants were routinely screened for dementia at the initial visit and follow-up examinations using a three-step protocol. Screening was done using the MMSE and the Geriatric Mental Schedule (GMS) organic level [2]. Those with MMSE < 26 or GMS organic level > 0 subsequently underwent an examination and informant interview using the Cambridge Examination for Mental Disorders of the Elderly (CAMDEX) [34]. Additionally, the total cohort was continuously monitored for dementia through computerized linkage between the study database and digitized medical records. The current sample included all participants with MMSE > 26 at the time of their first visit within the age window of interest (60–75 y). Formal assessment of MCI did not begin until 2005 in the Rotterdam Study. For the present analyses, we therefore developed a pragmatic diagnosis of MCI during follow-up, requiring a MMSE score < 26 or a drop of at least three points from the baseline visit in the 60–75-y age window, plus answering yes to a question about memory concerns.
For SALSA, participants over 60 y were sampled from six counties including census tracts with at least 5% Hispanic population in the Sacramento Valley of California in 1998–1999 and were followed approximately every 12–15 mo until 2008. Detailed methods are described elsewhere [29,35]. In brief, dementia assessment included screening with both the Modified Mini-Mental State Examination (3MS) [36] and a word list learning task from a standard battery [30]. Those scoring below the 20th percentile (using age-, education-, sex-, and language-adjusted norms) on either test (or for follow-up visits, dropping three points in word list learning) were further evaluated using the Informant Questionnaire on Cognitive Decline in the Elderly (IQCODE) [37,38] and, if this gave additional support for decline, were evaluated by a neurologist and categorized as cognitively unimpaired, memory-impaired (based on testing alone, without IQCODE corroboration), cognitively impaired not demented (CIND) [39], or having dementia. Given the requirement for both a cognitive testing abnormality and confirmation from an informant, CIND was treated as equivalent to MCI [35].
Participants in the NACC cohort were volunteers ascertained from various sources at 34 Alzheimer’s Disease Centers in the United States. We used the March 2016 data freeze for the present analyses, so these data reflect study visits between September 2005 and March 2016. The participants were evaluated according to the Uniform Data Set protocol [40], with each participant and a collateral informant interviewed by the study clinician to rate the Clinical Dementia Rating (CDR) [41] and with the administration of a battery of neuropsychological tests [42]. A diagnosis was made at each visit by the study clinician following standard criteria [40], but there were no study-wide standardized cutoffs on the CDR, MMSE, or other neuropsychological tests. Follow-up visits were conducted approximately annually.
Definition of predictor variables
Education was reported in years for the SALSA and NACC cohorts and in categories of less than high school, high school, some college, or college graduation for the Rotterdam and Framingham cohorts. Education data for the SALSA and NACC cohorts were translated into these categories as follows: <12 y, less than high school; 12 y, high school; 13–15 y, some college; and ≥16 y, college graduation.
To assess cognitive performance across cohorts, we used the cognitive screening test available for each site (MMSE for the Rotterdam Study, the Framingham Heart Study, and NACC, and 3MS for SALSA). To enable comparisons of relative performance within each cohort, we standardized based on the test score at the baseline visit (in the age 60–75-y age window) within each cohort, centering the raw scores around their sample mean and then dividing the centered scores by their standard deviation.
Memory concerns at NACC were based on a global clinician-rated variable asking whether the participant believed that he or she had a problem with memory. Memory concerns in the Rotterdam Study were based on three questionnaire items asking (1) whether the participant was worried about his or her memory, (2) whether the participant ever lost track of what he or she was doing in the midst of an activity, and (3) whether the participant experienced word-finding difficulties. A positive answer to any of these questions qualified as memory concerns.
Family history of dementia was defined as having at least one parent with dementia for the Rotterdam Study, and at least one first-degree relative with dementia for NACC.
For all cohorts, vascular risk was defined as follows. We obtained the sum of major vascular risk factors measured in each cohort: coronary artery disease or angina or stroke, hypertension, high cholesterol, diabetes, atrial fibrillation, and current smoking. After reviewing the distribution of the count of these risk factors (range 0–6), we categorized the participants as high risk (3–6 risk factors), moderate risk (1–2 risk factors), and low risk (0 risk factors) to provide a reasonable distribution across the three levels. We considered using the Framingham Stroke Risk Profile [43] or similar cardiovascular risk scores [44,45]. However, these were designed to predict risk within a specified time frame and thus had substantial age components, which complicated analyses in our regression models that already included age.
Statistical analysis
We performed all analyses first for MCI or dementia (“MCI/dementia”), then for dementia alone. For the purposes of this trial, the MCI/dementia outcome was critically relevant, in that incident dementia was unlikely during the trial period, while there was tangible risk for MCI. Analyses for dementia only were performed as well because dementia is a more robust outcome than MCI.
We estimated 5-y and “lifetime” (i.e., to age 80–85 y) cumulative incidence by APOE-e4 dose and 5-y baseline age stratum (age 60–64, 65–69, 70–75 y). We chose three age strata as a tradeoff between addressing the steeply changing risk with age and not overly subdividing the limited numbers of APOE-e4/e4 homozygote individuals, which left the number of APOE-e4/e4 individuals per age stratum too small for stable estimates in the SALSA cohort. These age strata were determined specifically as follows, based on the date of the baseline visit (within the 60–75-y age window): 60–64 y, 60 ≤ age < 65; 65–69 y, 65 ≤ age < 70; and 70–75, 70 ≤ age ≤ 75. We considered further stratification on sex, but the sample size did not support such stratification.
For the stratified analyses of the two longer-term studies, the Framingham Heart Study and the Rotterdam Study, individuals could contribute to multiple baseline age strata; we used the first visit within each age window as the baseline in these analyses. Lifetime estimates were computed as 20-y cumulative incidence for the age 60–64-y stratum, as 15-y for the 65–69-y stratum, and as 10-y for the 70–75-y stratum; these estimates were computed only for the two longer-term cohorts to minimize extrapolation.
Stratified cumulative incidence curves by age stratum and APOE-e4 dose were estimated, adjusting for loss to follow-up other than death and for the competing risk of mortality [46]; loss to follow-up other than death is treated as censored. In the presence of competing risks, the naïve Kaplan–Meier estimator, which treats failure from competing causes as censored observations, overestimates the cumulative incidence of the event of interest [47]. We used the “cmprsk” package in R software [48] to estimate the cumulative incidence for each age stratum by APOE-e4 dose stratum. Following the suggestion of Lin [49], we used the transformation log[−log(1 − x)] to construct the confidence interval for cumulative incidence. The transformation not only ensures that the boundaries of cumulative incidence are contained in [0,1], but also improves the coverage accuracy [49].
We used the same competing risks analytic framework to assess the effects of age and APOE-e4 dose plus additional covariates on the cumulative incidence of MCI/dementia and of dementia alone in order to inform personalized risk assessment and to understand differences across the cohorts. We used subdistribution hazard regression models [50] because they directly link the regression coefficients with the cumulative incidence function (in contrast to cause-specific hazards regression [51], where the direct link cannot be made [52]; in preliminary analyses we also fit these models, and results were very similar). These analyses were also performed using the “cmprsk” package in R software [48].
For each cohort and for each outcome, we first fit univariable models for baseline age, sex, APOE-e4 dose, education, standardized cognitive screening test score, subjective memory concerns, family history of dementia, and vascular risk score. Then, we ran simple multivariable models for each outcome including only APOE-e4 dose and demographic factors (age, sex, and education). Last, we ran larger multivariable models also including standardized cognitive screening score plus subjective memory concerns and family history of dementia if available for the cohort. The vascular risk score was not included in the full model because findings were inconsistent and primarily null in the univariable models, whether we used our low/moderate/high vascular risk levels described above or the Framingham Stroke Risk Profile.
Missing data on covariates was generally minimal, <2% for all covariates in all cohorts except family history in the Rotterdam Study (11.5%), vascular risk in SALSA (8.9%), and education in the Framingham Heart Study (3.3%). As these figures were small, participants with missing data were simply omitted from regression analyses in which the relevant missing variable was included.
For the Rotterdam Study, the exact date of dementia diagnosis was used if available; otherwise, the midpoint of the interval between visits was used as the onset time of MCI or dementia at a study visit (conducted at 4-y intervals) for both cumulative incidence estimates and subdistribution hazard regression. In addition, as a sensitivity analysis, we repeated our survival curves and regression models treating the onset of MCI or dementia as interval censored in addition to adjusting for competing risk, using the “MIICD” package in R software to estimate the cumulative incidence, and results were extremely similar except for somewhat larger confidence intervals.
Unlike in the stratified analyses, in the regression analyses, each participant was used only once. Typically, the baseline visit for the regression analyses was the first visit within the eligible age window of 60–75 y. For the Framingham Heart Study, MMSE was not available at visits prior to 1981 (as described above). Thus, for the regression analyses, we reset the baseline visit as the first visit at which MMSE was available. This had the additional benefit of increasing the range of baseline ages within the cohort.
Meta-analyses were conducted for the 5-y cumulative incidence estimates for all four cohorts and then for only the three population-based cohorts. Meta-analyses could not be conducted for the lifetime estimates because they were computed for only two cohorts. As there was considerable heterogeneity among the studies, a random-effects meta-analysis based on the DerSimonian–Laird method [53] was used. This analysis was performed using the “metafor” package in R software.
Because the primary goal was estimating cumulative incidence and understanding differences across cohorts and individuals rather than hypothesis testing, these analyses are reported with confidence intervals rather than statistical significance, and no adjustments are made for multiple comparisons.
The study was planned in summer of 2014 and conducted through fall of 2016. The original analysis plan, developed in the initial months of the study, specified using multiple cohort studies, including NACC as well as three population-based cohorts; stratified analyses in the three age and APOE-e4 dose groups; and regression models including demographic factors, cardiovascular risk, and baseline cognitive performance or symptoms, with survival models accounting for competing risk of death. The idea was that the stratified curves would provide general estimates, and the regression models more individualized estimates (and in any event insight into anticipated differences across cohorts). As noted above, after initial exploration, we decided to use specifically subdistribution hazard regression rather than cause-specific hazard regression models because these offer greater interpretability in the context of risk prediction. In preparation for our first presentation of the findings to the Generation Study team in April 2015, we also decided to make a table of estimated 5-y cumulative incidence to allow easier comparison across the studies. After this initial meeting, we also added the lifetime risk estimate, which we thought would be informative for potential participants. We performed the initial regression models separately by cohort in order to get an idea of how best to present the data given somewhat different variables available in each cohort, and then settled on a final analysis plan in spring 2016 that included univariable and two nested models as consistent as possible across the four cohorts. The NACC dataset was acquired in summer 2014 (the June 2014 data freeze) and then updated in summer 2016 (the March 2016 data freeze). The SALSA dataset was acquired in fall 2014, and the Framingham Heart Study dataset in spring 2015. Rotterdam Study analyses were conducted by Rotterdam Study investigators (F. J. W. and M. A. I.) beginning in spring 2015, in close collaboration with the rest of the group and sharing R code used with the other cohorts to ensure consistency.
Results
Composition of the cohorts
Table 1 presents the composition of the four cohorts. The cohorts differed considerably in size and duration of follow-up, with SALSA much smaller than the other cohorts, and long-term follow-up available only in the Framingham Heart Study and Rotterdam Study. Other substantial differences were seen in educational attainment, with mean years ranging from less than 8 y in SALSA to nearly 16 y in NACC, and sex, with 33.6% men in NACC compared to 42%–45% in the three population-based cohorts. The four cohorts also differed markedly in APOE-e4 allele frequency, ranging from 7.5% in SALSA to 17.8% in NACC. NACC also had a 58.3% fraction with a family history of dementia, compared to 18.6% in the Rotterdam Study, the only other site that assessed it.

Stratified cumulative incidence estimates
Fig 1 shows the cumulative incidence of MCI/dementia stratified by baseline age group and APOE-e4 dose; Fig 2 shows the corresponding curves for dementia alone. These figures show 8.5 y of follow-up for all four cohorts on the same scale, to facilitate comparison. Figs 3 and 4 show lifetime (to age 80–85 y) cumulative incidence curves for MCI/dementia and dementia alone for the two longer-term cohorts.




Table 2 shows the 5-y cumulative incidence of MCI/dementia for all four cohorts, and Table 3 the lifetime (to age 80–85 y) cumulative incidence across the two longer-term cohorts; Tables 4 and 5 show the corresponding data for dementia alone.


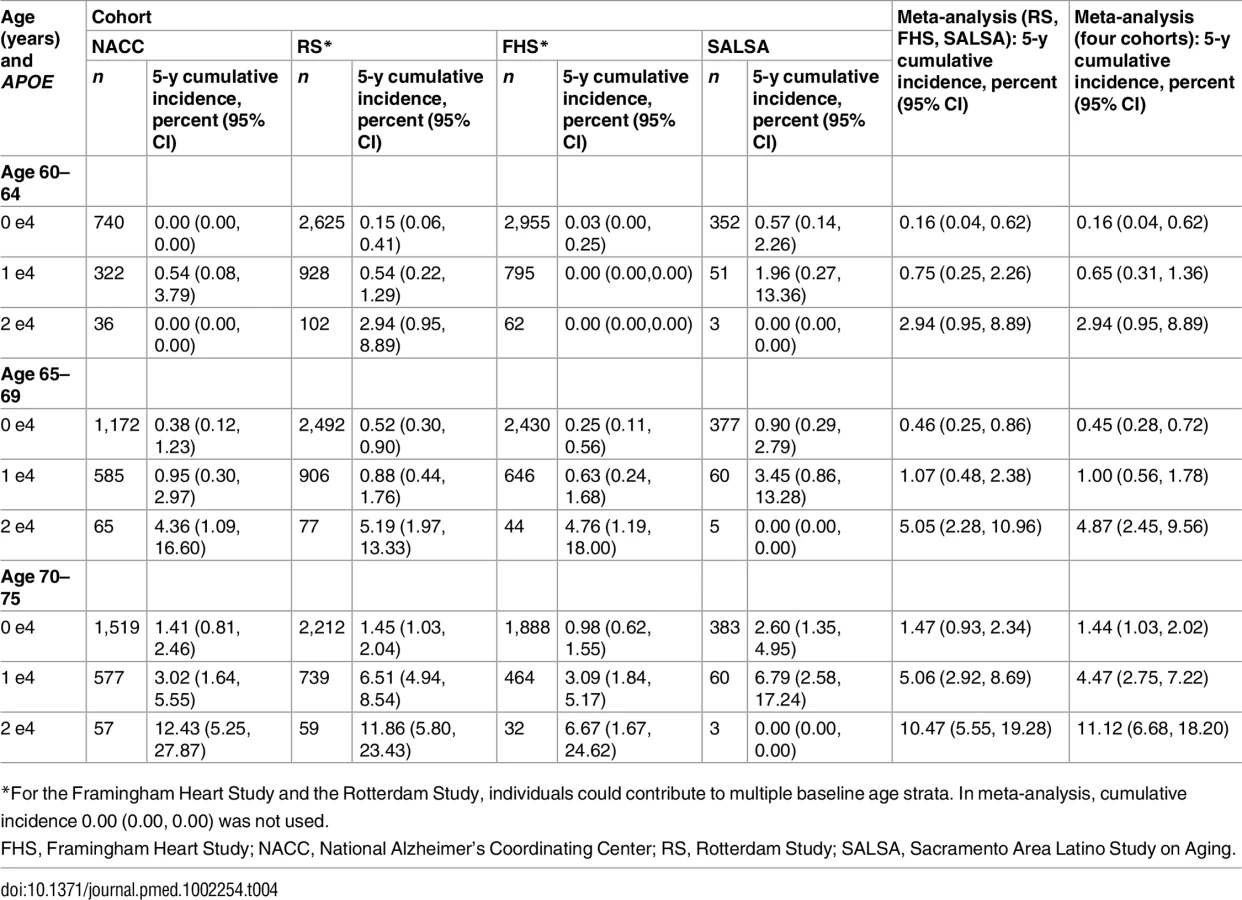

Overall, within each cohort, risk of MCI/dementia increased with increasing age and APOE-e4 dose. However, absolute risks differed substantially across the cohorts, particularly between NACC and the population-based cohorts. Especially for the MCI/dementia outcome, the NACC cohort typically had higher risk for any genotype at any age. Differences among the population-based cohorts were smaller, particularly for longer-term follow-up and the dementia outcome.
Five-year cumulative incidence of MCI/dementia was low in the youngest age stratum, particularly in the cohort studies, although somewhat higher for APOE-e4-positive individuals, especially homozygote individuals (23% in NACC and 5%–6% in Framingham and Rotterdam). Five-year incidence of MCI/dementia was higher in the highest age stratum, particularly among APOE-e4/e4 homozygote individuals (38% in NACC and 18%–23% in Framingham and Rotterdam). Five-year incidence of dementia alone was negligible at younger ages, even in APOE-e4/e4 homozygote individuals, but rose among older individuals, particularly among those with APOE-e4/e4 (12% in NACC and 7%–12% in Framingham and Rotterdam). The meta-analyses of the 5-y cumulative incidence estimates for the MCI/dementia outcome (Table 2) showed consistent increases in incidence by gene dose within age strata and by age stratum within gene dose, and were higher when the NACC estimates were included. These pooled estimates ranged from a low of 1.46% for individuals aged 60–64 y with no copies of APOE-e4 in just the population-based cohorts to a high of 26.70% for individuals aged 70–75 y with two copies of APOE-e4/e4 in all four cohorts.
Estimated only for the Rotterdam Study and the Framingham Heart Study, lifetime incidence, whether for MCI/dementia or for dementia alone, was consistent in the two cohorts (it was also consistent across age strata, but it should be noted that the strata are not independent and that older age strata included individuals who survived and did not experience the outcome in earlier strata). Lifetime incidence rose consistently with APOE-e4 dose: for MCI/dementia (Table 3), it ranged from 11.94%–15.57% for those with no copies of APOE-e4 to 37.47%–46.66% for APOE-e4/e4 homozygote individuals; for dementia alone (Table 5), it ranged from 5.26%–6.83% for no copies to 30.76%–40.26 for homozygote individuals.
Subdistribution hazard regression analyses
Results of the subdistribution hazard regression analysis are presented in S1 Appendix Tables A and B (univariable analyses), S1 Appendix Tables C and D (multivariable analyses modeling APOE-e4 dose and demographics), and S1 Appendix Tables E and F (additionally including family history of dementia and cognitive variables). Overall, the regression results were fairly consistent across the four cohorts, even in the small SALSA cohort, and considerably more consistent than the cumulative incidence results.
The univariable results (S1 Appendix Table A for MCI/dementia and S1 Appendix Table B for dementia) were fairly consistent across the two outcomes (although for some variables in some cohorts the hazard ratios [HRs] were somewhat higher for dementia alone), so we provide details in the text for the MCI/dementia outcome only. There was substantially higher risk of MCI/dementia with increasing age (HR 1.08–1.16 per year of age), increasing APOE-e4 dose (for one copy, HR 1.51–2.23; for two copies, HR 2.63–3.57), and lower education (HR 1.41–1.86 for less than high school compared to high school). Family history of dementia also had a nominally significant effect in both cohorts in which it was measured (HR 1.16–1.27). On the other hand, male sex, which was protective in the population-based cohorts (although only nominally significantly so in the Rotterdam cohort, HR 0.83–0.90), carried risk in NACC (HR 1.36). Subjective memory concerns carried risk in both cohorts that assessed them (HR 1.71–2.62). Higher standardized baseline cognitive screening test score (MMSE or 3MS) was consistently protective across all cohorts for both outcomes (HR 0.58–0.80 per standard deviation above the mean), except for the MCI/dementia outcome in the Rotterdam cohort. Vascular risk score had a variable and generally nonsignificant effect across all four cohorts.
The simple multivariable models including APOE and demographic factors (S1 Appendix Table C for MCI/dementia and S1 Appendix Table D for dementia alone) did not appreciably change the results for age and APOE-e4 dose, although there was some attenuation of associations for sex and education. In the more complex model (S1 Appendix Tables E and F), again the picture was similar, with attenuation for sex and education. It is noteworthy that standardized cognitive screening test score and subjective memory concerns (where available) generally showed substantial, nominally significant hazard ratios, even controlling for education, and that (where available) family history of dementia, even when controlling for APOE-e4 dose, also had an impact.
Discussion
Overall findings
Of 16,844 participants included from all four cohorts, 392 (2.3%) had the APOE-e4/e4 genotype, highlighting its low prevalence. Nonetheless, the expected age - and APOE-e4-dose-related increases in cumulative incidence and relative hazard in the regression models are readily apparent, even to some extent in the very small SALSA cohort. However, the striking differences in estimated cumulative incidence, particularly for the MCI/dementia outcome, between the population-based cohort studies and the highly ascertained NACC cohort (see below) suggest that overall APOE-e4-associated incidence is somewhat lower than the modeled findings previously available in the literature. Comparing 5-y cumulative incidence from the meta-analyses of the three population-based cohorts to that from NACC, in the youngest age stratum, cumulative incidence ranged across the three APOE-e4 doses from 1.46% to 5.60% in the population-based cohorts versus 7.94% to 23.06% in NACC, and in the oldest age stratum from 5.71% to 20.58% in the population-based cohorts versus 15.16% to 38.34% in NACC. Similarly, viewing the cumulative incidence for APOE-e4/e4 genotype across the three age strata, cumulative incidence ranged from 5.60% to 20.58% in the three population-based cohorts versus from 23.06% to 38.34% in NACC. The NACC findings were largely similar to those of the prospective analyses of Bonham et al. [24] in the same cohort, although Bonham et al. [24] focused on the relative risk of APOE-e4 across different age ranges, used different age categories (unrelated to the Generation Study), and did not incorporate several important variables in the models (i.e., family history, subjective memory concerns, and baseline cognitive performance). Moreover, the authors did not perform their analyses in a competing risk framework, which is vital to avoid overestimation of cumulative incidences in aging populations [54].
Differences in cumulative incidence estimates across the sites
Variability related to ascertainment and assessment methods has been reported previously for MCI and dementia prevalence [55,56]. Such variability is not unique to MCI and dementia, but can occur in a variety of settings, and is a particular problem for common disorders like MCI in which a subtle gradation from the normal makes rates especially sensitive to thresholding (e.g., attention deficit hyperactivity disorder, major depression, osteoarthritis).
Overall, as might be expected, absolute risk is more vulnerable to methodological differences than relative risk, especially over shorter time intervals and for the MCI/dementia outcome rather than the dementia alone outcome. This is underscored by the generally similar relative hazards across the regression analyses. These regression findings also contribute to an understanding of the variation across the cohorts.
Among the three population-based cohort studies, there are known and unknown differences in race and ethnicity, education, and screening and assessment methods. Nonetheless, these three cohorts were generally similar—within expected sampling variation—in their estimates of cumulative incidence for most age and APOE strata. The difference between the population-based cohort studies and NACC, on the other hand, is striking. The NACC cohort is a volunteer cohort, and as such would not be expected to represent the general population (although it may be representative of potential trial participants, as discussed below). Individuals join this cohort for a variety of reasons, but concerns about family history and their own memory are likely to play a role. This probably contributes to the relatively high APOE-e4 allele frequency and reported family history of dementia in this cohort seen in Table 1, although some of the difference in family history likely represents measurement issues (see below). Since family history increases risk beyond the APOE-e4 effect in these and other data [57,58], the high frequency of positive family history likely contributed to some of the observed differences in incidence. Another potential source of difference is the very high level of educational attainment within the NACC cohort. While higher education is associated with lower risk of dementia overall, more educated individuals with memory concerns actually have higher risk of developing dementia than their counterparts with less education [59], and this may be particularly true for the highly educated individuals who form a substantial fraction of the NACC cohort. Another issue is the high proportion of women in the NACC cohort; differences in the reasons that men and women volunteer for this cohort may underlie the increased risk of MCI and dementia for men observed in the regression analyses. Last, the NACC samples serve a variety of needs across the different Alzheimer’s Disease Centers in the United States; there is often substantial dropout and variable effort to retain participants, and decisions by participants and center staff are not likely to be random with respect to cognitive and other variables. While the population-based cohort studies also have some dropout, systematic ongoing efforts to retain participants and continuous surveillance even for those who do not attend study visits guarantee low attrition.
Beyond these differences in ascertainment, demographics and other attributes, and follow-up, there are differences in assessment between NACC and the three population-based cohorts that should be noted. The population-based cohorts evaluate cognition with a screening procedure typically followed by more formal clinical evaluation of participants who screen positive. While direct clinical evaluation of all participants at each NACC site is a strength, there are procedural differences across sites, quality control is limited, and the reliability of NACC diagnosis is not well established. In addition, the high educational level of NACC participants is not well captured by available norms, and a subset of individuals may have declined substantially but nonetheless may be viewed as cognitively unimpaired. This is a particular concern because within any group of normal individuals, those who are already declining are more likely to continue to do so [60–62]. Thus, baseline cognitive symptoms and preexisting subthreshold decline, both likely to be more frequent in the highly educated NACC cohort, have a substantial impact on short-term onset of MCI and even dementia. This phenomenon may underlie the higher risk of cognitive decline noted earlier for more versus less educated individuals among those with subjective cognitive concerns [59].
Of course, it is likely that there is some insensitivity to MCI and even dementia in the population-based cohort studies as well as differential loss to follow-up, but on balance the volunteer nature of the NACC cohort, the limited quality control across the NACC sites, and the consistency of the population-based cohort findings tend to favor the lower cumulative incidence found in the population-based cohorts.
Comparison to modeled estimates from the literature
One could argue that previously available modeled estimates for APOE-e4-associated absolute risk for dementia [17,19] are high (50%–67%), and thus favor the NACC estimates instead. Our estimates of lifetime risk for dementia for APOE-e4/e4 individuals from the Framingham Heart Study and the Rotterdam Study are in the 31%–40% range. While we did not estimate lifetime cumulative incidence for NACC given the short mean duration of follow-up, it would be expected to be considerably higher than the 5-y estimates—in the oldest APOE-e4/e4 homozygote individuals, 38.3% for MCI or dementia and 12.4% for dementia alone. However, there are some biases in the modeled estimates that overall are more likely to yield over - rather than underestimates of risk.
For the Cupples et al. estimates used in the REVEAL study [17], risk curves for incidence were derived from relatives and spouses in a family sample ascertained from a clinical population [18]; these incidence rates could be expected to be higher than those in the general population. In addition, the relative risks by sex, age, and genotype were applied from a large meta-analysis done primarily in clinically ascertained, younger-onset families [14], again yielding higher estimates [14,63]. In addition, the competing risk of death was not addressed in the cumulative incidence estimates, which also would tend to bias estimates upward. Moreover, the Cupples model does not account for the correlation among observations in the family sample used for incidence, which again might lead to bias [64].
For the Genin et al. estimates used by 23andMe [19], relative risks from a European GWAS [21] were applied to incidence estimates from the Rochester [22] and PAQUID [23] cohorts. The relative risk estimates come from cases and controls, with younger cases (with a greater APOE-e4 effect) overrepresented. In addition, these models assumed that the controls in GWAS samples were representative of the overall population. This likely does not hold with a very common disease like dementia (which occurs in over 10% of those over 65 y and 35% or more of those over 85 y [1]) because at higher ages those without dementia are fundamentally a selected sample. This also would tend to bias the estimates upward.
Insights from the regression models
Overall, the substantial effects of age and APOE-e4 dose were consistent across the univariable and basic and more complex multivariable models, persisting even when other demographic factors as well as cognitive variables and family history were taken into account. Education also exhibited a dose response, but behaved less consistently, as much illustrating as illuminating the profound differences in education across these four cohorts.
The effect of sex is even less consistent, perhaps reflecting ascertainment and cultural differences across disparate cohorts; findings in the literature are also inconsistent [23,65,66]. Some studies suggest also that APOE-e4 behaves differently by sex, with a greater effect in women [67,68]. If we had had sufficient sample size, we would also have stratified our risk estimates on sex or considered including an interaction term in our regression models. However, in the population-based cohorts for the MCI/dementia outcome, there was strong attenuation of the effect estimates of sex when adjusting for educational attainment, suggesting that lack of educational attainment in women of older birth cohorts may partly explain the difference. However, for dementia only in the same cohorts, a nominally statistically significant higher risk in women persisted even after adjustment for demographics and other risk indicators. Conversely, in NACC, there was a higher risk in men, which we believe is likely related to ascertainment differences by sex in this convenience sample, as noted above. Overall, potential sex differences deserve particular focus in future studies given the complex relationships among sex, education, vascular risk factors, birth cohort, longevity, and genetics.
Also of potential relevance, both to potential participants wishing to understand their absolute risk and to investigators designing clinical trials, both cognitive performance and subjective memory concerns were associated with an increased hazard of MCI or dementia. All in all, these associations suggest that relatively simple individual characteristics might be used to further refine individual risk stratification beyond age and APOE genotype.
Implications for study design and genetic counseling
For the purposes of the Generation Study and other prevention trials, absolute cumulative incidence, both during the 5-y duration of the trial and over the remaining lifetime, is critical, but the differences across these cohort studies make it difficult to offer precise estimates, even with meta-analyses. In an ideal world, estimates would be tailored to the population entering the trial or, better still, the specific individuals, and would take into account not only explicit inclusion criteria but also any other measureable or predictable characteristics that might predict willingness to volunteer. A review of the first registrants on the GeneMatch registry, which serves as the primary US recruiting site for the Generation Study APOE-e4/e4 trial, shows that registrants differ from the general population beyond the explicit entry criteria. The population of 13,704 registrants enrolled thus far is relatively young (mean age 62.7 y, standard deviation 5.2) and women are overrepresented (80%). Among the 4,978 registrants who were asked about race/ethnicity, 92% are white. The frequency of the APOE-e4/e4 genotype among registrants is higher than in the general population, at 4.47%, and the APOE-e4 allele frequency is 20.4%; among the 3,456 registrants asked about whether they had a family history of dementia or Alzheimer disease, 70.1% said yes. While education was not measured, the high percent of females and individuals with a significant family history (and the high APOE-e4 frequency) suggests a population that may be more like NACC. However, data on education, cognitive performance, and subjective memory concerns are not available. Moreover, over time, if there are broader recruiting efforts in order to reach the target sample size, volunteers could gradually become more reflective of the general population, and lower risks might be expected.
In the genetic counseling setting, any risk information would need to give a broad range of estimates to reflect uncertainty within cohorts and variation across cohorts. Because risk for disease is ongoing, and the lifetime risks were more stable than the 5-y risks in our analyses, we thought the lifetime risks were more informative for genetic disclosure. However, such risks may be less salient to some of those considering enrollment in trials at younger ages. The Generation Study elected to disclose the following “lifetime” risks of MCI or dementia to its potential participants: 30%–55% for individuals with APOE-e4/e4; 20%–25% for individuals with APOE-e3/e4 and -e2/e4 (with a note that risk might be lower for those with APOE-e2/e4); and 10%–15% for individuals with APOE-e3/e3, -e3/e2, and -e2/e2 (with a note that risk might be lower for those with APOE-e2/e3 and -e2/e2). These values are consistent with our findings, but use round numbers for intelligibility, and broader ranges to reflect statistical and other sources of uncertainty. The regression models are insufficiently precise for “personalized medicine” incidence estimates based on sex, education, or other factors, but they do allow for qualitative adjustments to overall stratified risk estimates. Relative risks by APOE genotype or APOE-e4 dose have limited relevance in the setting of the prevention trial, but may provide context. If these are provided, risk should be compared to the general population (based on a weighted average across the three possible APOE-e4 doses rather than the typical “no APOE-e4” base category used in regression models), which would more fairly allow a participant to put his or her own risk in the context of friends and acquaintances of unknown genotype. On the basis of our regression findings (S1 Appendix Table E), for APOE-e4/e4 homozygotes, the adjusted relative risk for MCI/dementia is 2.7 for NACC, 3.4 for the Framingham Heart Study, and 2.4 for the Rotterdam Study, so disclosing a relative risk of about 3-fold compared to the general population would make sense. Use of pictographs as a visual aid for risk communication could be useful, given their ability to visually represent both absolute and relative risk information simultaneously [69]. In addition, there is a robust literature on genetic risk communication that can inform best practices in cases where APOE information is disclosed to asymptomatic individuals [70].
Limitations
One major limitation of this study is that APOE-e4/e4 samples are small despite the large size of the initial cohorts, particularly for SALSA. This limits the stability of stratified cumulative incidence estimates (only partially addressed by the meta-analyses) as well as regression coefficients for APOE-e4 dose. This issue is further complicated by missing data (likely not missing at random) and likely differential dropout. Second, while the four cohorts are heterogeneous in sex distribution and education, there is little ethnic and racial diversity, so the findings are less relevant to participants of non-European background. Third, variations in the definitions of the exposure and outcome variables may hamper comparisons among cohorts. As noted above, each cohort uses different criteria to define unimpaired at baseline, and to screen, assess, and diagnose new onset cases. Different psychometric tests are applied, and even the same test performs differently across different groups; education - and/or age-adjusted norms can compensate for this, but may introduce other problems in interpretation. Other variation may come from differences in definitions (e.g., family history is based on a single question about parents only in the Rotterdam Study versus a detailed set of questions about each parent and sibling in NACC) or in how information is acquired (being positive for memory concerns is based on a yes answer to any one of three questionnaire items in the Rotterdam Study versus an overall clinical impression about the participant’s attitude in NACC). Moreover, some variables, notably level of education, may be defined similarly but have different meanings within different cultural contexts. Nevertheless, as we have shown, relative risk estimates are consistent despite this variation. Fourth, regression models for MCI or dementia are limited because of confounding and omitted predictors, and are complicated by multicollinearity of exposure and outcome variables that represents confounding, effect modification, and true signal.
Conclusion
Prospective cohort studies can be used to inform study design, power, and informed consent in clinical trials among cognitively unimpaired individuals. While trial designers and participants may be most interested in absolute risk over relatively short intervals, absolute risk is less robustly estimated than relative risk, and short-term risk less robustly estimated (and more sensitive to the definition and operationalization of cognitively unimpaired at baseline) than long-term risk. Estimation that serves informed consent and optimal trial design will require matching the cohort used to estimate risk as closely as possible to trial participants.
Overall, the estimates for APOE-associated risk of MCI or dementia were lower in our study than previously reported, and there is reason to believe that the risk estimates obtained in the population-based cohorts more accurately reflect the general population than those obtained in NACC. However, these lower risks may less accurately match the likely trial population. In general, such estimates are also sensitive to variation in sampling, assessment, and modeling. Rigorous attention to sampling, assessment, and statistical methods is critical to developing the best possible answers for clinical trial design.
Supporting Information
Zdroje
1. Alzheimer’s Disease International. World alzheimer report 2016. London: Alzheimer’s Disease International; 2016 [cited 2016 October 26]. Available from: http://www.alz.co.uk/research/WorldAlzheimerReport2016.pdf.
2. de Bruijn RF, Bos MJ, Portegies ML, Hofman A, Franco OH, Koudstaal PJ, et al. The potential for prevention of dementia across two decades: the prospective, population-based Rotterdam Study. BMC Med. 2015;13 : 132. doi: 10.1186/s12916-015-0377-5 26195085
3. Hsu D, Marshall GA. Primary and secondary prevention trials in Alzheimer disease: looking back, moving forward. Curr Alzheimer Res. 2016 Sep 30.
4. Sperling RA, Rentz DM, Johnson KA, Karlawish J, Donohue M, Salmon DP, et al. The A4 study: stopping AD before symptoms begin? Sci Transl Med. 2014;6(228):228fs13. doi: 10.1126/scitranslmed.3007941 24648338
5. Martinez-Lapiscina EH, Clavero P, Toledo E, San Julian B, Sanchez-Tainta A, Corella D, et al. Virgin olive oil supplementation and long-term cognition: the PREDIMED-NAVARRA randomized, trial. J Nutr Health Aging. 2013;17(6):544–52. doi: 10.1007/s12603-013-0027-6 23732551
6. Ngandu T, Lehtisalo J, Solomon A, Levalahti E, Ahtiluoto S, Antikainen R, et al. A 2 year multidomain intervention of diet, exercise, cognitive training, and vascular risk monitoring versus control to prevent cognitive decline in at-risk elderly people (FINGER): a randomised controlled trial. Lancet. 2015;385(9984):2255–63. doi: 10.1016/S0140-6736(15)60461-5 25771249
7. Reiman EM, Langbaum JB, Tariot PN, Lopera F, Bateman RJ, Morris JC, et al. CAP—advancing the evaluation of preclinical Alzheimer disease treatments. Nat Rev Neurol. 2016;12(1):56–61. doi: 10.1038/nrneurol.2015.177 26416539
8. Reiman EM, Langbaum JB, Tariot PN. Alzheimer’s Prevention Initiative: a proposal to evaluate presymptomatic treatments as quickly as possible. Biomark Med. 2010;4(1):3–14. doi: 10.2217/bmm.09.91 20383319
9. Alzheimer’s Prevention Registry. GeneMatch: connecting Alzheimer’s prevention studies with eligible volunteers. Phoenix: Alzheimer’s Prevention Registry; 2016 [cited 2016 November 10]. Available from: https://www.endalznow.org/genematch.
10. Slooter AJ, Cruts M, Kalmijn S, Hofman A, Breteler MM, Van Broeckhoven C, et al. Risk estimates of dementia by apolipoprotein E genotypes from a population-based incidence study: the Rotterdam Study. Arch Neurol. 1998;55(7):964–8. 9678314
11. Qiu C, Kivipelto M, Aguero-Torres H, Winblad B, Fratiglioni L. Risk and protective effects of the APOE gene towards Alzheimer’s disease in the Kungsholmen project: variation by age and sex. J Neurol Neurosurg Psychiatry. 2004;75(6):828–33. doi: 10.1136/jnnp.2003.021493 15145993
12. Goldman JS, Hahn SE, Catania JW, LaRusse-Eckert S, Butson MB, Rumbaugh M, et al. Genetic counseling and testing for Alzheimer disease: joint practice guidelines of the American College of Medical Genetics and the National Society of Genetic Counselors. Genet Med. 2011;13(6):597–605. doi: 10.1097/GIM.0b013e31821d69b8 21577118
13. Yu JT, Tan L, Hardy J. Apolipoprotein E in Alzheimer’s disease: an update. Annu Rev Neurosci. 2014;37 : 79–100. doi: 10.1146/annurev-neuro-071013-014300 24821312
14. Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, Mayeux R, et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA. 1997;278(16):1349–56. 9343467
15. Roberts JS, Cupples LA, Relkin NR, Whitehouse PJ, Green RC. Genetic risk assessment for adult children of people with Alzheimer’s disease: the Risk Evaluation and Education for Alzheimer’s Disease (REVEAL) study. J Geriatr Psychiatry Neurol. 2005;18(4):250–5. doi: 10.1177/0891988705281883 16306249
16. Roberts JS, Christensen KD, Green RC. Using Alzheimer’s disease as a model for genetic risk disclosure: implications for personal genomics. Clin Genet. 2011;80(5):407–14. doi: 10.1111/j.1399-0004.2011.01739.x 21696382
17. Cupples LA, Farrer LA, Sadovnick AD, Relkin N, Whitehouse P, Green RC. Estimating risk curves for first-degree relatives of patients with Alzheimer’s disease: the REVEAL study. Genet Med. 2004;6(4):192–6. 15266206
18. Lautenschlager NT, Cupples LA, Rao VS, Auerbach SA, Becker R, Burke J, et al. Risk of dementia among relatives of Alzheimer’s disease patients in the MIRAGE study: what is in store for the oldest old? Neurology. 1996;46(3):641–50. 8618660
19. Genin E, Hannequin D, Wallon D, Sleegers K, Hiltunen M, Combarros O, et al. APOE and Alzheimer disease: a major gene with semi-dominant inheritance. Mol Psychiatry. 2011;16(9):903–7. doi: 10.1038/mp.2011.52 21556001
20. andMe. Alzheimer’s disease (APOE variants): established research report on 2 reported markers. Mountain View (California): 23andMe; 2016 [cited 2016 November 10]. Available from: https://www.23andme.com/en-ca/health/i_alzheimers/techreport/.
21. Lambert JC, Heath S, Even G, Campion D, Sleegers K, Hiltunen M, et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet. 2009;41(10):1094–9. doi: 10.1038/ng.439 19734903
22. Rocca WA, Cha RH, Waring SC, Kokmen E. Incidence of dementia and Alzheimer’s disease: a reanalysis of data from Rochester, Minnesota, 1975–1984. Am J Epidemiol. 1998;148(1):51–62. 9663404
23. Letenneur L, Gilleron V, Commenges D, Helmer C, Orgogozo JM, Dartigues JF. Are sex and educational level independent predictors of dementia and Alzheimer’s disease? Incidence data from the PAQUID project. J Neurol Neurosurg Psychiatry. 1999;66(2):177–83. 10071096
24. Bonham LW, Geier EG, Fan CC, Leong JK, Besser L, Kukull WA, et al. Age-dependent effects of APOE epsilon4 in preclinical Alzheimer’s disease. Ann Clin Transl Neurol. 2016;3(9):668–77. doi: 10.1002/acn3.333 27648456
25. Bennett DA, Schneider JA, Arvanitakis Z, Wilson RS. Overview and findings from the Religious Orders Study. Curr Alzheimer Res. 2012;9(6):628–45. 22471860
26. Miech RA, Breitner JC, Zandi PP, Khachaturian AS, Anthony JC, Mayer L. Incidence of AD may decline in the early 90s for men, later for women: the Cache County study. Neurology. 2002;58(2):209–18. 11805246
27. Bachman DL, Wolf PA, Linn RT, Knoefel JE, Cobb JL, Belanger AJ, et al. Incidence of dementia and probable Alzheimer’s disease in a general population: the Framingham Study. Neurology. 1993;43(3 Pt 1):515–9.
28. Hofman A, Brusselle GG, Darwish Murad S, van Duijn CM, Franco OH, Goedegebure A, et al. The Rotterdam Study: 2016 objectives and design update. Eur J Epidemiol. 2015;30(8):661–708. doi: 10.1007/s10654-015-0082-x 26386597
29. Wu CC, Mungas D, Petkov CI, Eberling JL, Zrelak PA, Buonocore MH, et al. Brain structure and cognition in a community sample of elderly Latinos. Neurology. 2002;59(3):383–91. 12177372
30. Mungas D, Reed BR, Crane PK, Haan MN, Gonzalez H. Spanish and English Neuropsychological Assessment Scales (SENAS): further development and psychometric characteristics. Psychol Assess. 2004;16(4):347–59. doi: 10.1037/1040-3590.16.4.347 15584794
31. Beekly DL, Ramos EM, van Belle G, Deitrich W, Clark AD, Jacka ME, et al. The National Alzheimer’s Coordinating Center (NACC) database: an Alzheimer disease database. Alzheimer Dis Assoc Disord. 2004;18(4):270–7. 15592144
32. Farmer ME, White LR, Abbott RD, Kittner SJ, Kaplan E, Wolz MM, et al. Blood pressure and cognitive performance. The Framingham Study. Am J Epidemiol. 1987;126(6):1103–14. 3687920
33. Petersen RC, Smith GE, Waring SC, Ivnik RJ, Tangalos EG, Kokmen E. Mild cognitive impairment: clinical characterization and outcome. Arch Neurol. 1999;56(3):303–8. 10190820
34. Roth M, Tym E, Mountjoy CQ, Huppert FA, Hendrie H, Verma S, et al. CAMDEX. A standardised instrument for the diagnosis of mental disorder in the elderly with special reference to the early detection of dementia. Br J Psychiatry. 1986;149 : 698–709. 3790869
35. Haan MN, Mungas DM, Gonzalez HM, Ortiz TA, Acharya A, Jagust WJ. Prevalence of dementia in older latinos: the influence of type 2 diabetes mellitus, stroke and genetic factors. J Am Geriatr Soc. 2003;51(2):169–77. 12558712
36. Teng EL, Chui HC. The Modified Mini-Mental State (3MS) examination. J Clin Psychiatry. 1987;48(8):314–8. 3611032
37. Jorm AF, Jacomb PA. The Informant Questionnaire on Cognitive Decline in the Elderly (IQCODE): socio-demographic correlates, reliability, validity and some norms. Psychol Med. 1989;19(4):1015–22. 2594878
38. Jorm AF. The Informant Questionnaire on Cognitive Decline in the Elderly (IQCODE): a review. Int Psychogeriatr. 2004;16(3):275–93. 15559753
39. Ebly EM, Hogan DB, Parhad IM. Cognitive impairment in the nondemented elderly. Results from the Canadian Study of Health and Aging. Arch Neurol. 1995;52(6):612–9. 7763211
40. Morris JC, Weintraub S, Chui HC, Cummings J, Decarli C, Ferris S, et al. The Uniform Data Set (UDS): clinical and cognitive variables and descriptive data from Alzheimer Disease Centers. Alzheimer Dis Assoc Disord. 2006;20(4):210–6. doi: 10.1097/01.wad.0000213865.09806.92 17132964
41. Hughes CP, Berg L, Danziger WL, Coben LA, Martin RL. A new clinical scale for the staging of dementia. Br J Psychiatry. 1982;140 : 566–72. 7104545
42. Weintraub S, Salmon D, Mercaldo N, Ferris S, Graff-Radford NR, Chui H, et al. The Alzheimer’s Disease Centers’ Uniform Data Set (UDS): the neuropsychologic test battery. Alzheimer Dis Assoc Disord. 2009;23(2):91–101. doi: 10.1097/WAD.0b013e318191c7dd 19474567
43. D’Agostino RB, Wolf PA, Belanger AJ, Kannel WB. Stroke risk profile: adjustment for antihypertensive medication. The Framingham Study. Stroke. 1994;25(1):40–3. 8266381
44. Goff DC Jr, Lloyd-Jones DM, Bennett G, Coady S, D’Agostino RB, Gibbons R, et al. 2013 ACC/AHA guideline on the assessment of cardiovascular risk: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2014;129(25 Suppl 2):S49–73.
45. Thomsen T. HeartScore: a new web-based approach to European cardiovascular disease risk management. Eur J Cardiovasc Prev Rehabil. 2005;12(5):424–6. 16210927
46. Gray RJ. A class of K-sample tests for comparing the cumulative incidence of a competing risk. Ann Stat. 1988;16 : 1141–54.
47. Putter H, Fiocco M, Geskus RB. Tutorial in biostatistics: competing risks and multi-state models. Stat Med. 2007;26(11):2389–430. doi: 10.1002/sim.2712 17031868
48. R Core Team. R: a language and environment for statistical computing. Vienna: R Foundation for Statistical Computing; 2015.
49. Lin DY. Non-parametric inference for cumulative incidence functions in competing risks studies. Stat Med. 1997;16(8):901–10. 9160487
50. Fine JP, Gray RJ. A proportional hazards model for the subdistribution of a competing risk. J Am Stat Soc. 1999;94 : 496–509.
51. Prentice RL, Kalbfleisch JD, Peterson AV Jr, Flournoy N, Farewell VT, Breslow NE. The analysis of failure times in the presence of competing risks. Biometrics. 1978;34(4):541–54. 373811
52. Haller B, Schmidt G, Ulm K. Applying competing risks regression models: an overview. Lifetime Data Anal. 2013;19(1):33–58. doi: 10.1007/s10985-012-9230-8 23010807
53. DerSimonian R, Laird N. Meta-analysis in clinical trials. Control Clin Trials. 1986;7(3):177–88. 3802833
54. Andersen PK, Geskus RB, de Witte T, Putter H. Competing risks in epidemiology: possibilities and pitfalls. Int J Epidemiol. 2012;41(3):861–70. doi: 10.1093/ije/dyr213 22253319
55. Corrada M, Brookmeyer R, Kawas C. Sources of variability in prevalence rates of Alzheimer’s disease. Int J Epidemiol. 1995;24(5):1000–5. 8557432
56. Kochan NA, Slavin MJ, Brodaty H, Crawford JD, Trollor JN, Draper B, et al. Effect of different impairment criteria on prevalence of “objective” mild cognitive impairment in a community sample. Am J Geriatr Psychiatry. 2010;18(8):711–22. 21491632
57. Jarvik G, Larson EB, Goddard K, Schellenberg GD, Wijsman EM. Influence of apolipoprotein E genotype on the transmission of Alzheimer disease in a community-based sample. Am J Hum Genet. 1996;58(1):191–200. 8554056
58. Huang W, Qiu C, von Strauss E, Winblad B, Fratiglioni L. APOE genotype, family history of dementia, and Alzheimer disease risk: a 6-year follow-up study. Arch Neurol. 2004;61(12):1930–4. doi: 10.1001/archneur.61.12.1930 15596614
59. van Oijen M, de Jong FJ, Hofman A, Koudstaal PJ, Breteler MM. Subjective memory complaints, education, and risk of Alzheimer’s disease. Alzheimers Dement. 2007;3(2):92–7. doi: 10.1016/j.jalz.2007.01.011 19595922
60. Dickerson BC, Sperling RA, Hyman BT, Albert MS, Blacker D. Clinical prediction of Alzheimer disease dementia across the spectrum of mild cognitive impairment. Arch Gen Psychiatry. 2007;64(12):1443–50. doi: 10.1001/archpsyc.64.12.1443 18056553
61. Blacker D, Lee H, Muzikansky A, Martin EC, Tanzi R, McArdle JJ, et al. Neuropsychological measures in normal individuals that predict subsequent cognitive decline. Arch Neurol. 2007;64(6):862–71. doi: 10.1001/archneur.64.6.862 17562935
62. Knopman DS, Beiser A, Machulda MM, Fields J, Roberts RO, Pankratz VS, et al. Spectrum of cognition short of dementia: Framingham Heart Study and Mayo Clinic Study of Aging. Neurology. 2015;85(19):1712–21. doi: 10.1212/WNL.0000000000002100 26453643
63. Blacker D, Haines JL, Rodes L, Terwedow H, Go RC, Harrell LE, et al. ApoE-4 and age at onset of Alzheimer’s disease: the NIMH genetics initiative. Neurology. 1997;48(1):139–47. 9008509
64. Williams RL. Product-limit survival functions with correlated survival times. Lifetime Data Anal. 1995;1(2):171–86. 9385099
65. Andersen K, Launer LJ, Dewey ME, Letenneur L, Ott A, Copeland JR, et al. Gender differences in the incidence of AD and vascular dementia: the EURODEM Studies. EURODEM Incidence Research Group. Neurology. 1999;53(9):1992–7. 10599770
66. Gao S, Hendrie HC, Hall KS, Hui S. The relationships between age, sex, and the incidence of dementia and Alzheimer disease: a meta-analysis. Arch Gen Psychiatry. 1998;55(9):809–15. 9736007
67. Mielke MM, Vemuri P, Rocca WA. Clinical epidemiology of Alzheimer’s disease: assessing sex and gender differences. Clin Epidemiol. 2014;6 : 37–48. doi: 10.2147/CLEP.S37929 24470773
68. Payami H, Zareparsi S, Montee KR, Sexton GJ, Kaye JA, Bird TD, et al. Gender difference in apolipoprotein E-associated risk for familial Alzheimer disease: a possible clue to the higher incidence of Alzheimer disease in women. Am J Hum Genet. 1996;58(4):803–11. 8644745
69. Fagerlin A, Zikmund-Fisher BJ, Ubel PA. Helping patients decide: ten steps to better risk communication. J Natl Cancer Inst. 2011;103(19):1436–43. doi: 10.1093/jnci/djr318 21931068
70. Lautenbach DM, Christensen KD, Sparks JA, Green RC. Communicating genetic risk information for common disorders in the era of genomic medicine. Annu Rev Genomics Hum Genet. 2013;14 : 491–513. doi: 10.1146/annurev-genom-092010-110722 24003856
Štítky
Interní lékařstvíČlánek vyšel v časopise
PLOS Medicine
2017 Číslo 3
- Proces hojení ran krok za krokem a co ho může zkomplikovat
- Limity glykovaného hemoglobinu a význam dalších glykovaných proteinů
- Korelace dávky levothyroxinu s titrem autoimunitních protilátek u primární hypotyreózy
- Plicní hypertenze – syndrom mnoha tváří – vyžaduje přesnou diagnostiku a specializovanou léčbu
- Rána vizitkou (nejen) chirurga
Nejčtenější v tomto čísle
- Effectiveness of an intervention to facilitate prompt referral to memory clinics in the United Kingdom: Cluster randomised controlled trial
- , , and mutations in early-onset Alzheimer disease: A genetic screening study of familial and sporadic cases
- Challenges and opportunities in understanding dementia and delirium in the acute hospital
- Early diagnosis of mild cognitive impairment and mild dementia through basic and instrumental activities of daily living: Development of a new evaluation tool
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy