
Klinické nálezy u členů rodiny s výskytem izolované aniridie v důsledku PAX6 mutace
Clinical Findings in Family with Aniridia due the PAX6 Mutation
Background:
inborn isolated aniridia is rare bilateral impairment of several eye structures manifesting mainly by absence of iris, photophobia and decreased visual acuity. There are also others ocular symptoms associated with aniridia such as nystagmus, strabismus, eyelid ptosis, amblyopia, serious refractive errors, anisometropia, corneal changes, impairment of the lens, chamber angle dysgenesis, optic nerve and macular hypoplasia and congenital or secondary glaucoma. The most frequent aetiology of this eye dysgenesis is mutation in PAX6. Aim of this report is to describe ocular findings in the family with familial aniridia (MIM #106210), to debate their severity, prognosis and therapy options.
Material and methods:
assessment of previous medical history and actual ophthalmological findings in 4 persons of 3 generation family with aniridia. According to the compliance, the patients underwent these tests: assessment of the visual acuity, intraocular pressure, refraction test, slit-lamp examination and biomicroscopy, pachymetry test and OCT examination. The genetic counselling was performed with subsequent PAX6 mutation analysis.
Results:
all of the examined aniridia family members showed severe symptoms of the disease, the aniridia and photophobia were present. Positive age related correlation showed progressive visual acuity decrease to the practical blindness due to aniridia-associated keratopathy, secondary glaucoma and cataract. DNA analysis revealed presence of p.Gln180X PAX6 mutation in all of the affected persons. The mutation leads to shortened and therefore non-functional protein.
Conclusions:
PAX6 mutations leading to premature termination of protein translation are frequently associated with severe symptoms of aniridia and small intrafamilial variability of ocular impairment. This fact is also well demonstrated in members of family described by this report, the symptoms are severe and progressing with age. Therapy is difficult and often with partial success, such in case of secondary glaucoma in young girl from this family. Any eye surgery must be individually judged due to risk of several post-operative complications. And more, the poor vision in aniridia patients is progressively worsening in time to practical blindness.
Key words:
aniridia, PAX6, macular hypoplasia, glaucoma
Autoři:
L. Godavová 1; M. Godava 2; J. Sabová 3; G. Kolářová 1; Š. Mohlerová 1
Působiště autorů:
Ústav lékařské genetiky, FN Olomouc
přednostka
doc. MUDr. Ishraq Dhaifalah, Ph. D.
; Oční oddělení, Vojenská nemocnice
Olomouc
primářka MUDr. Šárka Mohlerová
1; Ústav lékařské genetiky a fetální
medicíny, LF UP Olomouc
2; Sekce lidské genetiky
a celogenomového sekvenování
Synlab Genetics, s. r. o., Praha
vedoucí garant
MUDr. Soňa Peková, Ph. D.
3
Vyšlo v časopise:
Čes. a slov. Oftal., 70, 2014, No. 4, p. 138-144
Kategorie:
Původní práce
Souhrn
Úvod:
vrozená izolovaná aniridie je vzácné postižení struktur obou očí projevující se především chyběním duhovky, fotofobií a sníženým viděním. V klinickém obrazu může být přítomný nystagmus, strabismus, ptóza víčka, amblyopie, vyšší refrakční vada, anisometropie, rohovkové změny, patologie čočky, dysgeneze struktur komorového úhlu, hypoplazie makuly a zrakového nervu, kongenitální nebo sekundární glaukom. Tento typ dysgeneze oka je nejčastěji podmíněn mutací PAX6. Cílem naší práce bylo popsat oftalmologické nálezy členů rodiny s výskytem dědičné aniridie (MIM #106210), diskutovat jejich závažnost, prognózu a možnosti terapie.
Materiál a metodika:
zhodnocení předchozích klinických záznamů a oftalomogické vyšetření proběhlo u 4 osob z tří generací rodiny s výskytem izolované vrozené aniridie. Dle možností a compliance byla u pacientů provedena tato vyšetření: vízus, nitrooční tenze, refrakce, vyšetření štěrbinovou lampou, biomikroskopické vyšetření, pachymetrie, OCT. Dále bylo provedeno klinicko genetické vyšetření a následně sekvenace PAX6.
Výsledky:
u všech vyšetřených osob byla zjištěna plně vyjádřená závažná symptomatologie, byla pozorována přítomnost aniridie a fotofobie. Pozitivní korelaci s věkem vykazoval progresivní pokles vizu k praktické slepotě v důsledku keratopatie asociované s aniridií, sekundárním glaukomem a kataraktou. DNA analýza u všech postižených osob odhalila přítomnost mutace p.Gln180X v PAX6. Tato mutace vede k tvorbě zkráceného atak nefunkčního PAX6 proteinu.
Závěr:
mutace v genu PAX6, které vedou k předčasné terminaci tvorby bílkoviny, jsou spojené často s těžkým průběhem onemocnění as malou variabilitou závažnosti postižení intrafamiliárně. Tento fakt dokumentuje irodina popsaná v této práci, kdy projevy upacientů jsou závažné aprogredují s věkem. Terapie je svízelná, často jen parciálně úspěšná, jako je tomu iv případě sekundárního glaukomu mladé pacientky. Každý operační zákrok je nutno individuálně vždy velmi pečlivě uvážit. Navíc utéto nemoci dochází kneúprosné progresi očního nálezu spostupným zhoršováním itak již většinou chabé zrakové ostrosti až do praktické slepoty.
Klíčová slova:
aniridia, PAX6, hypoplazie makuly, glaukom
Úvod
Izolovaná kongenitální aniridie patří mezi vzácná onemocnění, odhadovaná incidence je přibližně 1/65000–1/96000 (11). Dvě třetiny případů aniridie se vyskytují familiárně, jedna třetina se vyskytuje sporadicky (19). Izolována kongenitální aniridie představuje panokulární anomálii s bilaterálním výskytem, u které dominuje úplná nebo parciální absence duhovky a další anomálie v důsledku vadného vývoje komorového úhlu, rohovky, čočky, sítnice i zrakového nervu. Pacienti mají snížený vizus, světloplachost, často se vyskytuje i nystagmus a strabismus (25).
Aniridie je výsledkem abnormální interakce neuroektodermu v oblasti prosencefala a přilehlého ektodermu, a to v důsledku mutací v PAX6. Protein PAX6 patří do rodiny transkripčních regulátorů, má důležitou roli ve vývoji oka, může ale také ovlivňovat i vývoj mozku a pankreatu. K jeho expresi dochází v neuroektodermu prosencefala již v době indukce oční ploténky. Po jejím rozdělení na dvě části za vzniku očních váčků se exprese PAX6 omezuje na přední okraj těchto váčků. PAX6 tak následně indukuje v ektodermu přilehlém k očnímu váčku vznik plakody čočky. Pokračující exprese PAX6 v součinnosti s dalšími transkripčními faktory má důležitou roli i při vývoji čočky (29). Exprese PAX6 je přítomná také v rohovce, spojivce, ciliárním epitelu a retině, pokračuje i postnatálně (20).
Izolovaná aniridie v důsledku afekce PAX6 vykazuje autosomálně dominantní dědičnost, riziko výskytu této nemoci pro potomky postižené osoby je teoreticky 50 %. PAX6 se nachází na chromosomu č. 11, v oblasti 11p13. V tomto genu se mohou vyskytovat různé typy mutací, v jejichž důsledku dochází k tvorbě kratšího proteinu nebo proteinu s odlišnou sekvencí aminokyselin. Tyto vedou k porušení funkce popř. tvorby PAX6 proteinu (7). Defektní produkt genu vede k poruše morfogeneze zejména duhovky. Osoby s vrozenou aniridii mají plně funkční pouze jednu alelu PAX6, druhá alela je ve své funkci (parciálně nebo úplně) postižena mutací – jedná se tedy o heterozygotní konstituci. Na základě kazuistik popisujících rodiny, kdy u obou rodičů byla přítomná aniridie, se předpokládá, že přítomnost 2 mutací v PAX6 (homozygot, složený heterozygot) není viabilní (12).
Penetrance onemocnění je plná, expresivita je ale různá, důsledkem toho je větší variabilita klinických projevů a to někdy i v jedné rodině. U pacientů se tak objevuje aniridie (totální nebo parciální) a další související oční symptomy, nebo pouze jiné přidružené okulární abnormality. Rozlišujeme totální aniridii a parciální aniridii. I když minimálně, tak rudimenty duhovkové tkáně jsou gonioskopicky většinou patrné i u totální aniridie.
U totální aniridie se vyskytuje široká zornice sahající až k okraji rohovky a díky tomu je přehledný celý ekvátor čočky a někdy i závěsný aparát s ciliárními výběžky. Patologicko-anatomicky nalézáme většinou rudimentární tkáň duhovky, chybějící svaly, pigmentový epitel je ektropizovaný a ciliární výběžky bývají kratší (21, 25).
Incidence glaukomu u aniridie je přibližně 6–75%. Obvykle se objevuje již v preadolescentním nebo časném adolescentním věku (19). Glaukom se vyvíjí v důsledku abnormalit odtokových cest komorového úhlu, které brání odtoku komorové tekutiny přes Schlemův kanál. U pacientů s aniridií a současně přítomným glaukomem popsali Margo a spol. (15) abnormality irido – korneálního úhlu, přičemž u většiny těchto pacientů periferní zbytky duhovky překrývaly trabekulární trámčinu. V případě kongenitálního glaukomu jsou drenážní odtokové cesty obturovány membránou obsahující cévy pocházející z rudimentů duhovky. U sekundárního glaukomu jsou odtokové cesty blokovány zbytkovou duhovkovou tkání. Schlemův kanál může chybět u obou typů glaukomů (4, 8, 25).
Rohovka u pacientů s aniridií měří o 100 μm více než je průměrná tloušťka rohovky (3). Typická je přítomnost sekundárních rohovkových změn již v časném věku (první změny se objevují již v prvním deceniu). PAX6 mutace má za následek změny v rohovkové expresi cytokeratinu, buněčné adhezi a expresi glykokonjugátu. Toto společně s deficitem kmenových limbálních buněk se podílí na fragilitě rohovky a na s aniridií asociované keratopatii (AAK). Incidence keratopatie se uvádí kolem 20 % (4, 27). U aniridie je produkce slz normální, ale slzy postrádají normální viskozitu a dochází k snadnějšímu trhání slzného filmu. Protektivní hodnota slzného menisku je snížena, tím je otevřena cesta pro iritační procesy na povrchu oka (4, 11, 14, 25).
Na čočce jsou někdy zbytky vaskulární tuniky (tunica vasculosa lentis), perzistující pupilární membrána a ojediněle kolobom čočky, v nízkém procentu se vyskytuje ektopie čočky, subluxace a dislokace čočky. Přítomné opacity v čočce se můžou vyskytovat již vrozeně. U více než poloviny pacientů (50–85 %) se vyvíjí katarakta (19). Typicky může začínat již v dětství nebo postihuje mladé dospělé.
Na porušené funkci sítnice se může podílet její defektní vývoj během embryogeneze a následně během života fototoxické působení. Typická je hypoplazie makuly, která je histologicky charakterizována nahrazením struktury fovey souvislou vrstvou gangliových buněk. Foveální hypoplazie se manifestuje redukovaným foveálním reflexem, makulární hypopigmentací, abnormální vaskulární remodelací i snížením avaskulární zóny. OCT nálezy zachycují centrální foveolární ztluštění a menší makulární objem, nebývá zachována foveolární deprese (11, 13, 17, 22, 26, 31).
Nystagmus není univerzálním nálezem, ale vyskytuje se přibližně u 85–92 % pacientů s aniridií. Příčinou jeho vzniku je samotná hypoplazie makuly s kongenitálně způsobeným snížením vizu (25).
Materiál a metodika
Vyšetřené byly 4 osoby z jedné rodiny s výskytem aniridie ve třech generacích (obr. 1). Jednalo se o tyto osoby: bratr a sestra, jejich matka a otec matky. V této rodině je fenotyp nemoci závažně vyjádřen. Tyto osoby absolvovaly vyšetření oftalmologické i genetické.

V rámci genetického vyšetření byla rodině poskytnuta genetická konzultace a na základě informovaného souhlasu pak DNA analýza – analýza přítomnosti mutace v PAX6 pomocí přímé sekvenace.
Při vyšetření oftalmologem bylo použito obvyklého vyšetřovacího postupu. Nejdříve se určila zraková ostrost bez korekce i s korekcí a proběhlo změření refrakce autorefraktometrem. Následně bylo u pacientů provedeno měření nitrooční tenze a vyšetření pomocí štěrbinové lampy. Pachymetrie byla provedena u mladší pacientky s vrozenou aniridií. Fotodokumentaci fundu bylo možné pořídit u obou nejmladších pacientů s transparentní rohovkou. Vyšetření OCT se s obtížností povedlo u nejmladší pacientky (sestra) v rámci pokusu o vyšetření papil zrakového nervu a makuly.
Výsledky
DNA analýza odhalila přítomnost mutace c. 538C>T / p.Gln180X v PAX6, nacházející se v exonu č. 8. Tato mutace byla nalezena u všech 4 osob s výskytem kongenitální aniridie. Extraokulární postižení jedinců z této rodiny nebylo anamnesticky zaznamenáno.
Pětasedmdesátiletý muž (pacient I-1, obr. 1) je trvale dispenzarizován pro vrozenou totální aniridii, mírný horizontální nystagmus, sekundární glaukom a plně vyvinutou AAK s vaskularizovaným leukomem (obr. 2). Při vyšetření štěrbinovou lampou je při netransparentní rohovce možno jen tušit rudimenty duhovkové tkáně. Dle anamnézy a dokumentace je pacient afakický. Vízus pravého oka s afakickou korekcí i bez korekce je vpravo 0,5 m prsty, levé oko je slepé. Další symptomatologii u pacienta nebylo možné posoudit ani objektivně ani dle dokumentace.
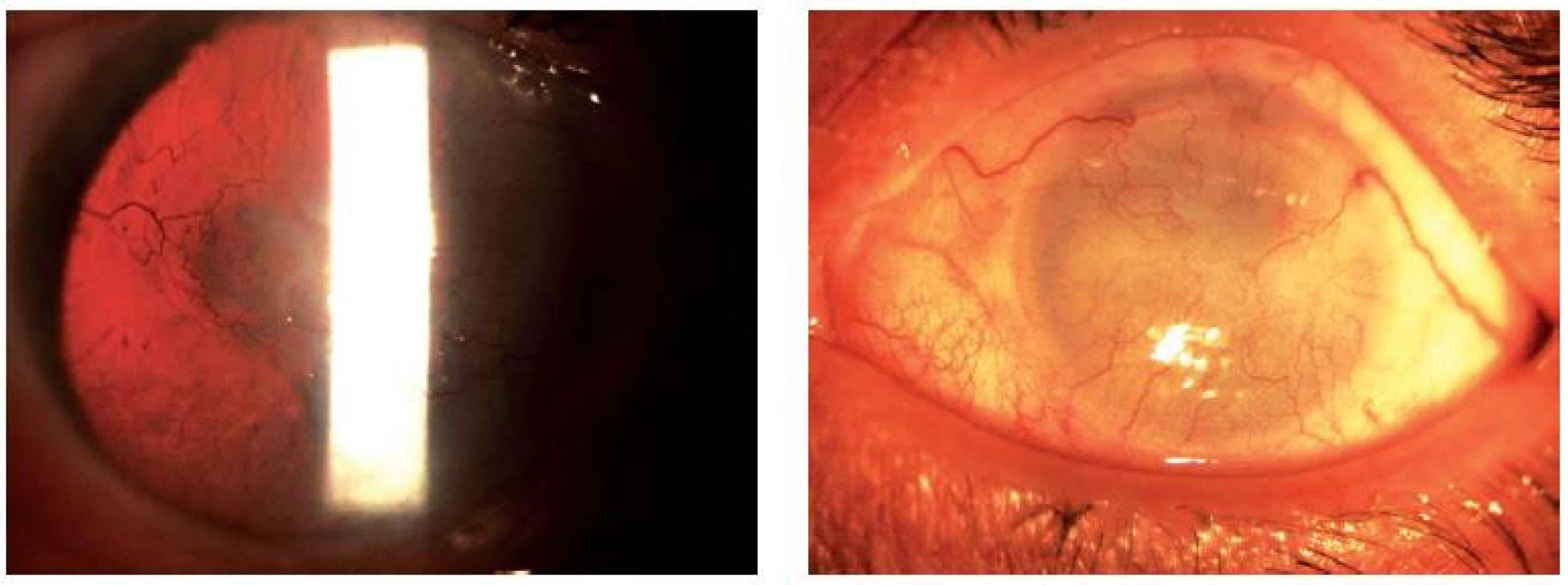
U pětatřicetileté matky (pacientka II-1, obr. 1) s vrozenou bilaterální aniridií s fotofobií je přítomný významný konvergentní strabismus a navíc i horizontální nystagmus a vyšší stupeň AAK (obr. 3). Rohovka je již několik let i dle starší dokumentace se sníženou transparencí a vaskularizací a neumožňuje tak prohlédnout nitrooční struktury a oční pozadí. Zraková ostrost pravého a levého oka se drží na 0,03 a 0,02. Refrakci a bezkontaktní tonometrii nebylo možné změřit. Nitrooční tenze dle Schiötze byla 3,5 / 5,5 a 3,5 / 5,5. Dle starší dokumentace a dle vlastní brýlové korekce byla u pacientky zjištěna myopická refrakční vada (-3,75dsf. bilaterálně, ve staré dostupné dokumentaci není údaj o refrakci nalezen). V šestnácti letech se dle dokumentace u pacientky objevilo počínající kalení čočky, v sedmnácti byla zahájená lokální antiglaukomová terapie betablokátorem pro sekundární glaukom. Poslední zmínka o očním pozadí je v starší dokumentaci z roku 1998 (ve dvaceti letech pacientky), kdy byl nalezen bledý disk zrakového nervu. Další zhodnocení očního pozadí již v té době nebylo možné.

Osmiletý chlapec (pacient III-1, obr. 1) měl již po porodu diagnostikovanou vrozenou totální bilaterální aniridii s fotofobií. I u tohoto pacienta byla zjištěna přítomnost těžké myopie s myopickým astigmatismem (refrakce pravého oka -5,25 D sf. -1,75 D cyl. ax. 22°, refrakce levého oka -7,0 D sf. -2,0 D cyl. ax. 173°), objektivně mírný konvergentní strabismus. Zraková ostrost s korekcí i bez korekce je bilaterálně 0,15. Při vyšetření štěrbinovou lampou byla zjištěna absence duhovky, na rohovce perilimbálně byla přítomná jemná cévní kresba, 1 cévka na pravém oku prostupovala přes limbus směrem k centru (obr. 4). Pomocí štěrbinové lampy byly nalezeny i ojedinělé opacity vpravo v čočce, vlevo naznačená zonulární katarakta. Na očním pozadí je bilaterálně přítomná papila s centrální drobnou exkavací – bez zřetelných glaukomových změn, chybějící foveolární reflex a prořídnutí pigmentového epitelu sítnice (RPE) hlavně v periferii (obr. 6). Oční tenze se drží stabilně – kolem 15 Torrů (gonioskopie, OCT, pachymetrie vzhledem k horší spolupráci a soustředění se společně se špatnou fixací nebylo možné provést).

Sedmiletá dívenka (pacientka III-2, obr. 1) byla porozena z II. fyziologické gravidity, porod v termínu per S.C. z oční indikace matky. Již po porodu byla zjištěna oční vada a v batolecím věku byla vyšetřena v celkové anestezii. V současné době byla přítomná vrozená totální bilaterální aniridie, fotofobie, refrakční vada (myopia gravis s myopickým astigmatismem) a počínající zadní polární katarakta více na pravém oku (obr. 5). Vízus pravého oka s korekcí i bez korekce 0,1 byl bilaterálně, refrakční vada vpravo: -7,25 D sf. -1,5 D cyl. ax. 52° a vlevo: -9,0 D sf. -2,0 D cyl. ax. 125°. Vyšetření štěrbinovou lampou prokazuje počínající perilimbální prošednutí na rohovce a jemnou vaskularizaci periferie rohovky v rámci AAK.

V šesti letech byl dle dokumentace diagnostikován sekundární glaukom a nasazená lokální monoterapie betablokátorem. Pro neuspokojivé nitrooční tenze byla po půl roce zvolena fixní dvojkombinace (brinzolamid s timololem). Nitrooční tenze se vzhledem k silným rohovkám pacientky (678–680 µm dle pachymetrie) pohybuje mezi 20–25 torrů. Obtížněji pořízené OCT skeny papil (špatná fixace pacientky) odhalily menší papily (1,23 mm2 vpravo a 1,17 mm2 vlevo), hodnoty neuroretinálniho lemu a podíl cup/disc svědčil pro pokročilé glaukomové změny se sníženým RNFL. Vzhledem k špatné fixaci a kvalitě skenů byla horší i validita samotného vyšetření. Dle OCT skenů makuly bylo zjištěno foveolární ztluštění 279 µm vpravo a 233 µm vlevo, makulární objem 6,56 mm3 vpravo a 11,08 mm3 vlevo. Při biomikroskopickém vyšetření je bilaterální nález exkavací 0,8–0,9 se zbytkovým neuroretinálním lemem zejména vlevo, makula je bez foveolárního reflexu a sítnice je hlavně v periferii prořídlá s prosvítající kresbou cévnatky (obr. 7, viz foto fundu). Gonioskopické vyšetření nebylo provedeno pro fotofobii a obtížnou spolupráci.

Diskuse
Za výskyt izolované aniridie jsou odpovědné mutace v PAX6¸ ojediněle se může jednat o mutaci v ultrakonzervované oblasti genu ELP4, 150 kb distálně od PAX6 (2).
V současné době je známo přes 286 PAX6 mutací, přičemž cca 90 % z nich bylo spojených s výskytem izolované aniridie nebo souvisejících fenotypů. Mutace se mohou vyskytovat v rozsahu celého PAX6, nejčastěji se vyskytují tzv. non-sense mutace, v důsledku kterých dochází k předčasnému ukončení tvorby bílkoviny. Víc než polovina těchto non-sense mutací vzniká v exonech 8–11. Celkově téměř 90 % PAX6 mutací vedoucích k izolované aniridii má charakter non-sense nebo mění čtecí rámec (28).
Non-sense mutace jsou velmi často příčinou těžšího postižení s menší variabilitou fenotypu. V této rodině byla v exonu č. 8 PAX6 nalezena mutace c.538C>T, v důsledku které glycín v pozici 180 je zaměněn za Stop kodon, tedy dochází hned k ukončení tvorby bílkoviny. Tato mutace byla popsána dosud pouze jedenkrát v rodině s výskytem aniridie (16). V námi popsané rodině je v souladu s charakterem non-sense mutací v PAX6 spojena se závažným fenotypovým dopadem s nevýraznou variabilitou symptomů.
U mnoha rodin s aniridií je zraková ostrost menší než 20/60 (0,33) a nižší než 20/200 (0,1) u více než 60 % v rámci kongenitální foveální hypoplazie nebo sekundárního glaukomu (25). Mezi refrakční vady nacházené u aniridiků patří myopie, hypermetropie, astigmatismus, častá je i anisometropie. Haploinsuficience v důsledku heterozygotní mutace PAX6 je často spojena s vysokou refrakční vadou, speciálně krátkozrakostí (10). U členů rodiny s totální aniridií popsané v této práci se zraková ostrost pohybuje od slepoty po nejlepší vidění s korekcí 0,2. Prognóza výrazného zhoršení zrakové ostrosti je zejména u pacientů z rodin se závažně vyjádřenou symptomatologií (fenotypovou expresivitou) nemoci.
Keratopatie asociována s aniridií je zachycena v jednotlivých stadiích onemocnění dle věku pacientů u všech námi vyšetřených osob s aniridií. AAK se v této rodině vyskytuje v časnějším věku a průběh onemocnění s progresivním zhoršením vizu probíhá rychleji.
OCT papil a makul u nejmladší pacientky svědčilo pro glaukomové postižení papil. U této dívenky bylo také nalezeno foveolární ztluštění 279 µm vpravo a 233 µm vlevo, makulární objem 6,56 mm3 vpravo a 11,08 mm3 vlevo. Holmström a kolektiv (13) testovali vhodnost OCT vyšetření v diagnostice foveální hypoplazie u dětí s albinismem a s aniridií (i speciálně volených a pořízených skenů v případě obtížné fixace a nystagmu). U těchto pacientů pomocí OCT prokázali centrální foveolární ztluštění (vyjádřeno mediánem foveálního minima 259 μm) a menší makulární objem (medián makulárního objemu 7,00–7,01 mm3) proti kontrolní skupině dětí (medián fovealního minima: medián 167 µm, medián makulárního objemu 7,10 μm). Konstatovali, že konfirmace foveální hypoplazie může být nápomocná v diagnóze izolované kongenitální aniridie u pacientů s mírně vyjádřeným fenotypem. Ve srovnání jediného použitelného OCT nálezu (nejmladší pacientka z námi popisované rodiny) s výše uvedenou prací je centrální foveolární ztluštění srovnatelné, přičemž makulární objem je ještě menší.
Vzhledem k tomu, že se často vyskytují patologické změny rohovky, je důležitá aplikace lokální léčby za použití kapek bez konzervačních látek. I jakýkoliv chirurgický zákrok sebou přináší riziko vzniku tzv. fibrózního syndromu u aniridie a zhoršení keratopatie. Syndrom aniridické fibrózy je charakterizován výskytem extenzivní progresivní retrolentikulární a retrokorneální fibrózní membrány. Při rozhodování a volbě terapeutického postupu je proto nutné mít na paměti všechna rizika, aby se dosáhlo co nejdéle možného nejlepšího vidění. I nošení speciálních kontaktních čoček může také zhoršovat nález na rohovce. U pokročilých změn na rohovce je volba keratoplastiky ke zlepšení vidění z dlouhodobého hlediska otázkou do diskuse vzhledem k možné rekurenci nálezu. V potaz je také nutno brát vysoké procento výskytu hůře kompenzovaného glaukomu a tím postupně se horšící zorné pole a vidění. Momentem překvapení může být nález na zrakovém nervu a sítnici při vyšetření očního pozadí po opětovném zprůhlednění optických médií po úspěšné keratoplastice nebo operaci katarakty.
U pětatřicetileté leté matky je náročná rozvaha při volbě zákroku k obnovení transparence rohovky. U této pacientky již v minulosti byl diagnostikován sekundární glaukom, ale oční pozadí nebylo od 20 let vyšetřeno. Je pouze otázkou, jakou zrakovou ostrost a na jak dlouhou dobu zákrokem získáme. I samotná AAK se zhorší často po chirurgických zákrocích na limbu nebo po aplikaci lokálních antimetabolitů v léčbě aniridia-asociovaného glaukomu (19). Z dlouhodobého hlediska je proto u této pacientky nejistý přínos keratoplastiky nebo transplantace limbálních buněk.
Závěr
Aniridie představuje závažné panokulární postižení. Sedmdesát procent diagnostikovaných jedinců s izolovanou aniridií má postižené rodiče. U izolované familiární aniridie je 50% riziko výskytu tohoto onemocnění pro dítě postiženého rodiče. Výjimečně se u těchto pacientů mohou objevovat i non-okulární senzorické projevy, neurologické abnormality popř. postižení slinivky. U pacientů s izolovanou aniridii byla popsána redukce čichu. Poruchy chování a opoždění vývoje jsou raritní (1, 5, 6, 11, 18, 24, 30). Vzhledem k možné výraznější variabilitě fenotypu je důležité genetické vyšetření, jehož význam se znásobňuje zejména v raném dětském věku, kdy pomáhá odlišit pacienty s WAGR (Wilmsův tumor, aniridie, abnormality genitálu, mentální retardace) syndromem, zatím se manifestujících pouze aniridií, od pacientů s mutací v PAX6. Jedná se o progredující onemocnění, terapie je často svízelná zejména u pacientů se závažně vyjádřenou symptomatologií. U takovýchto pacientů je nutné pečlivě zvažovat výběr očních zákroků vzhledem ke zvýšenému riziku následných komplikací. I když se uvádí, že symptomy u aniridie jsou nezvratné a s postupujícím věkem progredující, tak v současné době probíhající výzkumy skýtají pro postižené jistou naději. Aniridie je spojená s nedostatečnou produkcí PAX6 (v důsledku tzv. haploinsuficience PAX6), proto se Gregory-Evans a spol. (9) na myším modelu pokusili postnatálně ovlivnit dávku Pax6. Specifické topické podávání atalurenu a gentamycinu nejen zastavilo progresi onemocnění v důsledku Pax6 mutace, ale do jisté míry i zvrátilo některé změny na rohovce, čočce a retině.
Do redakce doručeno dne 16. 6. 2014
Do tisku přijato dne 18. 7. 2014
MUDr. Marek Godava, Ph.D.
Ústav lékařské genetiky, FN Olomouc
I. P. Pavlova 6
775 20 Olomouc
GodavaM@seznam.cz
Zdroje
1. Bamiou, D.E., Free, S.L., Sisodiya, S.M.: Auditory interhemispheric transfer deficits, hearing difficulties, and brain magnetic resonance imaging abnormalities in children with congenital aniridia due to PAX6 mutations. Arch Pediatr Adolesc Med, 2007; 161(5): 463–469.
2. Bhatia, S., Bengani, H., Fish, M. et al.: Disruption of autoregulatory feedback by a mutation in a remote, ultraconserved PAX6 enhancer causes aniridia. Am J Hum Genet, 93 : 1126-1134, 2013.
3. Brandt, J.D., Casuso, L.A., Budenz, D.L.: Markedly increased central corneal thickeness: an uncrecognized finding in congenital aniridia. AM J. Ophthalmol, 2004; 137(2): 348–350.
4. Socialstyrelsen. Congenital aniridia. [online]. 2012-06-19 [citováno 2014-05-15]. Dostupné z: .
5. Ellison-Wright, Z., Heyman, I., Frampton, I. et al.: Heterozygous PAX6 mutation, adult brain structure and fronto-striato-thalamic function in a human family. Eur J Neurosci, 2004; 19(6): 1505–1512.
6. Free, S.L., Mitchell, T.N., Williamson, K.A. et al.: Quantitative MR image analysis in subjects with defects in the PAX6 gene. Neuroimage, 2003; 20(5): 2281–2290.
7. Glaser, T., Jepeal, L., Edwards, J.G. et al.: PAX6 gene dosage effect in a family with congenital cataracts, aniridia, anophthalmia and central nervous system defects. Nat Genet, 1994; 7(4): 463–471.
8. Grant, W.M. and Walton, D.S.: Progressive changes in the angle in congenital aniridia, with development of glaucoma. Trans Am Ophthalmol Soc, 1974; 72 : 207–228.
9. Gregory-Evans, C.Y., Wang, X., Wasan, K.M.: Postnatal manipulation of Pax6 dosage reverses congenital tissue malformation defects. J Clin Invest, 2014; 124(1): 111–116.
10. Hewitt, A.W., Kearns, L.S., Jamieson, R.V. et al.: PAX6 mutations may be associated with high myopia. Ophthalmic Genet, 2007; 28(3): 179–182.
11. Hingorani, M., Hanson, I., van Heyningen V.: Aniridia. Eur J Hum Genet, 2012; 20(10): 1011–1017.
12. Hodgson, S.V, Saunders, K.E.: A probable case of the homozygous condition of the aniridia gene. J Med Genet, 1980; 17(6): 478–480.
13. Holmström, G., Eriksson, U., Hellgren, K.: Optical coherence tomography is helpful in the diagnosis od foveal hypoplasia. Acta Ophthalmol, 2010; 88(4): 439–442.
14. Jastaneiah, S., Al-Rajhi, A.A.: Association of aniridia and dry eyes. Ophthalmology, 2005; 112(9): 1535–1540.
15. Margo, C.E.: Congenital aniridia: a histopathologic study of the anterior segment in children. J Pediatr Ophthalmol Strabismus, 1983; 20(5): 192–198.
16. Martha, A., Strong, L.C., Ferrell, R.E. et al.: Three novel aniridia mutations in the human PAX6 gene. Hum Mutat, 1995; 6(1): 44–49.
17. McGuire, D., Weinreb, R., Goldbaum, M.: Foveal hypoplasia demonstrated in vivo with optical coherence tomography. Am J Ophthal, 2003; 135(1): 112–114.
18. Mitchell, T.N., Free, S.L., Williamson, K.A. et al.: Polymicrogyria and absence of pineal gland due to PAX6 mutation. Ann Neurol, 2003; 53 : 658–663.
19. Nelson, L.B., Spaeth, G.L., Nowinski, T.S. et al.: Aniridia. A review. Surv Ophthalmol, 1984; 28(6): 621–642.
20. Nishina, S., Kohsaka, S., Yamaguchi, Y. et al.: PAX6 expression in the developing human eye. Br J Ophthalmol, 1999; 83(6): 723–727.
21. Rush, C.C.: Congenital Aniridia. Trans Am Ophthalmol Soc. 1926;24 : 332-341.
22. Seefelder, R.: Die aniridie als eine entwicklungshemmung der retina. Albrecht Von Graefes Arch Klin Exp Ophthalmol, 1909; 70(1): 65–87.
23. Shaw M.W., Falls H.F., Neel J.V.: Congenital Aniridia. Am J Hum Genet, 1960; 12(4): 389–415.
24. Sisodiya, S.M., Free, S.L., Williamson, K.A. et al.: PAX6 haploinsufficiency causes cerebral malformation and olfactory dysfunction in humans. Nat Genet, 2001; 28(3): 214–216.
25. Traboulsi, E.I.: Genetic diseases of the eye. 2nd ed. (Oxford Monographs on Medical Genetics). China: Oxford University Press Inc., 2012. 923 s. ISBN 978-0195326147.
26. Tremblay, F., Gupta, S.K., De Becker, I. et al.: Effects of PAX6 mutations on retinal function: an electroretinographic study. Am J Ophthalmol, 1998; 126(2): 211–218.
27. Tseng, S.C., Prabhasawat, P., Barton, K. et al: Amniotic membrane transplantation with or without limbal allografts for corneal surface reconstruction in patients with limbal stem cell deficiency. Arch Ophthalmol, 1998; 116(4): 431-441.
28. Tzoulaki, I., White, I.M., Hanson, I.M.: PAX6 mutations: genotype-phenotype correlations. BMC Genet, 2005; 6 : 27.
29. Vacek, Z.: Embryologie.1. vyd. Praha: Grada Publishing, 2006. 256 s. ISBN 978-80-247-1267-3. Kapitola 8.8, Vývoj smyslových orgánů, s. 231–238.
30. Valenzuela, A., Cline, R.A.: Ocular and nonocular findings in patients with aniridia. Can J Ophthalmol, 2004; 39(6): 632–638.
31. Wu, L., Ma, Q., Chen, Y. et al.: Abnormalities of ERG in congenital aniridia. Yan Ke Xue Bao, 1991; 7(3): 151–152.
Štítky
OftalmologieČlánek vyšel v časopise
Česká a slovenská oftalmologie

2014 Číslo 4
- Selektivní laserová trabekuloplastika nesnižuje nitroční tlak více než argonová laserová trabekuloplastika
- Progresi glaukomu je třeba hodnotit strukturálními i funkčními parametry
- Ztráta centrálního vidění po filtrujících operacích glaukomu
- Od PGF-2 alfa-isopropyl esteru k latanoprostu: přehled vývoje Xalatanu
- Compliance u pacientů s glaukomem
Nejčtenější v tomto čísle
- Lymfangiom orbitopalpebrální oblasti
- Cytomegalovirová retinitida u pacienta se syndromem získané imunodeficience
- Supracor, laserová metoda korekce presbyopie: výsledky ročního sledování
- Klinické nálezy u členů rodiny s výskytem izolované aniridie v důsledku PAX6 mutace
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy