
Darierova choroba: současný pohled. Část II.
Darier Disease: Current View. Part II.
Darier disease is a rare genetic disorder, which is greatly influenced by the environmental factors. Apart from the skin involvement the disease could be accompanied by a range of certain complications and comorbidities.
The author presents a comprehensive review of this disease. First part summarizes history, epidemiology, course, prognosis, clinical presentation including its extracutaneous involvement, segmental Darier disease, atypical variants, dermoscopy, histopathology, complications and comorbidities. Second part thoroughly covers etiopathogenesis, differential diagnostics and current therapeutic options. The review also deals with an allelic disease called acrokeratosis verruciformis Hopfi.
Causal treatment of this genodermatosis is not known to date, nevertheless the deteriorated qualilty of patient’s life could be highly improved by lifestyle changes and by setting of the accessible treatment.
Keywords:
Darier disease – histopathology – differential diagnosis – therapy
Autoři:
M. Důra; J. Štork
Působiště autorů:
Dermatovenerologická klinika 1. LF UK a VFN, Praha, přednosta prof. MUDr. Jiří Štork, CSc.
Vyšlo v časopise:
Čes-slov Derm, 95, 2020, No. 3, p. 87-98
Kategorie:
Souborné referáty (doškolování lékařů)
Souhrn
Darierova choroba je vzácné, geneticky podmíněné onemocnění, které je silně ovlivňováno faktory vnějšího prostředí. Kromě dominujícího kožního postižení může být choroba doprovázena řadou komplikací a komorbidit.
Autor předkládá ucelený pohled na problematiku tohoto onemocnění. První část shrnuje historii, epidemiologii, průběh, prognózu, klinický obraz včetně mimokožního postižení, segmentální Darierovu chorobu, atypické varianty, dermatoskopii, histopatologický obraz, komplikace a komorbidity. Druhá část zevrubně pojednává o etiopatogenezi, diferenciální diagnostice a současných terapeutických možnostech. Článek též pojednává o alelické chorobě acrokeratosis verruciformis Hopfi.
Kauzální léčba této genodermatózy dosud není známa, avšak při vhodných režimových opatřeních a nastavení dostupné terapie může dojít k výraznému zlepšení kvality života, která je u pacientů vzhledem k charakteru onemocnění snížena.
Klíčová slova:
Darierova choroba – histopatologie – diferenciální diagnostika – terapie
ETIOPATOGENEZE
Molekulárně-biologický základ
Kauzálním genem v etiologii Darierovy choroby je gen ATP2A2, který leží na dlouhém raménku chromozomu 12, v lokusu 12q24.1. Kauzální gen byl identifikován v roce 1999, ale již v roce 1993 byl poprvé za kandidátní úsek označen lokus 12q23-24.1 [9, 49].
Gen ATP2A2, jehož délka činí 64 kb, má celkově 21 exonů. Je jím kódován protein SERCA2 – Ca2+-ATPáza sarkoplazmatického a endoplazmatického retikula. Jedná se tedy o ATP dependentní kalciovou pumpu zakotvenou v membráně endoplazmatického retikula (ER), která za pomoci energie získané při hydrolýze jedné molekuly ATP transportuje dva ionty Ca2+ z cytosolu do lumina ER.
Terciární proteinová struktura SERCA2 má tři části – transmembránovou, cytoplazmatickou a spojovací [28]. Transmembránová část (M doména) je podle sestřihové varianty proteinu (viz dále) tvořena deseti či jedenácti alfa-helikálními úseky (M1-M10/11), které prochází membránou ER (obr. 1). Úseky M1, M5, M6 a M8 se přímo účastní transportu Ca2+ do ER.
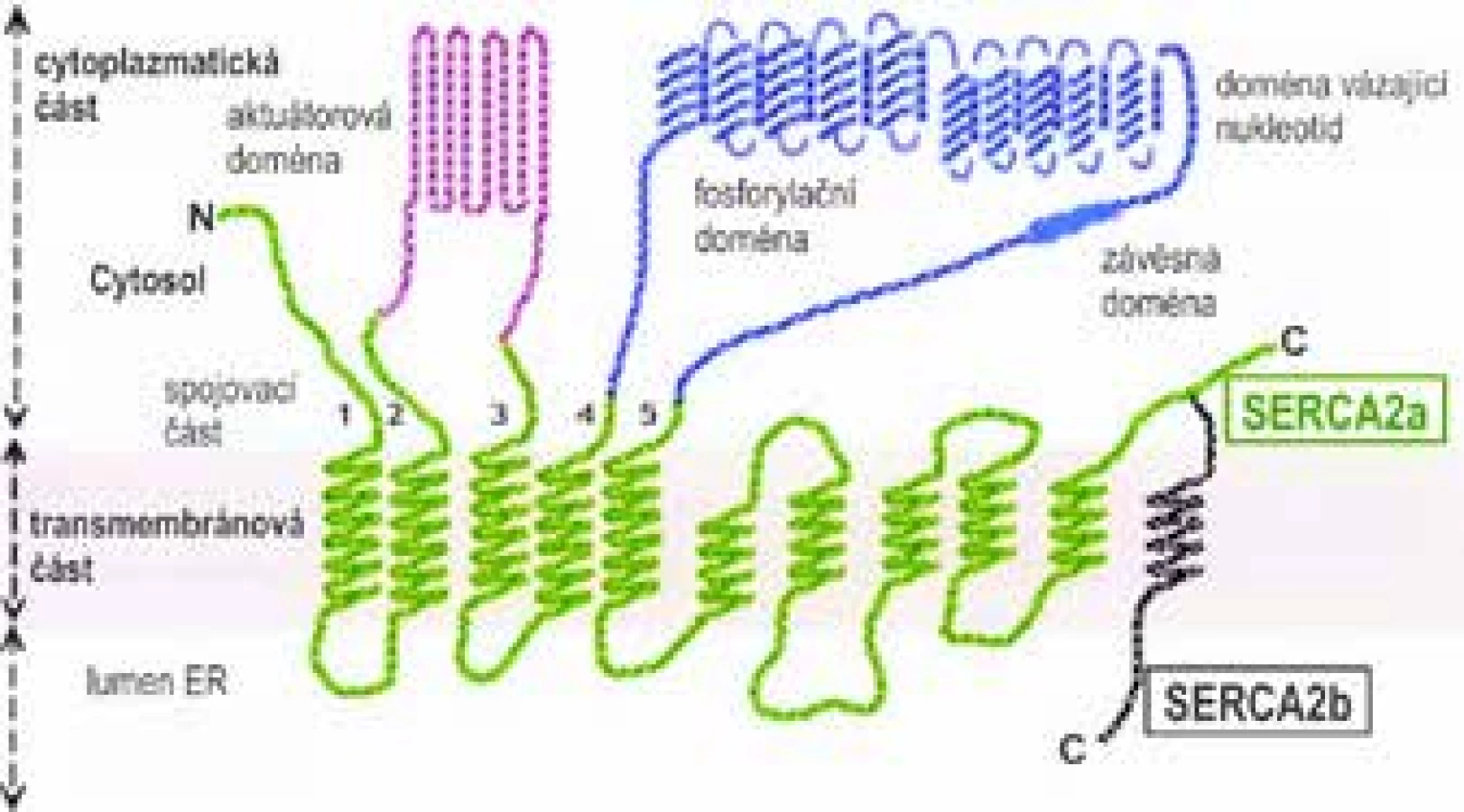
Největší cytoplazmatická část obsahuje tři globulární domény – fosforylační doménu (P doménu), doménu vázající nukleotid ATP (N doménu) a tzv. actuator doménu (A doménu, též zvanou β-strand domain). A doména je zodpovědná za následnou defosforylaci P domény. V primární struktuře proteinu leží sekvence aminokyselin pro A doménu mezi úseky M2S2 a M3S3 a pro doménu N a P mezi úseky M4S4 a M5S5. Aktivní centrum enzymu tvořené N a P doménou váže 2 ionty Mg2+ nutné k vlastní hydrolýze ATP. Aktivita SERCA2 je zpětnovazebně regulována hladinou Ca2+ v luminu ER.
Spojovací část (stalk region) připojuje transmembránovou část k cytoplazmatické, obsahuje pět alfa-helikálních úseků (S1-S5), které jsou prakticky pokračováním úseků M1-M5. Tzv. hinge domain připojuje N doménu cytoplazmatické části proteinu k úseku S5 spojovací části.
Díky alternativnímu sestřihu exonu 21 na 3´ konci primárního transkriptu SERCA2 vznikají čtyři dosud popsané enzymové izoformy – SERCA2a, SERCA2b, SERCA2c, SERCA2d, které se odlišují variabilitou v C-terminální transmembránové části SERCA2. S výjimkou SERCA2b jsou všechny izoformy tkáňově specifické, jsou exprimovány především ve svalových tkáních.
Izoforma SERCA2b (molekulová hmotnost 115 kDa, 1042 aminokyselin) není tkáňově specifická, je exprimována v řadě buněčných linií včetně keratinocytů a neuronálních buněk – zejména v pyramidálních buňkách hipokampu a v Purkyňových buňkách mozečku. Tento fakt podporuje pleiotropní hypotézu vzniku neuropsychiatrických komorbidit Darierovy choroby (viz kapitola Komorbidity v 1. části sdělení).
Etiopatogeneze Darierovy choroby
Darierova choroba (OMIM 124200) je geneticky podmíněné monogenní onemocnění způsobené mutací v genu ATP2A2 s autozomálně dominantním typem dědičnosti. Mutace je tedy v 50 % přenášena na potomka bez závislosti na pohlaví.
Dědičnost Darierovy choroby vykazuje vysokou penetranci (95 %), avšak silně variabilní expresivitu. U drtivé většiny nositelů mutace se objeví příznaky choroby, avšak jejich závažnost nese významné interindividuální rozdíly. Sporadické případy zaujímají 47 %, příčinou je zejména de novo mutace, vzácně může mít vliv i neúplná penetrance či neodhalení subklinických příznaků choroby u rodinných příslušníků [31].
Genetická analýza není pro diagnózu Darierovy choroby nutná. Preimplantační a prenatální genetická diagnostika je možná v případě úspěšné identifikace rodičovské mutace. Její interpretace je však problematická pro extrémně variabilní expresivitu téže mutace.
Mutace v genu ATP2A2 má za následek snížení či ztrátu funkce genového produktu („loss of function mutation“). Mutace v jedné alele postačuje k rozvoji klinických příznaků, jelikož jedna funkční alela genu nedostačuje k tvorbě dostatečného množství funkčního enzymu, dochází k tzv. haploinsuficienci. V případě některých mutací byla prokázána i inhibice plně funkčního enzymu SERCA2b interakcí s aberantním SERCA2b, kromě haploinsuficience tedy může být na vině i tzv. dominantně negativní efekt mutované alely.
Identifikováno bylo dosud více než 400 různých genových mutací v průběhu celého genu ATP2A2. Úseky se zvýšeným výskytem mutací („hotspots“) nebyly nalezeny. Identifikované typy mutací byly rozděleny do dvou skupin – mutace pravděpodobně narušující gen („likely gene disrupting“, LGD) a mutace protein alterující („protein altering“, PA) [29].
Při LGD mutaci dochází ke zkrácení genového produktu díky vzniku předčasného terminačního kodónu (PTC), případně transkripce není započata vůbec. Řadí se sem delece či inzerce s posunem čtecího rámce („frameshift mutation“), mutace odkrývající kryptická místa pro sestřih („splice site mutation“), nonsense mutace se vznikem terminačního kodonu („in-frame PTC mutation“) a mutace v iniciačním kodonu znemožňující začátek transkripce („loss of start codon mutation“).
PA mutace mají za následek zařazení chybné aminokyseliny bez zkrácení genového produktu, patří sem mis-sense mutace a inzerce či delece bez posunu čtecího rámce („in-frame mutation“).
Nejčastějším typem je missense mutace, tedy nesynonymní bodová substituce. Mutace jsou často unikátní pro jednu rodinu. Všechny dosud nalezené mutace jsou shromážděny v databázi Leiden Open Variation Database (LOVD)1.
1 https://databases.lovd.nl/shared/genes/ATP2A2
Ve 12–40 % případů se však žádnou mutaci nalézt nepodaří. Příčinou může být mutace v oblasti promotoru či v regulačních netranslatovaných oblastech, delece rozsáhlejšího úseku genu (např. celého exonu) či dysfunkce některého z transkripčních faktorů (např. Sp1), na kterém závisí exprese SERCA2 [31].
Výzkumy zabývající se genotypicko-fenotypickou korelací dosud nepřinesly přesvědčivé výsledky. I v rámci jedné rodiny mohou mít nositelé identické mutace velmi odlišný fenotyp. Existence fixní genotypicko-fenotypické korelace se však ani nepředpokládá zejména proto, že funkce SERCA2 je snížena při jakékoli patogenní mutaci. Bylo však zjištěno, že nositelé LGD mutací mají poněkud vyšší riziko vzniku závažných forem neuropsychiatrických komorbidit, tyto mutace byly častěji lokalizovány v úseku M4S4 [17]. U několika nepříbuzných rodin s výskytem akrálně hemoragického typu onemocnění byla nalezena missense mutace v exonu 15 (Asn767Ser), pozdější analýzy však tuto korelaci vyvrátily nálezem mutací v jiných úsecích [37].
Při mutaci s posunem čtecího rámce („frameshift“) dojde ke vzniku PTC a k ukončení transkripce. Vzniknuvší defektní mRNA je zčásti zlikvidována procesem „nonsense-mediated mRNA decay (NMD)”, zčásti je translatován defektní enzym SERCA2b. Ten je následně polyubikvitinován, avšak k jeho degradaci v proteazomu nedochází. Shlukuje se v nerozpustné agregáty (tzv. aggresom) v luminu ER. Přítomnost defektně sbaleného proteinu a deplece Ca2+ v ER způsobují stresovou odpověď ER dvojího typu – „unfolded protein response (UPR)“ a „ER overload response“. Zároveň dochází ke snížení exprese anti - apoptotických faktorů Bcl-2 a Bcl-XL. Následkem těchto pochodů dochází k apoptóze buňky.
Samotná oscilace hladiny intracelulárního Ca2+ je elementárním procesem nutným pro buněčnou proliferaci a diferenciaci. Porušená hladina Ca2+ má pravděpodobně za následek narušení fosforylační signální kaskády a narušení exprese transkripčních faktorů. Nízká hladina Ca2+ v ER má za následek kompenzatorní zvýšenou expresi Ca2+-ATPázy SPCA1 v Golgiho aparátu a nespecifického Na2+/Ca2+ iontového kanálu TRPC1 v plazmatické membráně.
Imunohistochemická studie prokázala aberantní předčasnou expresi involukrinu (markeru terminální keratinizace) v keratinocytech [25].
V patogenezi Darierovy choroby se dále uplatňuje dysfunkce mezibuněčných spojů, čímž se vysvětluje vznik akantolýzy a následné apoptózy keratinocytů. Apoptóza buňky z důvodu ztráty mezibuněčné adheze se též označuje jako anoikis.
Inhibice SERCA2 in vitro prokázala poruchu asemblace desmozómů, konkrétně redukci transportu desmozomálních cadherinů (desmogleinů a desmocollinu) a adaptorových proteinů (desmoplakinu, plakophilinu a plakoglobinu) k plazmatické membráně [21]. Dysfunkce desmozómů s sebou nese současnou deterioraci cytoskeletu, jelikož pomocí desmoplakinu jsou k desmozómu ukotvena cytokeratinová tonofilamenta.
Mislokalizace proteinů byla zjištěna imunofluorescenčně i v případě adhezních spojů (zonulae adherentes, adherens junctions), konkrétně E-cadherinů (transmembránových Ca-dependentních epiteliálních adherinů), cateninů a vinculinu [14]. Zonulae adherentes tvoří mezibuněčné spoje mezi keratinocyty navzájem a mezi keratinocyty a melanocyty, tvořící tak tzv. epidermální melaninovou jednotku. Při jejich dysfunkci dochází k poruše tvorby dendritických výběžků melanocytů, k nedostatečnému transportu melanozomů do keratinocytů a ve finále k apoptóze melanocytu. Na tomto faktu je postavena hypotéza vzniku varianty gutátní leukodermie, viz 1. část sdělení [16]. Tuto teorii podporuje nález patologické akumulace melanozomů v dendritických výběžcích melanocytů v ultrastrukturálním obrazu gutátní leukodermie [15].
V případě těsných spojení (zonulae occludentes, tight junctions) ve stratum granulosum nebyla u Darierovy choroby zjištěna signifikantní instabilita.
Negativní vliv slunečního záření je vysvětlován zvýšenou expresí COX-2 (cyklooxygenázy 2) vyvolanou UVB. Následná zvýšená produkce prostaglandinu E2 (PGE2) způsobuje redukci exprese SERCA2 [24].
Etiopatogeneze segmentální Darierovy choroby
Podstatou segmentální Darierovy choroby 1. a 2. typu je genomový mozaicismus. Genetická podstata segmentalismu 1. typu byla vysvětlena v roce 2000 [48]. Hypotéza vzniku segmentalismu 2. typu byla genetickou analýzou potvrzena nejprve u m. Hailey-Hailey v roce 2004, u Darierovy choroby v roce 2012 [13].
Segmentální Darierova choroba 1. typu vzniká somatickou postzygotickou mutací v genu ATP2A2 v časných fázích embryogeneze. Výsledkem je mozaika dvou buněčných linií – mutovaných a nemutovaných, přičemž v mutovaných buňkách se v budoucnu projeví příznaky Darierovy choroby. Experimentálně bylo zjištěno, že k segmentálnímu postižení dochází tehdy, pokud je postiženo více než cca 30 % buněk v dané lokalitě [20]. Rodinná anamnéza je u segmentální formy 1. typu zpravidla negativní.
Zastoupení buněčných typů nesoucích mutaci se odvíjí od momentu v průběhu embryogeneze, při kterém došlo k mutaci. Došlo-li k mutaci před rozdělením primitivního ektodermu (cca 14. den), bude mutace obsažena i v prekurzorech hematopoézy či gametogeneze [20]. V případě, že mozaicismus postihne část či veškeré gamety (tzv. gonozomální mozaicismus), mohou být potomci pacienta se segmentální Darierovou chorobou postiženi jejím klasickým typem.
Segmentální Darierova choroba 2. typu vzniká tehdy, pokud v embryu již nesoucím germinální heterozygotickou mutaci v genu ATP2A2 vznikne na druhé alele somatická, postzygotická mutace, čímž vzniká druhý zásah („second hit“) se ztrátou heterozygozity („loss of heterozygozity“, LOH). Postzygotická mutace může být typu delece (za vzniku hemizygotické buněčné linie), substituce (za vzniku homozygotické či složené heterozygotické linie), či mutace vzniká při somatické rekombinaci. Postzygotickou mutací vzniknou v embryu opět dvě buněčné linie, z nichž jedna linie nese jednu mutaci a v budoucnu se projeví příznaky klasické Darierovy choroby. Druhá buněčná linie, zasažena dvěma mutacemi, vytvoří závažnější příznaky choroby s lineárním uspořádáním podél Blaschkových linií.
Extrémně vzácný subtyp segmentalismu 2. typu, při němž se v oblasti superponovaného lineárního postižení střídají pruhy těžce postižené a zcela nepostižené kůže, vzniká na základě tzv. alelického „twin spot“ fenoménu (didymosis). Ten vzniká tehdy, dojde-li v průběhu vývoje heterozygotického embrya nesoucí germinální mutaci k fokální postzygotické somatické mitotické homologní rekombinaci sesterských chromatid. Při segregaci rekombinovaných alel vznikají v tomto místě dvě nové buněčné linie, z nichž obě jsou homozygotické – jedna nese mutaci na obou alelách a jedna nenese žádnou mutaci. Touto teorií je vysvětlen vznik linií těžce postižené a zcela nepostižené kůže [23, 45, 48].
Etiopatogeneze acrokeratosis verruciformis Hopfi
V roce 2003 byla u AKV poprvé identifikována heterozygotická mutace v identickém genu ATP2A2 [10]. Navíc u všech dosud diagnostikovaných rodin se objevila stejná missense mutace Pro602Leu v N doméně, přičemž tato mutace nebyla dosud nalezena u žádného pacienta s Darierovou chorobou [46]. V případě sporadické AKV byla identifikována missense mutace Ala698Val [5]. U některých nemocných byla genetická analýza nekonkluzivní. Genetická analýza však byla dosud provedena jen na velmi limitovaném počtu pacientů.
I přes diametrálně odlišný fenotyp je dnes toto onemocnění považováno za alelickou variantu Darierovy choroby. Příčina této diskrepance nebyla na molekulárně-genetické úrovni dosud uspokojivě objasněna.
DIFERENCIÁLNÍ DIAGNOSTIKA
Diferenciální diagnostika postižení kůže a kožních adnex
V diferenciální diagnostice Darierovy choroby stojí zejména dvě akantolytické neautoimunní dermatózy – morbus Hailey-Hailey a morbus Grover. Tyto choroby mohou napodobovat Darierovu chorobu jak klinicky, tak histopatologicky.
Morbus Hailey-Hailey (benigní chronický familiární pemfigus, OMIM 169600) byl poprvé popsán bratry Howardem a Hughem Haileyovými v roce 1939 [19]. Jedná se o genodermatózu s autozomálně dominantním typem dědičnosti, s téměř kompletní penetrancí a variabilní expresivitou. Nástup prvních příznaků se pohybuje mezi 15 a 40 roky. Primárním projevem je plihá vezikula, která rychle praská s následným vznikem erozí, krust a mokvání. Projevy mají tendenci šířit se centrifugálně. Výsev je typicky limitován do oblasti krku a flexur a je typický svými obdobími exacerbací a remisí. Hojení probíhá bez jizvení s případnými posuny pigmentu. Postižení nehtů podélnými bělavými proužky se vyskytuje asi u 75 % nemocných.
Popsány byly varianty papulózní, verukózní, anulární, vezikulobulózní i segmentální 1. a 2. typu. S Darierovou chorobou je m. Hailey-Hailey úzce spjat zejména kvůli příbuznému kauzálnímu genu ATP2C1 ležícímu na dlouhém raménku chromozomu 3, v lokusu 3q22.1. Jeho produktem je enzym SPCA1 – Ca2+/Mn2+ ATPáza v membráně Golgiho aparátu (secretory pathway Ca2+/Mn2+ ATPase).
V histologickém obrazu m. Hailey-Hailey výrazně převažuje akantolýza nad dyskeratózou. Corps ronds jsou nečetné, grains jsou zcela vzácné, z tohoto důvodu patří tato choroba do skupiny akantolytických dyskeratóz pouze okrajově. Akantolýza postihuje celou šíři epidermis, nemá suprabazální charakter, přirovnávána je k obrazu „rozpadající se zdi“. Porovnání Darierovy choroby a morbus Hailey-Hailey shrnuje tabulka 1.

Papulózní akantolytická dyskeratóza (akantolytická dermatóza vulvokrurální či anogenitální oblasti) je extrémně vzácná jednotka, která postihuje zejména dospělé ženy. Projevuje s výsevem svědivých papul v anogenitální a tříselné oblasti, výjimečně je postižen i hrudník. Histologicky nacházíme akantolytickou dyskeratózu a hyperkeratózu s parakeratózou. Na základě několika nálezů mutace v genu ATP2C1 je dnes onemocnění považováno za alelickou variantu m. Hailey-Hailey [55].
Groverova choroba (přechodná akantolytická dermatóza) je poměrně časté výrazně svědivé onemocnění zejména mužů středního věku. Poprvé bylo popsáno Ralphem Groverem v roce 1970 [18]. V oblasti hrudníku a horní poloviny zad se objevují mnohočetné drobné izolované papuly či papulovezikuly. Projevy mohou ustoupit po týdnech či měsících trvání (proto přechodná čili tranzientní dermatóza), mohou však přetrvávat i roky. U Groverovy choroby bylo taktéž popsáno lokalizované postižení, mimokožní projevy v dutině ústní, výskyt současných přechodných hypopigmentovaných makul či exacerbace choroby slunečním zářením. Histologicky je možno u Groverovy choroby rozlišit 4 varianty podle jejich podobnosti s jinými dermatózami: varianta podobná m. Darier, podobná m. Hailey-Hailey, podobná pemphigus vulgaris a varianta spongiotická. Kromě poslední varianty je jejich společným jmenovatelem fokální akantolýza, vilózní charakter papilárního koria a variabilní počet dyskeratotických buněk. Etiologie je zcela nejasná, možná genetická příčina Groverovy choroby dosud nebyla potvrzena.
Pityriasis rubra pilaris Devergie napodobuje Darierovu chorobu splývajícím výsevem hyperkeratotických papul, které jsou však folikulárně vázané a splývají do erytematoskvamózních ploch. Projevy typicky zanechávají ostrovy nepostižené kůže (fr. nappes claires). Dlaně a plosky jsou postiženy plošnou keratodermií. Choroba často ústí v erytrodermii s postižením obličeje a vznikem ektropií víček. Histologicky nacházíme akantotickou epidermis s psoriaziformní hyperplazií a hyperkeratózu s přítomností typických „šachovnicových“ úseků parakeratózy, variabilně může být přítomna akantolýza.
Keratosis follicularis spinulosa decalvans je vzácné, Darierově chorobě nepříbuzné, geneticky podmíněné onemocnění vázané na chromozóm X. Projevuje se difuzní folikulární keratózou a jizvící alopecií postihující kštici a obočí s počátkem příznaků v útlém dětství.
Keratosis follicularis squamosa Dohi je extrémně vzácná porucha keratinizace neznámé etiologie, která byla hlášena téměř výlučně z Japonska. Projevuje se asymptomatickým výsevem hnědavých šupících se makul s centrální folikulárně vázanou papulkou. Charakteristický dermatoskopický nález je přirovnáván k lotosovým listům na vodní hladině [30].
Prurigo pigmentosa je vzácná choroba neznámé etiologie, jejíž klinický obraz může přesvědčivě imitovat Darierovu chorobu. Projevy se objevují ve formě pruritických splývajících papul prakticky pouze v centrální části trupu, v dekoltu a na krku zejména u žen mladšího věku. Projevy spontánně ustupují se zanecháním retikulárních pigmentací a často recidivují. Histologický obraz se odvíjí od stadia choroby, v plně vyvinuté fázi má obraz spongiotické dermatitidy.
Retikulární erytematózní mucinóza (REM syndrom) se objevuje zejména u žen středního věku v oblasti dekoltu a centrální časti zad. Histologicky jsou zánětlivé změny lokalizovány v koriu s hojnou přítomností mucinu pod nezměněnou epidermis.
Klinicky může Darierovu chorobu napodobovat scabies, zejména jeho hyperkeratotická varianta (scabies norvegica).
Vzácná genodermatóza Cowdenové syndrom (syndrom mnohočetných hamartomů) se díky výskytu papul na obličeji, punktátnímu palmoplantárnímu postižení, akrálním keratózám a papilomům dutiny ústní klinicky přibližuje Darierově chorobě v mnoha znacích. Papuly obličeje jsou však histologicky fibrózní papuly či trichilemomy. Jedná se o autozomálně dominantní poruchu způsobenou mutací v genu PTEN s celoživotně zvýšeným rizikem vzniku mnoha typů benigních a maligních tumorů.
Dyskeratosis congenita je vzácná genodermatóza typická triádou příznaků – retikulárními hyperpigmentacemi na krku a trupu, dystrofiemi nehtů a leukokeratózou v dutině ústní s případnou tvorbou bul. Samotný název choroby je však poněkud zavádějící, jelikož dyskeratotické změny nejsou histologicky přítomny. Na rozdíl od Darierovy choroby se první příznaky objevují dříve a dystrofie nehtů jsou těžšího stupně. Identifikováno bylo několik kauzálních genů, z nichž všechny jsou součástí tzv. telomerázového komplexu.
Pachyonychia congenita je další vzácnou genodermatózou s dominující těžkou dystrofií nehtů doprovázenou plantární bolestivou keratodermií. Dnes je choroba rozdělována do několika typů, z nichž všechny jsou způsobeny mutacemi v genech pro cytokeratiny.
Erythrokeratodermia variabilis (Mendes da Costa syndrom) je vzácný typ ichtyózy s výskytem figurátních erytematózních a hyperkeratotických plaků od útlého dětství. Flexury zde však bývají ušetřeny.
V diferenciální diagnostice Darierovy choroby stojí dále choroby typické výsevem hyperpigmentovaných makulopapul predilekčně ve flexurách – acanthosis nigricans, papillomatosis confluens et reticularis (m. Gougerot-Carteaud), genodermatózy ze skupiny retikulárních pigmentovaných poruch (retikulární pigmentovaná anomálie flexur – m. Dowling-Degos se svou akantolytickou variantou m. Galli-Galli) a tzv. terra firma-forme dermatóza (lat. terra firma – suchá země). Posledně jmenovaná dostala svůj název podle podobnosti hnědavých hyperkeratotických rozpolíčkovaných ložisek na trupu a krku se suchou rozpukanou zemí, choroba dobře reaguje na lokálně aplikovaný alkohol a jeho efekt je i diagnostickým testem.
Postižení kůže u dětí s histiocytózou z Langerhansových buněk může díky distribucí lézí v seboroické lokalizaci napodobovat Darierovu chorobu s časným nástupem.
Postižení dlaní a plosek imituje skupinu získaných či vrozených palmoplantárních keratodermií (PPK). V případě výskytu punktátních jamek a papulek stojí v diferenciální diagnostice jejich specifická podskupina – punktátní PPK [52]. Na rozdíl od Darierovy choroby vykazují PPK většinou těžší postižení plosek nežli dlaní. Jamkovité punktátní hyperkeratózy dlaní a plosek jsou dále typické pro vzácnou genodermatózu Gorlin-Goltzův syndrom (syndrom névoidních bazaliomů) a výše zmíněný Cowdenové syndrom.
Postižení kůže při Darierově chorobě ve specifických lokalitách může napodobovat mnohá další kožní onemocnění. Postižení kštice imituje těžkou seboroickou dermatitidu. Zvláštní jednotkou v diferenciální diagnóze při postižení mamil a areol je tzv. névoidní hyperkeratóza, která se vyskytuje ve třech typech: jako součást epidermálního névu (první typ), jako součást jiného kožního onemocnění – typicky Darierovy choroby (druhý typ) či existuje izolovaně (třetí typ) [22]. Postižení mamil a areol je nutno též odlišit od mamární Pagetovy choroby. Vegetující ložiska na genitálu mohou imitovat condylomata accuminata či condylomata lata.
Postižení nehtů může napodobovat onychodystrofie z jiných příčin, ekzémovou onychopatii, onychomykózu, dermatofytom, dystrophia mediana canaliformis. Longitudinální erytronychii mohou vytvořit benigní tumory jako glomus tumor, onychopapilom, onychomatrikom či subunguální verukózní dyskeratom.
Diferenciální diagnostika postižení dutiny ústní
Nález v dutině ústní může připomínat stomatitis nicotinica, stomatitidu u uživatelů zubních protéz či intra - orální manifestaci lichen ruber planus.
Mezi geneticky podmíněná onemocnění v diferenciální diagnostice patří „white sponge nevus“ (Cannonova choroba), hereditární benigní intraepiteliální dyskeratóza (HBID) a intraorální příznaky dyskeratosis congenita a pachyonychia congenita. Ve všech těchto případech se tvoří bělavé plaky zejména na bukálních sliznicích a jazyku, příchod prvních příznaků bývá časnější, v případě HBID je postižena navíc spojivka.
Za zmínku ještě stojí multifokální epiteliální hyperplazie (Heckova choroba) projevující se mnohočetnými plošnými papilomy v dutině ústní vznikajícími od dětství. Souvisí s infekcí specifickými typy HPV.
Diferenciální diagnostika segmentální Darierovy choroby
V diferenciální diagnostice 1. typu segmentální Darierovy choroby figurují zejména epidermální névy, lichen striatus, lineární porokeratóza a segmentální m. Hailey-Hailey 1. typu, další jednotky jsou vzácné.
Dnes je rozlišováno na 10 subtypů epidermálních névů. V diferenciální diagnostice Darierovy choroby figuruje zejména ILVEN (inflamatorní lineární verukózní epidermální névus) a ADEN (akantolytický dyskeratotický epidermální névus).
ILVEN vzniká záhy po narození nejčastěji na dolní končetině. Histologicky vykazuje psoriaziformní obraz. ADEN se svým klinickým i mikroskopickým obrazem téměř překrývá se segmentální Darierovou chorobou, nejčastěji je však kongenitální. ADEN taktéž není doprovázen postižením nehtů či sliznic. Ač je některými autory stále považován za svébytnou jednotku, jeho postavení dnes není přesně determinováno [3].
Lichen striatus vzniká podél Blaschkových linií u dětí nejčastěji mezi 2.–3. rokem, má vysokou tendenci k spontánním remisím v horizontu několika měsíců od jeho vzniku. Histologicky je přítomen obraz lichenoidní dermatitidy.
Lineární porokeratóza je vzácnou formou porokeratózy s blaschkolineárním uspořádáním, vznikající nejčastěji v dětství. Klinicky však nalézáme typický periferní keratotický lem a histologicky obraz kornoidní lamely.
Relabující lineární akantolytická dermatóza se projevuje lokalizovaným recidivujícím výsevem papul a vezikul s histologickým obrazem m. Hailey-Hailey. Postavení této choroby doposud nebylo ukotveno. Je pravděpodobné, že se ve skutečnosti jedná o segmentální m. Hailey-Hailey 2. typu.
Izolované blaschkolineární verukózní ložisko na kůži tlustého typu svědčí pro keratodermia palmoplantaris striata typ I-III podle kauzálního genu. Typ I, způsobený mutací v desmogleinu 1, vykazuje zároveň akantolytické změny bez současné dyskeratózy.
Diferenciální diagnostika atypických variant
Varianta gutátní leukodermie je klinicky prakticky identická s pozánětlivými hypopigmentacemi či idiopatickou gutátní hypomelanózou, která se však vyskytuje zejména na končetinách u starších jedinců. Gutátní hypopigmentace ve spojení s punktátní palmoplantární keratodermií tvoří vzácnou jednotku Coleova choroba s nově identifikovaným zodpovědným genem ENPP1. Při výskytu depigmentací na krku a v dekoltu je nutno zvážit leucoderma syphiliticum (Venušin náhrdelník). V diferenciální diagnostice dále figuruje nově popsaný typ vitiliga, tzv. folikulární vitiligo.
V diferenciální diagnostice komedonického typu Darierovy choroby stojí zejména komedonické a nodulocystické akné, chlorakné, adnexální tumory (např. syringomy), tzv. nodulární elastóza s cystami a komedony (Favre-Racouchotův syndrom, který však vzniká na chronicky solárně změněné kůži) a vzácná genodermatóza familiární dyskeratotické komedony. Familiární dyskeratotické komedony se vyznačují vznikem mnohočetných komedonů na trupu a končetinách od časného dětství s histologickým nálezem dyskeratózy. Dědičnost je autozomálně dominantní, avšak jejich genetická podstata dosud nebyla identifikována.
Vezikulobulózní variantu Darierovy choroby mohou s úspěchem imitovat některé klinicky podobné autoimunně podmíněné bulózní dermatózy. Mezi ně patří pemphigus foliaceus (který v pozdních stadiích navíc vykazuje histologický nález dyskeratózy v úrovni stratum granulosum) a pemphigus vegetans, zejména jeho závažnější, tzv. Neumannův typ.
Diferenciální diagnostika acrokeratosis verruciformis Hopfi
V diferenciální diagnostice AKV figurují seboroické veruky, verrucae planae, lichen ruber planus, epidermodysplasia verruciformis, stucco keratosis, superficiální aktinická porokeratóza, papulózní granuloma annulare, akrální perzistentní papulózní mucinóza, Kitamurova retikulární akropigmentace (která je též doprovázena palmoplantárními punktátními jamkami přerušujícími dermatoglyfy) a skupina marginálních keratodermií (acrokeratoelastoidosis a keratoelastoidosis marginalis).
TERAPIE
Kauzální léčba Darierovy choroby dosud není známa. V terapii se v současné době uplatňují režimová opatření, lokální a systémová léčba a sporadicky další modality léčby.
Režimová opatření
Vhodné je volné vzdušné oblečení (např. bavlna). Naopak nevhodné je oblečení z umělého neprodyšného materiálu, které zhoršuje pocení a znemožní jeho odpařování. Nedílnou součásti péče o kůži je ochrana před sluncem vhodným oděvem a přípravky na ochranu proti UV záření na fyzikální či chemické bázi, a to zejména proti složce UVB.
Vzhledem k možnému postižení slinných žláz a zvýšené kazivosti je nutná správně vedená ústní hygiena.
Lokální terapie
Základem lokální terapie je užívání vyzkoušených emoliencií a lokálních antiseptických přípravků k prevenci či k redukci mikrobiální kolonizace a současného zápachu, nejčastěji jsou na bázi chlorhexidinu, hexamidinu či ethakridin laktátu. Pro běžnou hygienu postačují antiseptická tekutá mýdla.
Ke zmírnění mokvání a snížení bakteriální nálože je možno též vyzkoušet odpařující vysýchavé obklady s antiseptickým a adstringentním účinkem, např. ze světle růžového roztoku hypermanganu či z Jarischova roztoku. V úvahu přichází i vyzkoušená adstringencia na bázi fytofarmak.
Při mikrobiálním zánětu mírnějšího stupně jsou na místě antibakteriální či antimykotická externa. Při užívání HVLP či IPLP antibiotických extern je potřeba mít na mysli jejich potenciální riziko indukce rezistence.
K redukci deskvamace je s výhodou použití keratolytických extern s obsahem kyseliny salicylové, urey či kyseliny mléčné. Jejich koncentrace by měla být v IPLP individualizovaná pro každého pacienta, aby bylo dosaženo maximálního účinku bez iritace či vzniku pálení.
Lokální kortikosteroidy jsou v terapii Darierovy choroby oblíbenou modalitou řadící je do první linie lokální léčby. Užívány jsou středně silné a silné kortikosteroidy. Jejich přínos je však ambivalentní, při jejich aplikaci dochází k regresi zánětlivých změn, může však dojít ke zvýšení mikrobiální kolonizace a k podpoře vzniku infekčních komplikací. Měly by proto být používány s rozvahou a po nezbytně krátkou dobu s jejich postupným vysazováním.
Prokázaného efektu dosahují lokální retinoidy, a to jak u diseminovaného, tak segmentálního postižení [11]. Z nearomatických retinoidů 1. generace je to tretinoin (all-trans-retinová kyselina), z polyaromatických retinoidů 3. generace pak adapalen a tazatoren, který není v ČR registrován. Lokální retinoidy mohou působit negativně svým iritačním potenciálem, zejména v úvodu terapie. V případě vzniku iritace se doporučuje modifikovat léčbu podle individuální snášenlivosti (aplikace obden či současná aplikace středně silných nebo silných lokálních kortikosteroidů).
V několika publikovaných případech byl zaznamenán efekt lokálně aplikovaného 5-fluorouracilu (5-FU) v 1% krému v monoterapii nebo v kombinaci s perorálními retinoidy či lokálními kortikosteroidy. Bezpečnost se ukázala být dobrou, i přes aplikaci na více než 20 % povrchu těla nebyly metabolity 5-FU v plazmě detekovatelné [51].
Efekt byl zaznamenán při lokální terapii kalcineurinovými inhibitory – takrolimem (který byl v 0,1% masti podáván u pacientky se současnou atopickou dermatitidou) a pimekrolimem [39, 47].
Efekt 3% diklofenaku v gelu či lokálního takalcitolu (derivátu vitaminu D3) popsaly pouze izolované kazuistiky [1, 36].
Systémová terapie
Zlatým standardem systémové léčby Darierovy choroby je perorální retinoidní terapie, při níž může dojít ke zlepšení v 70–90 % případů.
První kazuistika úspěšné celkové léčby vitaminem A se objevila již v roce 1941, díky čemuž byla v této době choroba považována za projev hypovitaminózy [38]. Léčba etretinátem byla poprvé užita v roce 1978 [34].
V léčbě se uplatňuje nearomatický retinoid 1. generace izotretinoin (13-cis-retinová kyselina) a monoaromatický retinoid 2. generace acitretin, jehož SPC (informace k dubnu 2020) indikaci v léčbě Darierovy choroby přímo udává.
Mechanismus účinku retinoidů není u Darierovy choroby zcela objasněn, roli hraje pravděpodobně jejich antiproliferační, protizánětlivý a antiseboroický účinek. Je známo, že izotretinoin vykazuje v porovnání s acitretinem silnější supresivní účinek na proliferaci seboblastů mazových žláz a na jejich funkci, čehož je využíváno zejména při léčbě závažného akné.
Celková léčba retinoidy by měla být vyhrazena pro pacienty se závažnějším stupněm onemocnění, u kterých byla dosavadní lokální léčba neúspěšná. Léčba retinoidy je většinou dlouhodobá trvající měsíce až roky podle její efektivity a individuální snášenlivosti. V některých případech se osvědčuje zahájení léčby cyklicky před letním obdobím, kdy je očekávána exacerbace projevů [41]. Je však potřeba mít na paměti latenci v nástupu efektu léčby v řádu několika, většinou 2–4 týdnů, stejně tak při vysazení je nutno předpokládat zhoršení stavu v horizontu několika týdnů. U plánovaných operačních výkonů se léčba retinoidy obvykle přechodně vysazuje.
Iniciální dávka acitretinu se empiricky udává nižší než v jiných indikacích, cca 10 mg denně s postupným navýšením na klasickou dávku přibližně 0,5 mg/kg/den, eskalací léčby je možné předejít iniciální exacerbaci onemocnění [4]. Iniciální dávkování izotretinoinu je taktéž doporučováno poněkud nižší, cca 0,2 mg/kg/den s postupným navýšením na cílovou dávku 0,5–1,0 mg/kg/den [32].
Vedení léčby retinoidy včetně Programu prevence početí (PPP) se neliší od léčby v jiných dermatologických indikacích. Eliminační poločas acitretinu je přibližně 50 hodin, více než 99 % acitretinu je eliminováno do 36 dnů od ukončení dlouhodobé léčby, acitretin se tedy ve tkáních neakumuluje. Avšak při současném požívání alkoholu během léčby může dojít k esterifikaci acitretinu na etretinát, který se v tukové tkáni akumuluje a jehož eliminační poločas je přibližně 120 dní. U izotretinoinu toto nebezpečí nehrozí, jeho eliminační poločas je pouze 19 hodin a k tvorbě akumulujících se metabolitů nedochází. Z tohoto důvodu je účinná antikoncepce nutná po dobu 3 let od ukončení léčby acitretinem, ale pouze 1 měsíc od ukončení léčby izotretinoinem.
Dalším retinoidem s publikovanou účinností v terapii Darierovy choroby je alitretinoin. Jedná se o endogenní nearomatický retinoid 1. generace (9-cis-retinová kyselina). Kompletní remise při perorální léčbě alitretinoinem v dávce 30 mg denně byla popsána v monoterapii či se současnou lokální terapií 5-fluorouracilu u několika případů [53, 57]. V České republice není perorální alitretinoin registrován, v zahraničí se používá k terapii ekzému rukou.
Celková terapie retinoidy je zatížena známými nežádoucími účinky. Kromě jiných je to vzácná fotosenzitivní reakce a indukce depresí, kterými jsou pacienti s Darierovou chorobou ve vyšší míře ohroženi.
Celková kortikoterapie není u Darierovy choroby obecně doporučována, jelikož může stav kůže paradoxně zhoršit a zvýšit vnímavost k infekcím.
Pouze ve dvou publikovaných případech byla použita perorální léčba doxycyklinem 100 mg denně s výborným efektem [40, 50]. Hypotéza efektu tetracyklinových antibiotik vychází z etiologie choroby. Tetracykliny jsou vysoce lipofilní molekuly, které též působí jako chelatační látky, tzn. že vážou dvojmocné ionty včetně Ca2+ či Mg2+. Těmito vlastnostmi by mohly působit jako ionofor umožňující přenos Ca2+ přes membránu a tím ovlivnit intracelulární kalciový metabolismus, který je u Darierovy choroby alterován. Tetracykliny dále vykazují protizánětlivé účinky inhibicí chemotaxe granulocytů a inhibicí matrix metaloproteinázy 9 (MMP-9). Svým širokospektrým antibiotickým účinkem mohou účinně bránit bakteriální kolonizaci či vzniku infekčních komplikací. Efekt doxycyklinu byl též recentně potvrzen u m. Hailey-Hailey.
V literatuře se dále objevují izolované kazuistiky efektivní perorální terapie cyklosporinem, hormonální antikoncepcí (která může přinést úlevu u pacientek trpících exacerbacemi v závislosti na menstruačním cyklu) a magnéziem ve formě MgCl2 v dávce 300 mg denně [33]. Nízkodávkový naltrexon (5 mg denně), který byl s úspěchem vyzkoušen u m. Hailey-Hailey, přinesl u Darierovy choroby rozporuplné závěry [6]. Zaznamenáno bylo zlepšení kožních projevů při léčbě metotrexátem u pacienta se současným Sjögrenovým syndromem [54].
Sekundární bakteriální infekce si mohou vyžádat léčbu perorálními antibiotiky s dobrým průnikem do kůže (aminopeniciliny, klindamycin, doxycyklin, cefalosporiny). Jejich výběr se řídí výsledkem kultivačního vyšetření se stanovením citlivosti. Je potřeba mít na paměti interakci retinoidů a tetracyklinových antibiotik pro nebezpečí vzniku nitrolební hypertenze. K terapii či profylaxi recidivujících herpetických infekcí je užíván acyklovir, valacyklovir či v České republice dlouhodobě nedostupný brivudin [2]. V těžkých případech si terapie vyžádá parenterální antivirotickou terapii.
Úlevu od svědění mohou přinést systémově podaná antihistaminika. Jejich efekt však u Darierovy choroby dosud nebyl systematicky studován. Wang et al. (2019) udávají efekt cetirizinu v léčbě vezikulobulózní varianty, autoři zvažují potenciální roli eozinofilů v patogenezi vezikulobulózní Darierovy choroby [56].
Další modality léčby
Léčba chirurgická figuruje jako radikální možnost léčby izolovaných ložisek refrakterních na konvenční léčbu, hodí se zejména pro hyperkeratotický typ či segmentální Darierovu chorobu 1. typu. Použita byla prostá excize s případným graftingem, shave excize, kyretáž či dermabraze, která je vhodná i pro bradavičnaté projevy typu acrokeratosis verruciformis. Z alternativních metod byla použita kryochirurgie a elektrochirurgie (konkrétně elektrodesikace).
V několika případech byl prokázán léčebný efekt laseroterapie, konkrétně ablativním frakcionovaným CO2 laserem, erbium:YAG laserem, neablativním 1 550 nm erbium doped fiber laserem či 595 nm pulzním barvivovým laserem, který byl vyzkoušen na dosud největším vzorku 8 pacientů [8, 26, 43]. V několika případech byla po ošetření laserem zaznamenána generalizovaná infekce HSV a parciální relaps v horizontu několika měsíců.
Existuje několik teorií léčebného efektu ablativních metod. Jsou založeny na faktu, že ablativní léčba je úspěšná pouze v případě, je-li odstraněno i papilární korium. Destrukce vývodů potních žláz s následnou redukcí pocení pravděpodobně eliminuje provokující faktor pocení. Tuto teorii podporuje fakt, že jizvy po předchozích chirurgických výkonech bývají chorobou nepostiženy [7]. Je dále pravděpodobné, že úspěch chirurgické léčby segmentálního postižení 1. typu tkví v regeneraci epidermis preferenčně keratinocyty nesoucími nemutovaný gen ATP2A2.
Ve studii s 5 pacienty byla vyzkoušena fotodynamická terapie (PDT), u všech pacientů došlo minimálně k parciální léčebné odpovědi s žádnou či minimální rekurencí [12]. Zaznamenáno bylo rapidní zhoršení při užití fotochemoterapie PUVA.
Léčebný efekt radioterapie byl poprvé zaznamenán náhodně – při zevní radioterapii karcinomu prsu v terénu Darierovy choroby [42]. Později se objevily další případy úspěšné léčby RTG paprsky (Buckyho zářením), elektronovým svazkem, brachyterapií či celotělovým ozářením elektronovým svazkem (elektronová sprcha, total skin electron beam irradiation – TSEBI) [27, 44]. Léčba však byla často provázena vznikem závažné akutní radiodermatitidy.
Kompletní remise při sériové aplikaci botulotoxinu A bylo dosaženo v případech flexurálního postižení [35]. Mechanismem účinku bude pravděpodobně blokování činnosti potních žláz.
Budoucnost v léčbě
Ti pacienti, u nichž dochází kvůli nonsense mutaci ke vzniku „in-frame PTC“, by mohli v budoucnu profitovat z tzv. read-through therapy. Tato terapie je založena na farmakologickém ovlivnění ribozomu, který tak nabude schopnosti překlenout předčasný terminační kodón a pokračovat v translaci. Léky s „read-through“ indukčním potenciálem jsou mimo jiné např. aminoglykosidy (zejména gentamicin). Tato modalita je v současné době ve fázi klinických studií.
Genová terapie je pro svoji náročnost a potenciální toxicitu stále pouze experimentální modalitou léčby.
Psychosociální podpora
Součástí komplexní péče o pacienta s Darierovou chorobou je i psychosociální podpora. Dermatolog může v tomto smyslu hrát roli poradce a pomocníka při zvládání této choroby. V některých situacích nalezne uplatnění psycholog či psychiatr. Zároveň je potřeba brát v úvahu častější výskyt neuropsychiatrických komorbidit.
Pacienti mají možnost sdružovat se v neoficiální uskupení, v rámci nichž mohou sdílet své zkušenosti. Takovou mezinárodní skupinou je např. Darier Disease Support Group na sociální síti Facebook. Oficiální nadace či společnost určená pouze pro pacienty s Darierovou chorobou zatím nebyla vytvořena.
Do redakce došlo dne 15. 5. 2020.
Adresa pro korespondenci:
MUDr. Miroslav Důra
Dermatovenerologická klinika 1. LF UK a VFN
U Nemocnice 499/2
128 00 Praha 2
e-mail: miroslav.dura@vfn.cz
Zdroje
1. ABE, M., YASUDA, M., YOKOYAMA, Y. et al. Successful treatment of combination therapy with tacalcitol lotion associated with sunscreen for localized Darier‘s disease. J Dermatol, 2010, 37(8), p. 718–721.
2. ABRAHAM, S., JONES, A., TOUTOUS-TRELLU, L. et al. Linear Darier disease with herpes zoster superinfection treated successfully by brivudine. Br J Dermatol, 2006, 154(2), p. 365–367.
3. AKINSHEMOYIN VAUGHN, O., HINSHAW, M. A., TENG, J. M. Acantholytic dyskeratotic epidermal nevus. JAMA Dermatol, 2015, 151(11), p. 1259–1260.
4. BENÁKOVÁ, N., VAŠKŮ, V. Retinoidy v dermatologii. Čes-slov Derm, 2017, 92(3), s. 111–122.
5. BERK, D. R., TAUBE, J. M., BRUCKNER, A. L. et al. A sporadic patient with acrokeratosis verruciformis of Hopf and a novel ATP2A2 mutation. Br J Dermatol, 2010, 163(3), p. 653–654.
6. BOEHMER, D., EYERICH, K., DARSOW, U. et al. Variable response to low-dose naltrexone in patients with Darier disease: a case series. J Eur Acad Dermatol Venereol, 2019, 33(5), p. 950–953.
7. BROWN, V. L., KELLY, S. E., BURGE, S. M. et al. Extensive recalcitrant Darier disease successfully treated with laser ablation. Br J Dermatol, 2010, 162(1), p. 227–229.
8. CANNAROZZO, G., BONCIANI, D., SANNINO, M. et al. Dye laser treatment for Darier disease: results of a case series. Photomed Laser Surg, 2016, 34(7), p. 305–307.
9. CRADDOCK, N., DAWSON, E., BURGE, S. et al. The gene for Darier‘s disease maps to chromosome 12q23-q24.1. Hum Mol Genet, 1993, 2(11), p. 1941–1943.
10. DHITAVAT, J., MACFARLANE, S., DODE, L. et al. Acrokeratosis verruciformis of Hopf is caused by mutation in ATP2A2: evidence that it is allelic to Darier‘s disease. J Invest Dermatol, 2003, 120(2), p. 229–232.
11. DOGAN, S., KARADUMAN, A., ERKIN, G. et al. Effective treatment of linear Darier‘s disease with topical retinoids: case report and review of the literature. Acta Dermatovenerol Croat, 2011, 19(3), p. 206–209.
12. EXADAKTYLOU, D., KURWA, H. A., CALONJE, E. et al. Treatment of Darier‘s disease with photodynamic therapy. Br J Dermatol, 2003, 149(3), p. 606–610.
13. FÖLSTER-HOLST, R., NELLEN, R. G., JENSEN, J. M. et al. Molecular genetic support for the rule of dichotomy in type 2 segmental Darier disease. Br J Dermatol, 2012, 166(2), p. 464–466.
14. GLINOS, G., PASTAR, I., TOMIC-CANIC, M. et al. Mislocalization of adherens junction-associated proteins in a patient with Darier disease. Skin, 2018, 2(3), p. 194–201.
15. GOH, B., KUMARASINGHE, P., LEE, Y. Loss of melanosome transfer accounts for guttate leukoderma in Darier’s disease: electron microscopic findings. Pigment Cell Res, 2005, 18, p. 48.
16. GOH, B. K., KUMARASINGHE, S. P., NG, S. K. Two Singaporean cases of guttate leucoderma in Darier‘s disease. Clin Exp Dermatol, 2004, 29(3), p. 313–314.
17. GORDON-SMITH, K., GREEN, E., GROZEVA, D. et al. Genotype-phenotype correlations in Darier disease: A focus on the neuropsychiatric phenotype. Am J Med Genet B Neuropsychiatr Genet, 2018, 177(8), p. 717–726.
18. GROVER, R. W. Transient acantholytic dermatosis. Arch Dermatol, 1970, 101, p. 426–434.
19. HAILEY, H., HAILEY, H. Familial benign chronic pemphigus. Arch Dermatol, 1939, 39, p. 679–685.
20. HARBOE, T. L., WILLEMS, P., JESPERSGAARD, C. et al. Mosaicism in segmental Darier disease: an in-depth molecular analysis quantifying proportions of mutated alleles in various tissues. Dermatology, 2011, 222(4), p. 292–296.
21. HASHIMOTO, K., FUJIWARA, K., TADA, J. et al. Desmosomal dissolution in Grover‘s disease, Hailey-Hailey‘s disease and Darier‘s disease. J Cutan Pathol, 1995, 22(6), p. 488–501.
22. HUGHES, A., MEHREGAN, D. Nevoid hyperkeratosis of the nipple and areola in a man. JAAD, 2005, 52(3), p. 55.
23. ITIN, P. H., HAPPLE, R. Darier disease with paired segmental manifestation of either excessive or absent involvement: a further step in the concept of twin spotting. Dermatology, 2002, 205(4), p. 344–347.
24. KAMIJO, M., NISHIYAMA, C., TAKAGI, A. et al. Cyclooxygenase-2 inhibition restores ultraviolet B-induced downregulation of ATP2A2/SERCA2 in keratinocytes: possible therapeutic approach of cyclooxygenase-2 inhibition for treatment of Darier disease. Br J Dermatol, 2012, 166(5), p. 1017–1022.
25. KASSAR, S., CHARFEDDINE, C., ZRIBI, H. et al. Immunohistological study of involucrin expression in Darier‘s disease skin. J Cutan Pathol, 2008, 35(7), p. 635–640.
26. KATZ, T. M., FIROZ, B. F., GOLDBERG, L. H. et al. Treatment of Darier‘s disease using a 1,550-nm erbium-doped fiber laser. Dermatol Surg, 2010, 36(1), p. 142–146.
27. KITTRIDGE, A., WAHLGREN, C., FUHRER, R. et al. Treatment of recalcitrant Darier‘s disease with electron beam therapy. Dermatol Ther, 2010, 23(3), p. 302–304.
28. MARTONOSI, A. N., PIKULA, S. The structure of the Ca2+-ATPase of sarcoplasmic reticulum. Acta Biochim Pol, 2003, 50(2), p. 337–365.
29. NAKAMURA, T., KAZUNO, A. A., NAKAJIMA, K. et al. Loss of function mutations in ATP2A2 and psychoses: A case report and literature survey. Psychiatry Clin Neurosci, 2016, 70(8), p. 342–350.
30. NAKANO, M., KAMBE, N., SATOH, T. Dermoscopy of keratosis follicularis squamosa. Dermatol Reports, 2011, 3(2), p. e26.
31. NELLEN, R. G., STEIJLEN, P. M., VAN STEENSEL, M. A. et al. Mendelian disorders of cornification caused by defects in intracellular calcium pumps: mutation update and database for variants in ATP2A2 and ATP2C1 associated with Darier disease and Hailey-Hailey disease. Hum Mutat, 2017, 38(4), p. 343–356.
32. NICKLE, S. B., PETERSON, N., PETERSON, M. Updated physician‘s guide to the off-label uses of oral isotretinoin. J Clin Aesthet Dermatol, 2014, 7(4), p. 22–34.
33. OI-YEE LI, H., COLANTONIO, S., KANIGSBERG, N. Treatment of Darier‘s disease with oral magnesium: a case report. SAGE Open Med Case Rep, 2018, 6, 2050313X18795071.
34. ORFANOS, C. E., KURKA, M., STRUNK, V. Oral treatment of keratosis follicularis with a new aromatic retinoid. Arch Dermatol, 1978, 114(8), p. 1211–1214.
35. OSSORIO-GARCÍA, L., COLLANTES-RODRÍGUEZ, C., Villegas-Romero, I. et al. Vegetating Darier disease treated with botulinum toxin. JAMA Dermatol, 2018, 154(1), p. 106–108.
36. PALACIOS-ÁLVAREZ, I., ANDRÉS-RAMOS, I., SILVA, M. Y. et al. Treatment of Darier‘s disease with diclofenac sodium 3% gel. Dermatol Ther, 2017, 30(3), p. e12478.
37. PEĆINA-SLAUS, N., MILAVEC-PURETIĆ, V., KUBAT, M. et al. Clinical case of acral hemorrhagic Darier‘s disease is not caused by mutations in exon 15 of the ATP2A2 gene. Coll Antropol, 2003, 27(1), p. 125–133.
38. PECK, S. M., CHAGRIN, L., SOBOTKA, H. Keratosis follicularis (Darier‘s disesase): A vitamin A deficiency disease. Arch Derm Syphilol, 1941, 43(2), p. 223–229.
39. PÉREZ-CARMONA, L., FLETA-ASÍN, B., MORENO-GARCÍA-DEL-REAL, C. et al. Successful treatment of Darier‘s disease with topical pimecrolimus. Eur J Dermatol, 2011, 21(2), p. 301–302.
40. PETTIT, C., ULMAN, C. A., SPOHN, G. et al. A case of segmental Darier disease treated with doxycycline monotherapy. Dermatol Online J, 2018, 24(3).
41. PLZÁKOVÁ, Z., ŠTORK, J., ŠLAJSOVÁ, M. Darierova choroba a příbuzné dermatózy. Čes-slov Derm, 2010, 85(6), s. 309–316.
42. PODGORNII, A., CIAMMELLA, P., RAMUNDO, D. et al. Efficacy of the radiotherapy on Darier‘s disease: an indirect evidence. Case Rep Dermatol Med, 2013, 2013, 907802.
43. RASZEWSKA-FAMIELEC, M., DUDRA-JASTRZĘBSKA, M., BORZĘCKI, A. et al. Darier-White disease treated with fractional CO2 laser in two cases. Dermatol Ther, 2015, 28(4), p. 254–257.
44. RODRIGUEZ, L. M., KAZEMI, T., CHENG, C. E. et al. Focal multimodality radiation therapy: A promising treatment for recalcitrant Darier disease. Dermatol Ther, 2018, 31(4), p. e12641.
45. RODRÍGUEZ-PAZOS, L., GOMEZ-BERNAL, S., LOUREIRO, M. et al. Type 2 segmental Darier disease with twin spot phenomenon. J Eur Acad Dermatol Venereol, 2011, 25(4), p. 496–497.
46. RONAN, A., INGREY, A., MURRAY, N. et al. Recurrent ATP2A2 p.(Pro602Leu) mutation differentiates acrokeratosis verruciformis of Hopf from the allelic condition Darier disease. Am J Med Genet A, 2017, 173(7), p. 1975–1978.
47. RUBEGNI, P., POGGIALI, S., SBANO, P. et al. A case of Darier‘s disease successfully treated with topical tacrolimus. J Eur Acad Dermatol Venereol, 2006, 20(1), p. 84–87.
48. SAKUNTABHAI, A., DHITAVAT, J., BURGE, S. et al. Mosaicism for ATP2A2 mutations causes segmental Darier‘s disease. J Invest Dermatol, 2000, 115(6), p. 1144–1147.
49. SAKUNTABHAI, A., RUIZ-PEREZ, V., CARTER, S. et al. Mutations in ATP2A2, encoding a Ca2+ pump, cause Darier disease. Nat Genet, 1999, 21(3), p. 271–277.
50. SFECCI, A., ORION, C., DARRIEUX, L. et al. Extensive Darier disease successfully treated with doxycycline monotherapy. Case Rep Dermatol, 2015, 7(3), p. 311–315.
51. SCHMIDT, H., OCHSENDORF, F. R., WOLTER, M. et al. Topical 5-fluorouracil in Darier disease. Br J Dermatol, 2008, 158(6), p. 1393–1396.
52. SMIŽANSKÝ-BARI, L., DRLÍK, L., POCK, L. Keratodermia punctata palmaris et plantaris typ 1: popis případu matky a dcery. Čes-slov Derm, 2015, 90(6), p. 243–247.
53. SOENEN, A., SAINT-JEAN, M., DAGUZÉ, J. et al. Combination of alitretinoin and topical 5-fluorouracil in Darier disease. JAAD Case Rep, 2018, 5(1), p. 75–77.
54. TOPAL, I. O., KAMALI, G. H., GOKDEMIR, G. et al. Concomitant Darier‘s disease and Sjögren‘s syndrome. Indian J Dermatol Venereol Leprol, 2014, 80(6), p. 579.
55. VODO, D., MALCHIN, N., FURMAN, M. et al. Identification of a recurrent mutation in ATP2C1 demonstrates that papular acantholytic dyskeratosis and Hailey-Hailey disease are allelic disorders. Br J Dermatol, 2018, 179(4), p. 1001–1002.
56. WANG, J. F., LEDERHANDLER, M. H., BRINSTER, N. et al. Vesiculobullous Darier disease symptomatically responsive to cetirizine. J Drugs Dermatol, 2019, 18(2), p. 213–214.
57. ZAMIRI, M., MUNRO, C. S. Successful treatment with oral alitretinoin in women of childbearing potential with Darier‘s disease. Br J Dermatol, 2013, 169(3), p. 709–710.
Štítky
Dermatologie Dětská dermatologieČlánek vyšel v časopise
Česko-slovenská dermatologie

2020 Číslo 3
- Ingenol-mebutát rozšiřuje možnosti léčby aktinické keratózy
- Proces hojení ran krok za krokem a co ho může zkomplikovat
- Léčba chronické blefaritidy vyžaduje dlouhodobou péči
- Léčba zánětů spojivek a mazových žlázek víčka v primární péči
Nejčtenější v tomto čísle
- Darierova choroba: současný pohled. Část II.
- Rituximab v léčbě pemphigus vulgaris – popis případu
- Využití terapeutické aferézy v dermatologii
- Lokalizované edémy bérců
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy