
Výskyt molekulárně definovaných renálních karcinomů v retrospektivním souboru jednoho urologického centra
The incidence of molecularly defined renal cell carcinomas in a retrospective cohort from a single urology center
Aim: To evaluate the frequency of molecularly defined renal cell carcinomas (RCC) in a retrospective cohort of patients from a single urology center. Patients and methods: All patients diagnosed with a renal neoplasm at the Department of Urology, University Hospital Pilsen, between January 2018 and September 2025 were retrospectively identified. Particular attention was paid to cases that underwent diagnostic genetic testing. Ultimately, patients with a confirmed “molecularly defined RCC” were included. Clinical data, tumor type, stage, and disease course were evaluated. Results: Of a total of 1,589 renal tumors, molecular-genetic testing was performed in 73 cases. A “molecularly defined RCC” was identified in 17 patients (23.29% of those molecularly tested; 1.2% of all tumors). The most frequent entity was TFE3-
-rearranged RCC (N = 9), followed by fumarate hydratase-deficient RCC (N = 4), ELOC-mutated RCC (N = 2), TFEB-amplified RCC (N = 1), and succinate dehydrogenase-deficient RCC (N = 1). A proportion of patients presented with advanced-stage disease. In addition, one case of RCC with fibro(leio)myomatous stroma, an emerging entity according to the current WHO classification, was identified. Conclusion: Molecularly defined RCCs accounted for 1.2% of all renal tumors and represented a heterogeneous group dominated by TFE3-altered RCCs, with a frequent occurrence of advanced stages. Molecular-genetic testing confirmed this tumor type in 23.29% of targeted cases. Our data confirm that systematically indicated genetic testing is crucial for accurate classification, has a direct impact on prognosis, and is expected to be important for the selection of appropriate systemic therapy in the future. The results of this study highlight the necessity of accessible and systematically indicated molecular testing of suspected renal tumors as an integral part of modern diagnostic practice.
Keywords:
ELOC-mutated renal cell carcinoma – fumarate hydratase-deficient renal cell carcinoma – molecularly defined renal cell carcinoma – TFEB-altered renal cell carcinoma – TFE3-rearranged renal cell carcinoma
Autoři:
Petr Stránský jr. 1; Jiří Kolář 1; Tomáš Pitra 1; Jan Pernický 2; Taťána Bryndaková 3; Ondřej Fiala 4,5; Milan Hora 1; Petr Stránský 1; Michal Michal 3,6; Ondrej Ondič 3,6; Kristýna Pivovarčíková 3,6
Působiště autorů:
Urologická klinika LF UK a FN Plzeň
1; Klinika zobrazovacích metod LF UK a FN Plzeň
2; Šiklův ústav patologie, LF UK a FN Plzeň
3; Onkologická a radioterapeutická klinika LF UK a FN Plzeň
4; Biomedicínské centrum, LF UK Plzeň
5; Bioptická laboratoř, s. r. o., Plzeň
6
Vyšlo v časopise:
Ces Urol 2026; 30(2): 67-76
Kategorie:
Původní práce
doi:
https://doi.org/10.48095/cccu2026008
Souhrn
Cíl: Zhodnotit zastoupení molekulárně definovaných renálních karcinomů (RCC – renal cell carcinoma) v retrospektivním souboru pacientů jednoho urologického centra. Soubor pacientů a metodika: Retrospektivně byli vyhledáni všichni pacienti s diagnostikovaným novotvarem ledviny na Urologické klinice FN Plzeň v období od ledna 2018 do září 2025. Pozornost byla zaměřena zejména na případy s diagnostickým genetickým vyšetřením. Finálně byli zařazeni pacienti s potvrzeným „molekulárně definovaným RCC“. Hodnoceny byly klinické údaje, typ nádoru, stadium a průběh onemocnění. Výsledky: Z celkových 1 589 RCC bylo v 73 případech provedeno molekulárně-genetické vyšetření, u 17 pacientů (23,29 % molekulárně-
-geneticky testovaných; 1,2 % všech nádorů) byl prokázán „molekulárně definovaný typ RCC“. Nejčastější jednotkou byl RCC s přestavbou genu TFE3 (n = 9), dále FH (fumarát hydratáza) deficientní RCC (n = 4), ELOC mutovaný RCC (n = 2), TFEB amplifikovaný RCC (n = 1) a SDH (sukcinát dehydrogenáza) deficientní RCC (n = 1). U části pacientů bylo zjištěno pokročilé stadium onemocnění. Dále byl zaznamenán jeden případ RCC s fibro(leio)myomatózním stromatem, tzv. emerging entity dle aktuální WHO klasifikace. Závěr: Molekulárně definované RCC tvořily 1,2 % všech RCC a představovaly heterogenní skupinu s dominancí TFE3 alterovaných RCC a častým výskytem pokročilých stadií. Molekulárně-genetické vyšetření potvrdilo tento typ tumoru u 23,29 % cíleně testovaných případů. Naše data potvrzují, že systematicky indikované genetické testování je klíčové pro správnou klasifikaci, má přímý dopad na prognózu a v budoucnu předpokládáme také důležitost pro volbu adekvátní systémové terapie. Výsledky této práce zdůrazňují nezbytnost dostupného a systematicky indikovaného molekulárního testování suspektních RCC jako součásti moderního diagnostického postupu.
Klíčová slova:
ELOC mutovaný renální karcinom – fumarát hydratáza deficientní renální karcinom – molekulárně definovaný renální karcinom – TFEB alterovaný renální karcinom – TFE3 rearanžovaný renální karcinom
Úvod
V aktuálním pátém vydání WHO klasifikace nádorů ledvin z roku 2022 došlo v porovnání s předchozí verzí z roku 2016 ke změnám, mimo jiné k začlenění kategorie „molekulárně definované renální karcinomy“, která zahrnuje sedm renálních karcinomů (RCC – renal cell carcinoma) [1]. Do kategorie jsou řazeny:
- TFE3 rearanžovaný RCC;
- TFEB alterovaný RCC;
- ELOC mutovaný RCC;
- fumarát hydratáza (FH) deficientní RCC;
- sukcinát dehydrogenáza (SDH) deficientní RCC;
- ALK rearanžovaný RCC;
- SMARCB1 deficientní renální medulární karcinom.
U většiny těchto nádorů je k určení definitivní diagnózy nezbytné provedení molekulárně-genetického vyšetření s průkazem charakteristické genetické alterace definující danou nádorovou jednotku.
Zároveň jsou v aktuální WHO klasifikaci zmiňovány i jiné renální neoplazie, např. bifázický hyalinizující a psammomatózní RCC, RCC s fibro(leio)myomatózním stromatem (RCC-FMS), které též pro definitivní diagnózu vyžadují průkaz specifické genetické alterace. Ty jsou však zatím ve WHO zmiňovány pouze jako tzv. emerging entities, a to z důvodu nedostatečného počtu v literatuře popsaných případů v čase utváření klasifikace. Literární data věnující se těmto jednotkám však narůstají a lze předpokládat, že i tyto nádory budou ve WHO klasifikaci začleněny do kategorie „molekulárně definovaných RCC“.
Tato práce se zaměřuje na frekvenci výskytu těchto vzácných geneticky definovaných RCC na jednom urologickém pracovišti.
Materiál a metodika
Ve zdravotnickém informačním systému FN Plzeň byli retrospektivně vyhledáni pacienti s histologicky verifikovanou renální neoplazií v období od ledna 2018 do září 2025. Do analýzy byli zařazeni pacienti po operačním odstranění RCC (nefrektomie, parciální resekce), pacienti pouze s provedenou diagnostickou biopsií z primárního ložiska (punkce pod CT či ultrasonografickou (USG) kontrolou) nebo biopsií z metastázy (punkce pod CT či USG kontrolou či metastazektomie).
Ze všech identifikovaných případů byli následně vyselektováni pacienti, u nichž bylo patologem pro určení finální diagnózy indikováno molekulárně-genetické vyšetření, např. metodou fluorescenční in situ hybridizace (FISH), sekvenování nové generace (NGS) nebo polymerázové řetězové reakce (PCR – polymerase chain reaction). Molekulárně-genetické vyšetření nebylo rutinně indikováno u všech RCC, ale selektivně u případů, u nichž morfologický obraz a imunohistochemický profil, event. klinické údaje pacienta (zejména věk či přítomnost jiných neoplazií) vzbuzovaly podezření na některý z molekulárně definovaných subtypů RCC, případně u diagnosticky nejednoznačných či obtížně klasifikovatelných nádorů. Indikace genetického testování tak vycházela z komplexního patologického zhodnocení konkrétní léze. V současné době neexistují jednoznačná kritéria ani doporučení (ať už odborných patologických společností či WHO) pro indikaci genetického testování. Indikace genetického vyšetření je proto plně na zvážení vyšetřujícího patologa a na dostupnosti vyšetření v dané lokalitě (event. ovlivněna i finančními možnostmi pracoviště).
Z této skupiny byli následně vybráni pacienti s potvrzeným molekulárně definovaným RCC. U těchto pacientů byly dále dohledány podrobné klinické a patologické údaje zahrnující pohlaví, věk v době stanovení diagnózy, histologický typ a velikost primárního tumoru, způsob diagnostiky, použitou léčbu (pokud byla indikována) a délku sledování spolu se stavem pacienta při poslední kontrole.
Výsledky
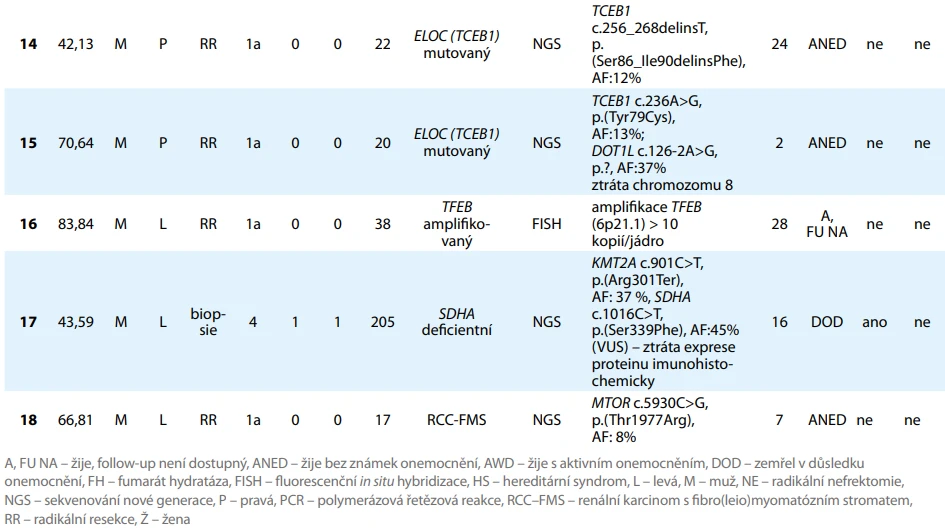
Za období od ledna 2018 do září 2025 bylo ve FN Plzeň histologicky verifikováno 1 589 primárních RCC, z toho 1 414 (88,99 %) nádorů operovaných a 175 (11,01 %) bioptovaných. U 73 (4,59 %) tumorů bylo v rámci histologického vyšetření doplněno molekulárně-genetické vyšetření, u 17 (23,29 % z celkového počtu provedených molekulárně-genetických vyšetření) případů byl prokázán molekulárně definovaný RCC. Celkem bylo nalezeno devět TFE3 rearanžovaných RCC, čtyři FH deficientní RCC, dva ELOC mutované RCC a po jednom TFEB amplifikovaném RCC a SDH deficientním RCC. Mimo jiné byl nalezen jeden případ RCC-FMS. Přehled počtu histologicky verifikovaných RCC v jednotlivých letech shrnuje tab. 1. Souhrnné informace o jednotlivých pacientech a přehled specifických genetických alterací je uveden v tab. 2.


TFE3 rearanžovaný RCC
Průměrný věk devíti pacientů s TFE3 RCC byl 45,46 (rozmezí 24,75–62,43) roku. Poměr žen a mužů (Ž : M) byl 1,25 : 1. Velikost primárního tumoru (u operovaných tumorů určeno dle makroskopického vyšetření patologem, u bioptovaných pak dle CT vyšetření) se pohybovala v rozmezí 18–260 mm (průměr 72,22 mm). Šest pacientů podstoupilo operační výkon (čtyři nefrektomii, dva parciální resekci), tři pacienti byli diagnostikováni na základě biopsie pod CT kontrolou (ve dvou případech z renální léze, v jednom případě z lymfatické uzliny nad levou klíční kostí). Typický radiologický nález ilustruje obr. 1.

U čtyř pacientů bylo již v době stanovení diagnózy přítomno lokálně pokročilé/generalizované onemocnění (klinické stadium III nebo IV). Průměrná doba sledování pacientů od stanovení diagnózy byla 30 (rozmezí 2–66) měsíců, zaokrouhleno na celé měsíce. Z toho tři pacienti byli při poslední kontrole bez známek lokální recidivy či generalizace onemocnění, tři pacienti zemřeli v důsledku onemocnění a tři pacienti jsou na živu s aktivním onemocněním (po relapsu nebo s primárně generalizovaným onemocněním).
Systémovou léčbu v průběhu sledování podstoupilo pět pacientů. Podrobnosti o systémové léčbě jednotlivých pacientů shrnuje tab. 3.

Fumarát hydratáza deficientní RCC
Průměrný věk pacientů s FH deficientním RCC byl 56,49 (rozmezí 44,00–72,06) roku, jednalo se o čtyři muže. Průměrná velikost primárního tumoru (u pacientů po operačním výkonu určeno patologem v rámci makroskopického vyšetření, u bioptovaných nádorů určeno dle CT vyšetření) činila 63,75 (rozmezí 15–150) mm. U dvou pacientů byl proveden operační výkon, v jednom případě byla diagnóza stanovena na základě core-punkční biopsie pod CT kontrolou z primárního ložiska a v jednom případě z histologického vyšetření vzdálené metastázy RCC.
V době stanovení diagnózy měli dva pacienti generalizované onemocnění, zbývající dva pacienti byli diagnostikováni ve stadiu lokálně ohraničeného tumoru. Průměrná délka sledování od stanovení diagnózy byla 31,5 měsíce (rozmezí 3–57 měsíců). Při poslední kontrole byl jeden pacient bez známek lokální recidivy či generalizace, tři pacienti zemřeli v důsledku onemocnění. Jeden z pacientů (v tab. 2 pacient č. 10) měl v době diagnózy postižení retroperitoneálních lymfatických uzlin, jater, sleziny, skeletu a nadledvin. Pacient č. 13 byl diagnostikován rovněž v generalizovaném stadiu onemocnění s popsaným postižením lymfatických uzlin nad a pod bránicí, levé nadledviny, mnohočetným postižením skeletu vč. páteře a stenózou páteřního kanálu a vícečetnými kožními metastázami. U pacienta č. 11 došlo k recidivě onemocnění v podobě generalizace do mediastinálních a retroperitoneálních lymfatických uzlin. Systémovou léčbu podstoupili dva pacienti (tab. 3).
Germinální testování klinickým genetikem bylo provedeno pouze u jednoho z pacientů, u tohoto pacienta byla potvrzena germinální mutace genu FH (a tedy potvrzena asociace se syndromem hereditární leiomyomatózy a RCC).
ELOC mutovaný RCC
V našem souboru byli identifikováni dva pacienti s ELOC mutovaným RCC. V obou případech se jednalo se o muže ve věku 24,13 a 70,64 (průměr 56,39) roku. Velikost primárních tumorů (dle makroskopického vyšetření patologem) činila 20 a 22 (průměr 21) mm.
V obou případech se jednalo o lokalizované onemocnění. Oba pacienti podstoupili radikální operační výkon. Délka sledování byla 24 měsíců, resp. 2 měsíce. Při poslední kontrole byli oba pacienti bez známek lokální recidivy či generalizace onemocnění.
TFEB alterovaný RCC
TFEB alterovaný RCC, konkrétně TFEB amplifikovaný RCC, byl v daném období diagnostikován pouze u jednoho muže ve věku 83,84 roku. Velikost primárního tumoru činila 38 mm, dle TNM klasifikace pT1a cN0 cM0. Nemocný podstoupil roboticky asistovanou resekci tumoru.
Pacient je 28 měsíců od operačního výkonu živ, bližší informace o jeho stavu nejsou známy, protože se pacient nedostavuje k dispenzárním vyšetřením.
SDH deficientní RCC
U jednoho muže byl diagnostikován SDH deficientní RCC ve věku 43,59 roku. Velikost primárního tumoru dosahovala 205 mm (dle CT) a onemocnění bylo v době diagnózy klinicky již v generalizovaném stadiu. Diagnóza byla stanovena na základě core-punkční biopsie tumoru pod CT kontrolou.
Pacient následně podstoupil tři linie systémové léčby a po 16 měsících od stanovení diagnózy zemřel (blíže tab. 3). Germinální testování klinickým genetikem nebylo provedeno.
RCC s fibro(leio)myomatózním stromatem
V našem souboru byl identifikován jeden pacient s RCC-FMS s prokázanou mutací genu MTOR. Přestože tato jednotka dle současné WHO není zařazena mezi „molekulárně definované RCC“, případ uvádíme jako tzv. emerging entitu, neboť i zde je molekulárně-genetické vyšetření klíčové pro definitivní klasifikaci.
Nádor byl náhodně zachycen při vyšetření pozitronovou emisní tomografií kombinovanou s výpočetní tomografií (PET/CT) u muže sledovaného pro adenokarcinom prostaty. Pacient podstoupil dvoudobou roboticky asistovanou parciální resekci tumoru obou ledvin. Na levé straně byl potvrzen RCC-FMS o velikosti 17 mm dle makroskopického vyšetření, ve druhé době na pravé straně světlobuněčný RCC, v obou případech dle TNM klasifikace pT1a cN0 cM0. Po 7 měsících sledování je pacient bez známek lokální recidivy či metastatického onemocnění.
Diskuze
WHO kategorie „molekulárně definované RCC“ sice zahrnuje vzácné jednotky, jejich přesná identifikace patrně má (a zejména lze předpokládat, že do budoucna bude mít) významné prognostické i terapeutické důsledky. Naše data ukazují, že výskyt těchto entit v běžné klinické praxi není zanedbatelný, a zároveň zdůrazňují potřebu rutinního využívání molekulárně-genetických metod u suspektních případů.
Věk a klinické stadium onemocnění v našem souboru molekulárně definovaných RCC se v řadě ohledů liší od „tradiční“ populace pacientů s RCC. Nejčastější formy RCC (zejména světlobuněčný a papilární RCC) dosahují vrcholu incidence mezi 60. a 70. rokem života, často v časnějších stadiích onemocnění [2]. Naproti tomu většina molekulárně definovaných RCC často postihuje mladší pacienty a řada z nich je zachycena ve vyšších klinických stadiích (výjimku představuje ELOC mutovaný RCC většinou s indolentním chováním) [1].
RCC s přestavbou TFE3 genu byl v našem souboru nejčastěji zastoupenou entitou. Pacienti měli průměrný věk 45,5 (medián 47,82) roku a vyrovnané pohlavní zastoupení s mírnou převahou žen. Recentní studie autorů popisují výskyt u mladších dospělých, s mediánem věku 33 let [3] nebo průměrem 44 ± 15 let [4]. Také v literárních kohortách převažovalo ženské pohlaví, přičemž agresivní průběh s časnými recidivami/metastázami byl popisován pouze v multicentrické studii autory Liu et al. [4]. Morfologický vzhled, biologické chování a prognóza tumoru je patrně ovlivněna typem fúzního partnera genu TFE3 [5]. Histologický obraz ilustruje obr. 2.

TFEB alterované RCC jsou velmi heterogenní a zásadní je rozlišení mezi translokovaným a amplifikovaným subtypem. V našem souboru byl identifikován jeden případ TFEB amplifikovaného RCC u pacienta vyššího věku s lokalizovaným onemocněním, který však nebyl dostupný k dalšímu sledování. V literatuře je TFEB amplifikovaný podtyp popisován jako vzácný, často agresivní a s výrazným metastatickým potenciálem [6]. Naproti tomu TFEB translokovaný RCC se typicky objevuje v nižším věku (medián 31 let u TFEB translokovaného RCC [7] vs. 65 let u TFEB amplifikovaného RCC [6,7]) a obvykle v nižším klinickém stadiu [6]. Tyto dvě jednotky je proto nutné striktně odlišovat.
ELOC mutovaný RCC, dříve označovaný jako TCEB1 mutovaný, byl v našem souboru zachycen dvakrát. Oba pacienti měli malé, lokalizované léze a byli při poslední kontrole bez známek recidivy. Publikované práce popisují tuto jednotku jako vzácnou, s převážně indolentním průběhem [8] a častějším výskytem u mužů [9], což odpovídá i našim pozorováním. Pro správnou identifikaci tumoru je nezbytné využití kombinace morfologických znaků (světlobuněčné elementy a objemné fibromyomatózní stroma), imunohistochemie, vč. průkazu cytokeratinu 7 (CK7), a molekulárně-genetického vyšetření [9], bez něhož nelze odlišit morfologicky shodné tumory s mutacemi v genech signální dráhy mTOR (mechanistic target of rapamycin) [10], jako je RCC-FMS.
FH deficientní RCC představuje vysoce agresivní jednotku, u níž je častý časný rozvoj metastáz a nepříznivá prognóza [11]. Mírně častější výskyt byl zaznamenán u mužů a věkový medián v době diagnózy byl 44 let [12]. V našem souboru měli všichni čtyři pacienti objemné primární tumory a u dvou došlo k úmrtí v důsledku nádorového onemocnění. Pozorovaný klinický průběh odpovídá dostupným literárním údajům, nicméně vzhledem k malému počtu pacientů nelze z našeho souboru vyvozovat širší prognostické závěry. Nádor může být hereditárně podmíněn (jako součást syndromu hereditární leiomyomatózy a RCC), může se však vyskytovat i sporadicky [13]. V případě diagnózy je silně doporučeno genetické poradenství pro pacienta a jeho potomky [1].
SDH deficientní RCC byl za sledované období v našem souboru zastoupen jediným případem s prokázanou mutací genu SDHA, pokročilostí tumoru a generalizovaným onemocnění a úmrtím do 16 měsíců po diagnóze. SDH deficientní RCC je vzácnou nádorovou jednotkou, obvykle diagnostikovanou v mladším věku (medián 43,5 roku) [14], často asociovanou s germinální mutací postiženého genu. Germinální mutace podjednotek SDH jsou asociovány také s dalšími tumory, zejména paragangliomy, feochromocytomy, gastrointestinálními stromálními tumory a adenomy hypofýzy [15,16]. Při stanovení diagnózy tohoto tumoru je vhodné vyšetření klinickým genetikem [1]. Může se chovat agresivně a riziko metastazování roste u případů s vyšším nukleárním stupněm a nádorovou heterogenitou [14].
RCC-FMS je v současné WHO zařazen jako vzácná „emerging entita“ [1,10]. Dostupná data naznačují, že tyto tumory jsou diagnostikovány spíše u mladších až středně starých dospělých, Shah et al. uvádějí průměrný věk 52 (rozmezí
33–69) let [17]. Neoplazie je asociována s diagnostickou alterací signální dráhy mTOR/TSC (mechanistic target of rapamycin/tuberous sclerosis complex) – mutace TSC1/TSC2/MTOR, přičemž obdobný histologický obraz (při odlišném genetickém pozadí) může být přítomen i u ELOC mutovaných RCC. Bez cíleného molekulárně-genetického vyšetření proto nelze tyto jednotky morfologicky ani imunohistochemicky spolehlivě odlišit [8–10]. Náš případ RCC-FMS s mutací genu MTOR, diagnostikovaný v časném stadiu pT1a a s příznivým průběhem onemocnění, je v souladu s dosud publikovanými literárními údaji a podporuje význam molekulárně-genetického vyšetření při klasifikaci těchto nádorů.
Otázka, zda WHO klasifikace „nepředběhla“ možnosti léčby, je v případě molekulárně definovaných RCC zcela oprávněná. U většiny těchto jednotek dosud neexistují specifické standardizované léčebné algoritmy, systémová léčba se proto v klinické praxi opírá zejména o obecná doporučení pro nesvětlobuněčné RCC a o limitovaná data pro jednotlivé podtypy [18]. Mělo by být zváženo zařazení pacientů do klinických studií [18]. Přesto význam přesné klasifikace těchto jednotek roste. Rozlišení TFE3 fúzních partnerů může mít prognostický dopad, stejně tak prognostický význam má odlišení TFEB amplifikovaných a TFEB rearanžovaných RCC. Identifikace ALK mutace umožňuje využití cílené terapie a alterace genů FH či SDH indikují genetické poradenství s ohledem na možnou hereditární asociaci a ovlivňují sledování probanda i rodinných příslušníků. Ačkoliv WHO klasifikace sama neurčuje léčebné postupy, přesnější diagnostické vymezení těchto jednotek vytváří předpoklad pro budoucí individualizaci péče. Současné algoritmy systémové onkologické léčby však zatím tento diagnostický pokrok plně nereflektují.
Praktický dopad přesné molekulární klasifikace se však neomezuje pouze na diagnostiku a systémovou léčbu, promítá se též do následné dispenzarizace pacientů. U těchto vzácných jednotek neexistují samostatná standardizovaná schémata sledování, v klinické praxi se v současnosti vychází z obecných doporučení pro nesvětlobuněčné RCC [18]. Přesná diagnóza může vést k modifikaci dispenzarizace – u agresivnějších jednotek (např. FH a SDH deficientní RCC nebo některé TFE3/TFEB alterované nádory) lze předpokládat potřebu intenzivnějšího a dlouhodobějšího sledování, zatímco u jednotek s popisovaným příznivým průběhem (např. ELOC mutovaný RCC) může být sledování méně zatěžující, avšak vzhledem k limitovaným datům je stále nutná opatrnost. U hereditárně asociovaných nádorů navíc přesná diagnóza rozšiřuje péči i o genetické poradenství, sledování rizikových rodinných příslušníků a event. sledování pacientů i jinými specialisty (neboť u těchto pacientů může docházet ke vzniku neoplazií i mimo urogenitální trakt).
Poměr mezi počtem molekulárně testovaných nádorů a počtem definitivně potvrzených molekulárně definovaných RCC v našem souboru jasně ukazuje, že samotná morfologie a imunohistochemie nejsou vždy zcela dostačující znaky pro finální diagnostiku renálních neoplazií. Vyšší počet genetických testů oproti počtu detekovaných molekulárně definovaných RCC odpovídá současné klinicko-patologické praxi, v níž je podezření na molekulárně definovaný RCC často založeno na subtilních či nespecifických morfologických a imunohistochemických znacích, značně je tato suspekce modifikována i znalostmi a subjektivními vjemy hodnotícího patologa. V našem souboru tvořil podíl prokázaných molekulárně definovaných RCC vůči všem provedeným molekulárně-genetickým testům 23,29 %. Je ovšem nutné upozornit na fakt, že část indikovaných genetických testů u RCC nebyla provedena z důvodu suspekce na některý z molekulárně definovaných RCC, ale v souvislosti s přítomností morfologicky a imunohistochemicky nezařaditelného tumoru. V těchto diagnosticky obtížných a nejasných případech je na našem patologickém pracovišti u primárně neklasifikovatelných RCC někdy indikováno genetické testování za účelem průkazu/vyloučení přítomnosti genetických alterací typických pro světlobuněčný RCC (VHL mutace, metylace promotoru VHL, či LOH 3p) tak, aby pro další onkologickou léčbu mohl být tumor zařazen do skupiny „clear cell“ vs. „non-clear cell“ RCC.
Je též nutné zdůraznit, že molekulárně-genetické testy představují finančně nákladné metody, a proto by měly být indikovány uvážlivě a cíleně. Na druhou stranu je v indikovaných případech jejich role zcela nezastupitelná. Umožňují přesnou klasifikaci, zabraňují chybné diagnóze a mají přímý dopad na prognózu, genetické poradenství i léčebné možnosti pacientů, což je relevantní i z hlediska budoucích diskuzí o dostupnosti a úhradě těchto metod. Naše zkušenosti s diagnostikou molekulárně-geneticky definovaných RCC potvrzují, že k diagnostice těchto jednotek je nezbytný komplexní přístup zahrnující morfologický obraz, imunohistochemické vyšetření i molekulárně-genetické metody. Bez jejich využití nelze diagnostiku považovat za kompletní.
Limitací této práce je malý počet pacientů (daný raritou těchto entit) a retrospektivní, monocentrický design studie. Při interpretaci výsledků je nutné zdůraznit, že uvedený podíl molekulárně definovaných RCC (1,2 % všech histologicky verifikovaných RCC) nepředstavuje skutečnou prevalenci těchto jednotek v neselektované populaci, ale především odráží výstup diagnostického algoritmu používaného na našem pracovišti. Doba sledování byla nehomogenní a u části pacientů nebyly dostupné kompletní follow-up informace. Srovnání zastoupení jednotlivých molekulárně definovaných RCC mezi studiemi je dále omezeno rozdíly v jejich designu a indikačních kritériích. Naše výsledky vycházejí ze záchytu těchto jednotek v běžné klinické praxi jednoho centra a poskytují relevantní real-world data o jejich frekvenci, věkovém spektru a biologickém chování.
Závěr
Molekulárně definované RCC tvořily v našem souboru 1,2 % všech histologicky verifikovaných RCC a představovaly biologicky i klinicky heterogenní skupinu s dominancí TFE3 rearanžovaných tumorů a s častým výskytem pokročilých stadií onemocnění u mladších pacientů. Molekulárně-genetické vyšetření vedlo k potvrzení molekulárně definovaného RCC u 23,29 % cíleně testovaných případů v našem souboru, což odráží jak jejich vzácnost, tak nezbytnost pečlivé indikace těchto metod. Naše data z běžné klinické praxe potvrzují, že správná diagnostika a léčba vyžadují úzkou mezioborovou spolupráci patologa, urologa, onkologa a případně klinického genetika. Přesná molekulární diagnóza je zásadní nejen pro stanovení prognózy a genetické poradenství, ale v budoucnu také pro potenciální volbu adekvátní systémové terapie. Výsledky této práce zdůrazňují nezbytnost dostupného a systematicky indikovaného molekulárního testování suspektních RCC jako klíčové součásti moderní diagnostiky. Současně poukazují i na potřebu multicentrických studií s většími soubory, které umožní detailnější pochopení biologického chování molekulárně definovaných RCC a optimalizaci léčebných postupů.
Střet zájmů: Autoři prohlašují, že zpracování tohoto článku nebylo podpořeno žádnou společností.
Prohlášení o podpoře: Univerzita Karlova Praha, Lékařská fakulta Plzeň (COOPERATIO program, SURG) a institucionální výzkum Fakultní nemocnice Plzeň (FNPl 00669806).
Zdroje
1. Amin MB, Gill AJ, Hartmann A et al. Tumours of the kidney. In: WHO Classification of Tumours Editorial Board (eds). Urinary and male genital tumours. ed. Lyon: IARC 2022 : 32–86.
2. Bex A, Ghanem YA, Albiges L et al. European Association of Urology Guidelines on renal cell carcinoma: the 2025 update. Eur Urol 2025; 87(6): 683–696. doi: 10.1016/j.eururo.2025.02.020.
3. Han W, Peng K, Shen Q et al. Clinical characteristics and survival outcomes of tfe3-rearranged renal cell carcinoma: a retrospective study compared with clear cell renal cell carcinoma using propensity score matching. World J Urol 2025; 43(1): 654. doi: 10.1007/s00345-025-05927-4.
4. Liu J, Wei J, Zhang Y et al. Analysis of clinicopathologic features and imaging findings of tfe3-rearranged renal cell carcinoma. Clin Genitourin Cancer 2025; 23(6): 102446. doi: 10.1016/j.clgc.2025.102446.
5. Tretiakova MS. Chameleon tfe3-translocation RCC and how gene partners can change morphology: accurate diagnosis using contemporary modalities. Adv Anat Pathol 2022; 29(3): 131–140. doi: 10.1097/PAP.0000000000000332.
6. Argani P, Reuter VE, Zhang L et al. TFEB-amplified renal cell carcinomas: an aggressive molecular subset demonstrating variable melanocytic marker expression and morphologic heterogeneity. Am J Surg Pathol 2016; 40(11): 1484–1495. doi: 10.1097/PAS.0000000000000720.
7. Argani P. Translocation carcinomas of the kidney. Genes chromosomes cancer 2022; 61(5): 219–227. doi: 10.1002/gcc.23007.
8. Hakimi AA, Tickoo SK, Jacobsen A et al. TCEB1-mutated renal cell carcinoma: a distinct genomic and morphological subtype. Mod Pathol 2015; 28(6): 845–853. doi: 10.1038/modpathol.2015.6.
9. Hes O, Michalová K, Pivovarčíková K. Novinky ve WHO klasifikaci renálních nádorů 2022. Čes-slov Patol 2022; 58(4): 187–191.
10. Trpkov K, Williamson SR, Gill AJ et al. Novel, emerging and provisional renal entities: the genitourinary pathology society (GUPS) update on renal neoplasia. Mod Pathol 2021; 34(6): 1167–1184. doi: 10.1038/s41379-021-00737-6.
11. Chen YB, Brannon AR, Toubaji A et al. Hereditary leiomyomatosis and renal cell carcinoma syndrome-associated renal cancer: recognition of the syndrome by pathologic features and the utility of detecting aberrant succination by immunohistochemistry. Am J Surg Pathol 2014; 38(5): 627–637. doi: 10.1097/PAS.0000000000000163.
12. Trpkov K, Hes O, Agaimy A et al. Fumarate hydratase-deficient renal cell carcinoma is strongly correlated with fumarate hydratase mutation and hereditary leiomyomatosis and renal cell carcinoma syndrome. Am J Surg Pathol 2016; 40(7): 865–875. doi: 10.1097/PAS.0000000000000617.
13. Pivovarcikova K, Martinek P, Grossmann P et al. Fumarate hydratase deficient renal cell carcinoma: chromosomal numerical aberration analysis of 12 cases. Ann Diagn Pathol 2019; 39 : 63–68. doi: 10.1016/j.anndiagpath.2019.02.008.
14. Gill AJ, Hes O, Papathomas T et al. Succinate dehydrogenase (SDH)-deficient renal carcinoma: a morphologically distinct entity: a clinicopathologic series of 36 tumors from 27 patients. Am J Surg Pathol 2014; 38(12): 1588–1602. doi: 10.1097/PAS.0000000000000292.
15. Gill AJ. Succinate dehydrogenase (SDH)-deficient neoplasia. Histopathology 2018; 72(1): 106–116. doi: 10.1111/his.13277.
16. Rogala J, Zhou M. Hereditary succinate dehydrogenase-deficient renal cell carcinoma. Semin Diagn Pathol 2024; 41(1): 32–41. doi: 10.1053/j.semdp.2023.11.001.
17. Shah RB, Stohr BA, Tu ZJ et al. “Renal cell carcinoma with leiomyomatous stroma” harbor somatic mutations of TSC1, TSC2, MTOR, and/or ELOC (TCEB1): clinicopathologic and molecular characterization of 18 sporadic tumors supports a distinct entity. Am J Surg Pathol 2020; 44(5): 571–581. doi: 10.1097/PAS.0000000000001422.
18. European Association of Urology. EAU Guidelines on renal cell carcinoma. Arnhem: EAU Guidelines Office 2026. [online]. Available from: https://uroweb.org/guidelines/renal-c
Štítky
Dětská urologie Nefrologie UrologieČlánek vyšel v časopise
Česká urologie

2026 Číslo 2
- I kondomový urinal může zabít
- Preskripce inkontinenčních pomůcek v praxi: pravidla platí i pro jejich kombinace
- Na výběru inkontinenčních pomůcek záleží − ale jak se mezi nimi neztratit?
- Metodika úhrady pomůcek pro inkontinentní pacienty
- Úspěšná operační terapie stresové inkontinence – kazuistika
Nejčtenější v tomto čísle
- Vzácné systémové komplikace intravezikální BCG terapie
- Invertovaný uroteliální papilom – benigní léze s potenciální vazbou na uroteliální karcinom
- Zkušenosti našeho pracoviště s rekonstrukcí panuretrální striktury močové trubice metodou podle Kulkarniho
- Editorial
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy