
Súčasná klasifikácia, diagnostika a prognóza primárnych monoklonových gamapatií (paraproteinémií)
Contemporary classification diagnostic and prognosis of primary monoclonal gammopathies (paraproteinemias)
Since the first reports of Waldenström and others (1940), who introduced diagnostics of polyclonal and monoclonal gammopathies based on paper electrophoresis and immunoelectrophoresis and since largest clinical studies of Kyle and co-workers, the concept of primary monoclonal gammopathies has been presently broaded in itsę clinical importance also due to the increased knowledge in molecular genetics. While implementation of immunofixation and Freelite method into biochemical analysis shown major variability of immunoglobuline molecule, histochemical, immunophenotype, cytogenetic and DNA analyses confirmed polymorphisms of plasma cells and their precursors due to genetic and epigenetic factors. The aim of the presented article is to give information on current classification of monoclonal gammapathies, changes in the opinions on the group of monoclonal gammopathies of undetermined significance (MGUS) and malignant gammapathies (multiple myeloma (MM), Waldenström macroglobulinemia (WM), primary systemic AL amyloidosis, in malignant lymphomas). The risk of malignant transformation of MGUS into myeloma in case of paraproteinemia IgG, IgA and into WM in case of IgM, respectively, is highlighted. Biochemical diagnostics of paraproteinemias using Freelite method for kappa/lambda chains of Ig has gained great importance. While immunofixation can be used for precipitation of paraproteins with complete Ig molecule only, Freelite is able to demonstrate also the free chains not bound to IgH epitopes. Biochemical diagnostics of paraproteinemias has role in the early diagnosis of smoldering WM, smoldering, non-secretory and light-chain myeloma and AL amyloidosis. Freelite method was also implemented into the international diagnostic algorithms of these conditions. The following part of the article is aimed for presentation of our experience with diagnostics in cohort of 1182 malignant gammopathies in years 1990–2009 (995 patients with MM, 96 with smoldering myeloma, 58 with WM and 33 with systemic AL amyloidosis). Conclusion: Demonstration of paraprotein, that has incidence of 1–3 % in healthy population at age 70 years has the role of premalignant factor.
Key words:
classification, monoclonal gammopathies, early diagnostics, paraproteinemias
Autoři:
A. Sakalová 1; D. Škultétyová 2; Z. Konečná 3; K. Weiglová 4; M. Hrubiško 1; M. Mistrík 5; L. Plank 6
Působiště autorů:
Katedra hematológie a transfuziológie Slovenskej zdravotníckej univerzity, Bratislava, 2Národný ústav srdcových a cievnych chorôb, Bratislava, 3Katedra Laboratórnej medicíny Slovenskej zdravotníckej univerzity, Bratislava, 4Medirex s. r. o., biochemické l
1
Vyšlo v časopise:
Transfuze Hematol. dnes,16, 2010, No. 4, p. 193-201.
Kategorie:
Souhrnné práce, původní práce, kazuistiky
Souhrn
Od prvých údajov Waldenströma a ďalších (1940), ktorí na základe papierovej elektroforézy a imunoelektroforézy zaviedli diagnostiku polyklonových a monoklonových gamapatií a dosiaľ najväčších klinických štúdií Kyleho a spolupracovníkov, sa v súčasnosti koncepcia primárnych monoklonových gamapatií podstatne rozšírila v jej klinickom význame a doplnila v hĺbke molekulovo-genetických poznatkov. Kým biochemické analýzy zavedením imunofixačnej elektroforézy a dôkazu ľahkých reťazcov Freelite metódou poukázali na veľkú variabilitu imunoglobulínovej molekuly, histochemické, imunofenotypizačné, cytogenetické a DNA analýzy potvrdili polymorfizmus plazmatických buniek a ich prekurzorov vplyvom genetických a epigenetických faktorov. Cieľom práce je informácia o súčasnej klasifikácii monoklonových gamapatií, o zmenách v názoroch na skupinu monoklonových gamapatií nejasnej príčiny (MGUS) a malígnych gamapatií (mnohopočetný myelóm (MM), Waldenströmová makroglobulinémia (WM), primárna systémová AL amyloidóza, pri malígnych lymfómoch). Zdôrazňuje sa riziko zhubnej transformácie MGUS do myelómu pri paraproteinémii IgG, IgA resp. do WM pri IgM. Veľký význam má zlepšená biochemická diagnostika paraproteinémií zavedením Freelite metódy pre dôkaz kappa/lambda reťazcov Ig. Kým imunofixáciou precipitujú iba paraproteíny s kompletnou Ig molekulou, Freelite zachytáva aj voľné reťazce nenaviazané na epitopy IgH. Biochemická diagnostika paraproteinémií je prínosom pre ich včasnú diagnostiku (WM, smoldering, nesekrečný a ľahkoreťazcový myelóm, AL amyloidóza) a metóda Freelite sa zaviedla aj do medzinárodných diagnostických algoritmov týchto chorôb. V ďalšej časti práce sa uvádzajú vlastné skúsenosti s diagnostikou súboru 1182 malígnych gamapatií v rokoch 1990–2009 (z toho 995 chorých na MM, 96 na smoldering myelóm, 58 na WM a 33 na systémovú AL amyloidózu. Záver: Dôkaz paraproteínu, ktorý sa vyskytuje v zdravej populácii v 1–3 % vo veku 70 rokov v 7–15 % má význam ako premalígny faktor.
Kľúčové slová:
klasifikácia, monoklonové gamapatie, včasná diagnostika paraproteinémie
Úvod
Monoklonová gamapatia (MG) je laboratórny nález zvýšenej hodnoty jednej imunoglobulínovej triedy (najčastejšie IgG) so súčasným znížením hodnôt iných tried (IgA, IgM, IgD, IgE). Pri fyziologických podmienkach je tvorba imunoglobulínov (Ig) humorálnou odpoveďou organizmu na antigénové stimulácie. Tvoria ich imunocyty (lymfoplazmatické a plazmatické bunky), ktoré si vytvorili počas diferenciácie z B lymfocytov zložitý systém tvorby protilátok (imunoglobulínov) špecificky reagujúcich na antigénové podnety. Podľa Waldenströma (1940) je štruktúra Ig natoľko adaptabilná, že reaguje na milióny rôznorodých antigénov (1). V súčasnosti sú na molekulovej úrovni preskúmané chemické štruktúry 5 Ig tried s rôznymi imunobiologickými funkciami. V obrane proti infekciám má dominantné zastúpenie trieda IgG so 4 podtriedami a 25 alotypovými variantami. U zdravých sa na tvorbe Ig podieľajú viaceré subpopulácie lymfocytov, preto sa nazývajú polyklonové Ig (2).
Monoklonový Ig (MIg, paraproteín) tvorí jeden klon lymfocytov pre ťažké reťazce a jeden klon pre ľahký reťazec kappa (κ) alebo lambda (λ). Táto zmena sa uskutočňuje na úrovni pre-B lymfocytov v štádiu, keď dochádza k preskupeniu génov pre ťažké a ľahké reťazce. Zmena polyklonovej tvorby na monoklonovú je výsledkom somatickej mutácie génov vplyvom epigenetických faktorov vonkajšieho prostredia. Tvorba paraproteínu je preto klonový proces rôznej etiológie. Pri primárnych MG je príčinou plazmocelulárna dyskrázia na podklade autonómnej transformácie a proliferácie plazmatických buniek, pri sekundárnych MG sa dokáže súvislosť s iným ochorením. Najpočetnejšiu skupinu primárnych tvorí MGUS (monoklonová gamapatia nejasného významu, ale aj z neznámej príčiny), na druhom mieste sú malígne gamapatie podmienené zhubným lymfoproliferatívnym procesom. Skupinu sekundárnych resp. reaktívnych MG, zapríčiňujú autoimunitné, infekčné, metabolické a toxické ochorenia (3, 4, 5).
Znalosti o klinickom význame MG nadväzovali na technické zdokonalenie elektroforetických metód ako aj na klinické skúsenosti z veľkých súborov pacientov, najmä z pracoviska prof. Kyleho (Mayo Clinic, Rochester) (4). Výsledky imunochemických analýz Ig metódou imunofixácie v klinickej praxi ukázali, že táto presná metóda reaguje iba s paraproteínmi, tvorenými kompletnými molekulami Ig a diagnostike unikali procesy zapríčinené nekompletnými molekulami Ig, predovšetkým paraproteinémie so zvýšením iba voľných ľahkých reťazcov. Tento nedostatok odstránila nefelometrická / imunoturbidimetrická metóda (latex assay, Freelite), ktorá viaže voľné ľahké reťazce v sére alebo v moči pomocou vysoko senzitívnych protilátok. Tieto sa môžu pri zhubných lymfoplazmocelulárnych procesoch (napr. ľahkoreťazcový podtyp MM) neprimerane zvýšiť v porovnaní ku ťažkým reťazcom a preto cirkulujú ako voľné, nenaviazané na epitopoch ťažkých Ig reťazcov. Tieto nízkomolekulové proteíny unikajú detekcii imunofixačnou elektroforézou. Freelite imunoelektroforéza sa zaviedla v roku 2000 v USA a postupne sa rozšírila do biochemických pracovísk. Bolo dokázané, že má vyššiu senzitivitu a tým aj selektívnejšiu záchytnosť, najmä pri chorobách z nadprodukcie ľahkých reťazcov (6, 7, 8).
Klasifikácia monoklonových gamapatií (paraproteinémií)
Laboratórny dôkaz paraproteínu nemožno považovať za fyziologický resp. nevýznamný nález, aj keď sa veľmi zriedka vyskytuje u zdravých vo veku < 50 rokov (v 1–3 %). V staršom veku sa v dôsledku poklesu imunitnej kontroly jeho frekvencia zvyšuje a vo veku > 75 rokov dosahuje 3–10 % v bielej populácii (prevalencia v čiernej rase je podľa niektorých autorov až 3x vyššia). Keďže sa v súčasnosti zisťuje, že u 20–25 % nosičov paraproteinémie po 1 až niekoľkých rokoch nastáva malígna transformácia, zmenili sa názory na význam MG, ba dokonca sa nález paraproteínu považuje za jeden z najvýznamnejších premalígnych ukazovateľov. Samotný klasický názov „paraproteinémia“ neznamená, že sa tvorí patologický imunoglobulín, ale vystihuje skutočnosť, že rôzne štruktúrne abnormality imunoglobulínovej molekuly sú prejavom polymorfizmu génov na molekulovej úrovni ako následok nekoordinovanej proteosyntézy (9).
Diagnostické kritéria a algoritmy podľa smerníc Medzinárodnej pracovnej skupiny pre MM a iné MG (IMWG) a ďalších autorov (9, 10, 12, 13)
Primárne – monoklonová gamapatia nejasného významu (MGUS monoclonal gammopathy of undetermined significance (tab. 1))

- mnohopočetný myelóm (s prevahou IgG paraproteínu) a jeho varianty (tab. 2, 3, 4)



- Waldenströmova makroglobulinémia s paraproteínom IgM (tab. 5)

- primárna systémová AL amyloidóza (tab. 6)

- pri malígnych lymfómoch (NHL lymfómy, CLL, heavy chain disease (zriedkavé), idiopatická kryoglobulinémia – zriedkavé).
Sekundárne – sú reaktívneho pôvodu (vírusové a bakteriálne infekcie, autoimunitné, nádorové procesy, reakcie štepu proti hostiteľovi po transplantáciach kostnej drene).
Klinická charakteristika primárnych monoklonových gamapatií
Monoklonová gamapatia nejasnej príčiny (MGUS)
Vyskytuje sa ako náhodný nález u zdravých. Keďže nie sú prítomné klinické príznaky choroby, je nález paraproteínu prekvapením. Najčastejšie sa zisťuje zvýšenie IgG > 20 g/l (norma < 17 g/l). Hodnoty paraproteínu IgA a IgM pri MGUS sa pohybujú medzi 5–15 g/l (norma < 5 g/l) (tab. 1). Paraproteín sa dokazuje v sére. V súčasnosti je dôležité pri štandardnom používaní imunofixačnej elektroforézy pre dôkaz množstva ťažkých reťazcov Ig (G, A, M, E, D), doplniť vyšetrenie ľahkých reťazcov κ a λ Freelite metódou. Ďalšími diagnostickými kritériami pre MGUS je normálne zastúpenie lymfocytov v kostnej dreni, chýbanie proteinúrie, dobrý klinický stav. MGUS sa donedávna považovala za benígny nález. Stanovisko ku prognóze sa zmenilo v dôsledku výsledkov dlhodobých klinických štúdií Kyleho, Rajkumara a ďalších, ktorí v roku 1998–1999 vyhodnotili súbor 1149 paraproteinémií a zistili, že MGUS sa vyskytovala v 63 % a 22 % tvorili mnohopočetný myelóm (MM) s jeho variantami, Waldenströmova makroglobulinémia v 2 %, AL amyloidóza v 10 % a iné lymfoproliferatívne ochorenia v 3 % (4). Ďalšie pozorovania v súbore 29 528 paraproteinémií sa v podstate nelíšili a potvrdili prevalenciu MGUS v 61 %, AL amyloidózy v 9 % a MM približne v 30 % (8). Na MGUS sa preto v súčasnosti pozeráme ako na rizikový faktor s možnosťou transformácie do Waldenströmovej makroglobulinémie pri IgM paraproteinémii alebo do mnohopočetného myelómu pri IgG, IgA type. Potrebná je preto dlhodobá dispenzarizácia a kontroly v približne 6 mesačných intervaloch (14, 15).
Waldenströmova makroglobulinémia (WM)
Zaraďuje sa do skupiny nehodgkinových lymfómov s výskytom 2–7 % z celkového počtu NHL, ktoré sú najčastejším zhubným hematologickým procesom (priemerný výskyt nových prípadov v USA cca 50 000). V porovnaní s výskytom MM je výskyt WM asi 10x zriedkavejší (0,34/100 000). Pre latentný priebeh s nevýrazným klinickým obrazom, ktorý je komplikovaný častými infekciami (bronchitídy, pyelonefritídy) je medián prežitia iba ±5 rokov. Vyskytuje sa vo veku > 50 rokov s miernou prevahou u mužov. Najčastejšie klinické príznaky sú: anémia > 50 %, hepatosplenomegália a lymfadenopatia v 15–20 %, periférna neuropatia v 20 %, hyperviskozita séra v 15 %. Príčinou WM je nádorová proliferácia lymfocytov, resp. lymfoplazmocytov-imunocytov, secernujúcich IgM paraproteín. Infiltrácia kostnej drene nádorovým procesom zapríčiňuje potlačenie normálnej krvotvorby, pokles imunitnej obrany s následnými klinickými komplikáciami. Mnohé štúdie poukazujú na genetickú predispozíciu (familiárny výskyt monoklonových gamapatií) avšak na etiopatogenéze ochorenia sa podieľajú aj získané faktory (autoimunitné choroby, alergická predispozícia, vírusové – najmä HCV, HIV, ricketsiózy a recidivujúce bakteriálne infekcie). Anémia býva normocytová, častejšie mikrocytová, ktorá je príčinou slabosti a kolapsov. Príčinou krvácania, najmä do kože a slizníc je trombocytopénia pri hypoplázii krvotvorby. Časté infekcie ohrozujú chorého respiračnými a kardiálnymi komplikáciami. Paraproteinémia triedy IgM so zvýšením > 15 g/l zhoršuje klinický stav život ohrozujúcim syndrómom zvýšenej viskozity, ktorý vedie ku poruchám cirkulácie najmä v mozgu, obličkách, k očným komplikáciam nebezpečným najmä pri zvýšenej krvácavosti. Asi u jednej tretiny sa môže WM transformovať do lymfocytovej leukémie a veľmi zriedka do mnohopočetného myelómu s refraktérnosťou na chemoterapiu a fatálnym priebehom (11, 12, 18). V súčasnej diagnostike okrem štandardného histomorfologického vyšetrenia kostnej drene značne prispieva imunofenotypizácia lymfoplazmocytov a pre prognostické posúdenie okrem kritérií Medzinárodného prognostického systému sú známe aj niektoré molekulovo-genetické markery (tab. 5) (16, 17, 18, 19, 23, 27, 30, 43, 44).
Mnohopočetný myelóm (MM)
Tvorí asi 10 % hematologických malignít, je druhým najčastejším hematologickým ochorením. Incidencia MM u belochov je 3,6–4/100 000 a v čiernej rase je výskyt MM na prvom mieste asi 30/100 000. V USA je ročný prírastok cca 15 000–19 000 chorých, prevalencia 63 000. V Anglii je incidencia cca 3 000 a na Slovensku cca 200–250 (9, 10, 14, 15, 22, 28, 29). MM je ochorením starších s mediánom výskytu 66–68 rokov s miernou prevalenciou mužov.
Podobne ako pri iných zhubných procesoch nie je známa presná príčina. Predpokladá sa proces somatickej mutácie génov vplyvom epigenetických faktorov (dlhodobá antigénová stimulácia vírusovými a bakteriálnymi infekciami, expozícia radiácii a iným fyzikálnym a chemickým faktorom), pričom sa pripúšťajú aj familiárne predispozície a zdôrazňuje sa význam dlhodobého nosičstva MGUS na transformácii do MM (11, 14, 21). Nádorová choroba plazmatických buniek a ich prekurzorov je zapríčinená ich nekontrolovanou monoklonovou transformáciou a proliferáciou v kostnej dreni, ale aj v iných orgánoch (najmä v kostiach, obličkách). Proces somatickej mutácie génov sa uskutočňuje v pre-B lymfocytoch (uzliny) na úrovni preskupenia génov pre ťažké (IgH) reťazce, ktorý vyúsťuje do génového polymorfizmu a početných chromozómových mutácií. V kostnej dreni sa uskutočňuje fáza nekoordinovanej proliferácie aktivovanej kontaktom transformovaných plazmocytov so stromálnymi bunkami kostnej drene a aktiváciou cytokínovej kaskády.
V klinickom obraze sa symptomatický MM charakterizuje príznakmi hematologickými (anémia, neskôr trombocytopénia, pancytopénia), biochemickými (monoklonová gamapatia s tvorbou predovšetkým paraproteínu IgG κ alebo λ a ďalších Ig tried), imunologickými, metabolickými a paraneoplastickými (hyperkalcémia, hyperviskózny syndróm, trombózy, krvácania z interakcie paraproteínov s koagulačnými faktormi a ďalšie).
Z orgánových komplikácií sú závažné predovšetkým kostné (tvorba osteolytických ložísk, patologické fraktúry, ale aj difúzna osteoporóza), obličkové (tubulo-intersticiálna nefropatia), neurologické (polyneuropatie, parézy). Kardiálne a hepatálne komplikácie sa častejšie vyskytujú pri MM s AL amyloidózou. Metabolické zmeny a tzv. cytokínová búrka súvisia s interakciou myelómových buniek so stromálnymi (monocyty, makrofágy, fibroblasty, osteoblasty, osteoklasty, endotelové a dendritické bunky). Následkom tejto interakcie makrofágy, ale aj myelomové bunky tvoria osteolyticky pôsobiace faktory rodiny TNF – nukleárny faktor kappa B-NFkB a jeho cirkulujúci aktivátor RANK-L (ligand aktivujúci receptor NFkB). Ich interakciou nastáva zmnoženie a aktivácia osteoklastov. Zvyšuje sa ďalej tvorba interleukínov (1, 3, 6 a ďalších), rastových faktorov (endotelový, fibroblastový a najmä skupina transformujúcich rastových faktorov-ß (TGF-ß) (14, 15).
Nádorový proces infiltruje kostnú dreň, odkiaľ makrofágy ale aj myelómové bunky cirkulujú do krvi a usídľujú sa v rôznych orgánoch. MM je asi v 90 % difúzny proces a 10–15 % tvoria varianty MM (solitárny – kostný a extramedulárny, nesekrečný, oligosekrečný) (tab. 4), POEMS (polyneuropatia, osteosklerotické ložiská, endokrinopatie, M-proteín, kožné prejavy) (tab. 6), plazmocelulárna leukémia.
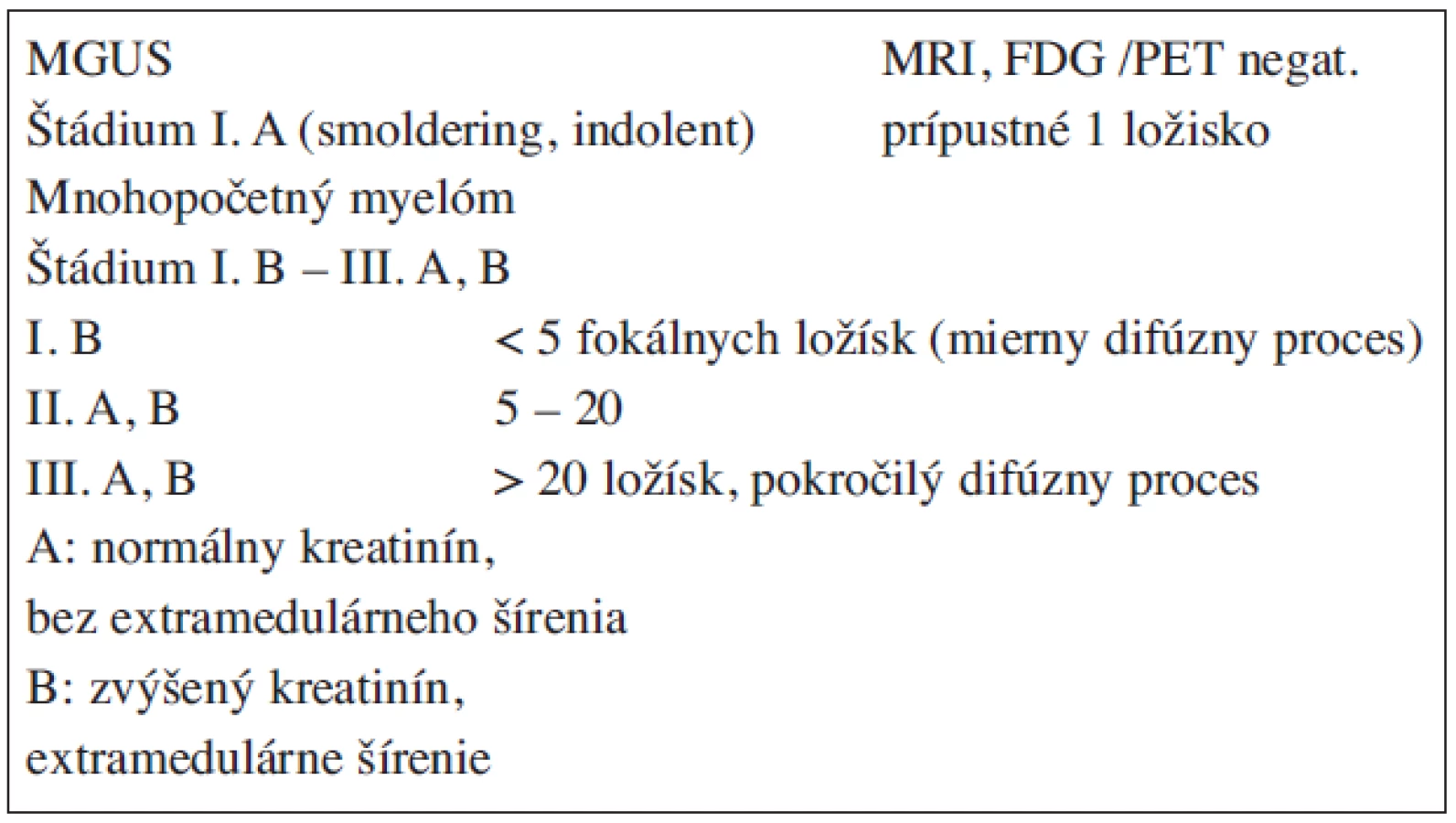
Mitotická aktivita nádorovej plazmatickej bunky je pomalá, preto tento zhubný proces môže prebiehať 2 a viac rokov latentne, bez klinických prejavov ako asymptomatický (smoldering MM), ktorému môže predchádzať MGUS (tab. 2) (13, 14, 15, 24, 25, 26, 28, 34). Kým riziko transformácie MGUS do MM je 0,6–3 % ročne a MGUS sa hodnotí ako premalígny proces, smoldering MM má laboratórne a molekulovo-genetické charakteristiky ako symptomatický MM s rizikom progresie ≥10 % ročne. Na význam rozpoznania smoldering MM poukazujú práce Kyleho et al., ktorí počas 5ročného sledovania 276 chorých zistili v skupine s 19% plazmocytózou prechod do MM až v 57 % (4). Je potrebné ďalej zdôrazniť, že sa pozmenilo aj určenie štádia podľa Durieho-Salmona v tom, že autori I. A štádium považujú za smoldering MM (tab. 7, 8) (12).

Včasná diagnostika tohto zákerného ochorenia a komplexná liečba môžu zlepšiť prognózu prežívania aj kvalitu života. Zlepšením diagnostiky a liečebných postupov (intenzívna chemoterapia s transplantáciou krvotvorných buniek, nové lieky – bortezomib, thalidomid, lenalidomid a ďalšie) sa v súčasnosti prognóza prežitia predĺžila na > 5–10 rokov a zvýšil sa najmä počet kompletných remisií (10, 20, 21, 22, 46, 47, 48, 51).
Systémová AL amyloidóza
Považuje sa za variant symptomatického MM asi v 25–30 %, ale môže prebiehať ako primárna monoklonová gamapatia podmienená transformáciou plazmatických buniek s amyloidogénou sekrečnou aktivitou bez príznakov MM, resp. v kombinácii so smoldering MM. Zriedka (± 5 %) postihuje iba jeden orgán (oblička, srdce), ale v 70–80 % má charakter systémového postihnutia (tab. 9).

Hlavnou charakteristikou tohto veľmi heterogénneho a nevyliečiteľného metabolického procesu je tvorba a ukladanie nerozpustnej bielkoviny – amyloidu do extracelulárnej matrix orgánov s ich následným zlyhaním. Súčasné ultraštruktúrové, imunochemické a genetické analýzy umožňujú klasifikáciu amyloidózy na:
Primárna AL (amyloid light) – z nadprodukcie, fibrilárnej agregácie a orgánového ukladania amyloidu tvoreného z ľahkých reťazcov kappa, ale najčastejšie lambda. Amyloidové ložiská sa tvoria v interstíciu orgánov a v stenách arteriol s ich následnou hyalinizáciou a sklerotizáciou.
Sekundárna AA – amyloidové fibrily tvoria zápalové non-imunoglobulínové proteíny (sérový amyloid-SAA, prealbumín-transtyretín a ďalšie amyloidogénne proteíny – tyreokalcitonín, inzulín, prolaktín, ß2-mikroglobulín, nátriuretický peptid). Ako sprievodný proces sa vyskytuje asi v 5 % pri reumatickej artritíde a pri zriedkavých chronických zápalových ochoreniach (TBC, osteomyelitis). So zlepšením liečby základných ochorení ich frekvencia značne poklesla.
Familiárna amyloidóza – je dedičným ochorením, ktoré je spôsobené rôznymi amyloidogénnymi proteínmi. Je známych 23–27 amyloidogénnych proteínov. Okrem spomínaných sú na molekulovej úrovni preskúmané niektoré varianty lyzozýmu, apolipoproteínov, cystatínu, gélsolínu, fibrinogénu. V súčasnosti pribudli pozorovania o familiárnom výskyte AL amyloidózy, preto sa pri diferenciálnej diagnostike AL amyloidózy zapríčinenej MM v špeciálnych prípadoch vyžaduje molekulovo genetické vyšetrenie (31, 32, 33, 34, 35, 36, 37, 41, 42, 45).
Klinický obraz sa prejavuje obrazom nefrotického syndrómu, kardiálnych príznakov (kolapsovité stavy, palpitácie, obraz restrikčnej kardiomyopatie), senzorickou neuropatiou, hepatomegáliou, makroglosiou, paraproteinémiou (lambda). Ak sa nemyslí na možnosť amyloidózy obyčajne sa diagnóza určí až v pokročilom štádiu. Na kombináciu MM a AL amyloidózy poukazuje nález paraproteínu v sére a v moči, pozitívny nález farbenia na amyloid (konžskou červeňou a iné) v kostnej dreni a v bioptických materiáloch. Pretože u väčšiny sa zisťuje nízka hodnota M proteínu v sére, ba dokonca sa ani nedokáže, veľký význam pri určení diagnózy a aj pri sledovaní odpovede na liečbu má metóda stanovenia ľahkých reťazcov. Ich pokles o > 50 % je znamením dobrej odpovede (13, 16).
Vlastné skúsenosti z dlhoročného sledovania malígnych gamapatií
Retrospektívne sme vyhodnotili súbor malígnych gamapatií diagnostikovaných a dlhodobo sledovaných od roku 1990 do roku 2009. Z celkového počtu 1 182 pacientov sa zistil symptomatický mnohopočetný myelóm u 995 (77,2 %), smoldering MM u 96 (15,1 %), Waldenströmova makroglobulinémia u 58 (4,0 %), AL amyloidóza u 33 (3,8 %). Výskyt MGUS sme nesledovali (tab. 10, 11).



Podrobné klinické charakteristiky, liečebné postupy, výsledky a prognostické závery boli uverejnené (24, 34, 36, 50, 51). Štúdia bola vykonávaná v spolupráci so zahraničnými centrami a podrobne štatisticky hodnotená (51, 52, 53, 54). Výskum sa uskutočnil so súhlasom etických komisií. Skupiny liečené intenzívnou chemoterapiou v kombinácii s autotransplantáciou krvotvorných buniek a skupiny liečené kombináciami thalidomidu, bortezomibu, lenalidomidu sa priebežne vyhodnocujú v spolupráci s Českou myelómovou skupinou (48, 53).
Súbor 995 chorých na MM delíme na dve 10-ročné etapy z dôvodov rozdielov v chemoterapii: V I. skupine 485 pacientov bola podávaná kombinovaná chemoterapia (VMCP, VBMCP), v II. skupine boli chorí liečení rôznymi chemoterapeutickými postupmi. Ak nespĺňali podmienky intenzívnej liečby v kombinácii s autológnou transplantáciou krvotvorných buniek podľa medzinárodných protokolov (74 chorých) bola podávaná okrem spomínaných chemoterapia VAD, CAD (cyklofosfamid, adriamycín, dexametazón) a novšie kombinácie (bortezomib, lenalidomid, thalidomid (48, 50, 10). Druhým dôvodom delenia súboru bolo sledovanie významu zlepšenej biochemickej diagnostiky zavedením dôkazu ľahkých reťazcov Ig Freelite metódou a porovnanie jej senzitivity so štandardnými elektroforetickými metódami (imunofixačná ELFO séra a moči).
Výsledky
Pri porovnávaní výsledkov biochemickej analýzy proteínov v skupine 510 chorých na MM sledovaných od r. 2000 nastal v porovnaní so skupinou 485 chorých (1990–2000) posun vo vyššej záchytnosti paraproteínov v hodnotách medzi 20–30 g/l, čo umožnilo detegovať oligosekrečné a smoldering podtypy vo včasnom štádiu MM. Vyššia záchytnosť dôkazu ľahkých reťazcov v sére a v moči sa odzrkadlila na vyššej záchytnosti najmä ľahkoreťazcových podtypov MM, na spresnení diagnózy AL amyloidózy a nesekrečných foriem MM.
Kým v prvej skupine sme podľa hodnôt paraproteínu mohli do I. štádia zaradiť zo 485 iba 84 pacientov (17,3 %), v skupine 510 chorých pozorovanej od r. 2000 sme zaradili 159 (30,9 %). Počet oligosekrečných foriem stúpol z 8 % na 13,1 %, nesekrečných z 2 % na 4 % a ľahkoreťazcových z 3,9 % na 8 % (tab. 12, 13).





Diskusia
Cieľom práce je informácia o súčasných poznatkoch v oblasti včasnej detekcie malígnych gamapatií, ktorých hlavným reprezentantom je mnohopočetný myelóm a upozorniť na súčasnú koncepciu v prognostickom posudzovaní MGUS donedávna považovanej za „benígnu“. Nie je možné podceňovať nález paraproteínu aj v nízkych hodnotách v populácii < 50 rokov, bez opätovnej kontroly a bez kompletného prešetrenia. MGUS predstavuje v súboroch sledovaných pre výskyt MG cca 60 % zastúpenie (4, 8). Z tejto skupiny môže 1–3 % prípadov ročne transformovať do MM resp. do WM alebo amyloidózy. Ak usúdime, že základom úspešnej liečby je včasná diagnóza, v plnej miere platí táto zásada dlhodobého sledovania MGUS. Včasnej diagnóze unikajú ďalej oligosekrečné, nesekrečné, ľahkoreťazcové, lymfoplazmocytové varianty MM a AL amyloidóza. Zavedením detekcie ľahkých reťazcov Ig metódou Freelite sa zvýšila senzitivita v odhalení týchto atypických ale prognosticky veľmi závažných malígnych gamapatií. Sledovanie voľných reťazcov sa stalo súčasťou medzinárodných diagnostických a prognostických algoritmov (12, 13, 28, 35, 39).
V práci sa ďalej zameriavame na posúdenie významu rôznych diagnostických kritérií a algoritmov pre MM a iné MG. Je potrebné zvážiť, ktoré by mali byť štandardnými pre prax a ktoré sú potrebné pre hlbšiu diferenciálnu diagnostiku. V mnohých publikovaných štúdiách z najvýznamnejších centier, ktoré sledujú klonalitu nádorových buniek sa kvantitatívne hodnoty pre plazmocytózu neuvádzajú a zdôrazňuje sa prínos imunofenotypizácie prietokovým cytometrom, alebo cytogenetiky FISH metódou (22, 49). Pre výskumné účely sa v súčasnosti používa klasifikácia molekulovo-genetická, ktorá využíva cytogenetické metódy a molekulovú genetiku (porovnávanie chromozómových aberácií – hyperploídia a translokácia IgH t(11;14)(q 32;q32) je spojená s dobrou prognózou, hypoploídia s t(4;14)(p16;q32) a ďalšie abnormality so zlou prognózou) so zastúpením génov zodpovedných za MM (MMSET, CCND, cMAF, MAFB v kombinácii s cyklínmi) (25, 26).
Skúsenosti niekoľkých rokov ukázali, že pre štandardnú klinickú diagnostiku MG je vhodná IMWG klasifikácia z roku 2003 s určitými doplnkami (9, 13, 16, 28, 31, 48). Pre určenie štádia pokročilosti MM ako štandardný zostáva staging Durieho a Salmona (1975) doplnený ako tzv. „staging plus“ (2003) (tab. 7, 8). Neoddeliteľnou súčasťou diagnostiky zostáva štandardná histomorfologická diagnostika a skórovacie systémy určujúce liečebnú stratégiu a odpoveď na liečbu (Tab. 14, 15). Bližšie sa nezaoberáme významnými pokrokmi v neinvazívnej diagnostike ani s problémami liečebných postupov, ktoré sa menia a doplňujú v súlade s vedeckým pokrokom. Potešiteľná je skutočnosť, že z literatúry už vymizla smutná definícia MM ako ochorenia s mediánom prežitia asi 3 rokov. Súčasná prognóza MM sa hodnotí podľa štádia, veku a iných prognostických faktorov, odpovede na liečbu s predpoveďou na prežitie nad 10 rokov od 15–30 % a v skupine chorých < 50 rokov nad 50 % (47, 48). V našom súbore liečenom v I. etape (1990–2000) sme pozorovali medián prežitia 88 mesiacov s prežívaním nad 10 rokov u 19 % chorých. V druhej skupine bol medián prežitia 94 mesaciov a dalšie výsledky sú totožné s medzinárodnými a priebežne sa vyhodnocujú (51). V skupinách 58 chorých na Waldenströmovú makroglobulinémiu a 33 chorých na systémovú AL amyloidózu sa medián prežitia pohyboval okolo 50 mesiacov (50, 51). Variantné formy MM (nesekrečný, solitárny plazmocytóm, plazmocelulárna leukémia) – bez známok monoklonovej gamapatie neuvádzame, pretože pri týchto by sa paraproteín nemal vyskytovať.


Záver
Zlepšená biochemická diagnostika má mimoriadny význam pre včasnú diagnózu malígnych gamapatií a pre zlepšenie prognózy ich liečiteľnosti.
Doručeno do redakce: 21. 12. 2009
Přijato po recenzi: 21. 6. 2010
Prof. MUDr. Adriena Sakalová, DrSc.
Katedra hematológie a transfuziológie
Slovenská zdravotnícka univerzita
Limbová 12
833 03 Bratislava - Kramáre
Slovensko
Zdroje
1. Waldenström J. Diagnosis and treatment of multiple myeloma. Grune Stratton ed., New York, 1970.
2. Whicher JT. Monoclonal proteins. In: Holborow and Reeves WG Immunology in Medicine 2nd Ed. Academic Press, London and New York, 1983; 655 : 531-557.
3. Kyle RA, Lust JA. Monoclonal gammopathies of undetermined significance. Semin Hematol 1989; 26 : 176-200.
4. Kyle RA, Rajkumar V. Monoclonal gammopathies of undetermined significance. Hematol Oncol Clin N Amer 1999; 13 : 1181-1202.
5. Adam Z, Tomíška M, Hájek R, et al. Monoklonální gamapatie nejasného významu (benigní monoklonální gamapatie). Vnitř Lék 2000; 46 : 286-296.
6. Bradwell AR, Carr-Smith HD, Mead GP, et al. Highly sensitive automated immunoassay for immunoglobulin free light chains in serum and urine. Clin Chem 2001; 47 : 673-680.
7. Bradwell AR, Harding SJ, Fournier NJ, et al. Assessment of monoclonal gammopathies by nephelometric measurement of individual immunoglobulin κ/λ ratio. Clin Chem 2009; 55 : 1646-1655.
8. Katzmann JA, Clark RJ, Abraham RS, et al. Serum reference intervals and diagnostic ranges for free κ and free λ immunoglobulin light chains: relative sensitivity for detection of monoclonal light chains. Clin Chem 2002; 48 : 1437-1444.
9. Kyle RA, Child JA, Durie BGM, et al. The International Myeloma Working Group criteria for the classification of monoclonal gammopathies, multiple myeloma and related disorders: a report of the International Myeloma Working Group. Br J Haematol 2003; 121 : 749-757.
10. Durie BGM, Kyle RA, Belch A, et al. Myeloma management guidelines: a consensus report from the Scientific Advisors of the International Myeloma Foundation. Hematol J 2003; 4 : 379-398.
11. Kyle RA, Kumar S. The significance of monoclonal gammopathy of undetermined significance. Haematologica 2009; 94 : 1641-1644.
12. Durie BGM, Harousseau JL, Miguel JS, et al. International uniform criteria for multiple myeloma. Leukemia 2006; 20 : 1467-1473.
13. Dispenzieri A, Kyle RA, Merlini G, et al. International Myeloma Working Group guidelines for serum-free light chain analysis in multiple myeloma and related disorders. Leukemia 2009; 23 : 215-224.
14. Pontet F. A database for 3000 monoclonal immunoglobulin cases and a new classification. Clin Chimica Acta 2005; 355 : 13-21.
15. Landgren O, Kyle RA, Pfeiffer RM, et al. Monoclonal gammopathy of undetermined significance (MGUS) and multiple myeloma: a prospective study. Blood 2009; 113 : 5412-5417.
16. Dimopoulos M, Kastritis E, Delimpassi S, Zomas A, Kyrtsonis Ch, Zervas K. The International prognostic scoring system for Waldenströmęs macroglobulinemia is applicable in patients treated with rituximab-based regimens. Haematologica 2008; 93 : 1420-1422.
17. Kyle RA, Rajkumar SV. Criteria for diagnosis, staging, risk stratification and response assessment of multiple myeloma. Leukemia 2009; 23 : 3-9.
18. Treon S. How I treat Waldenströmęs macroglobulinemia. Blood 2009; 114 : 2375-2385.
19. Gertz M, Fonseca R, Rajkumar V. Waldenströmęs macroglobulinemia. Oncologist 2000; 5 : 63-67.
20. Dispenzieri A, Zhang L, Katzmann JA, et al. Appraisal of immunoglobulin free light chain as a marker of response. Blood 2008; 111 : 4908-4915.
21. Weiss BM, Abadie J, Verma P, et al. A monoclonal gammopathy precedes multiple myeloma in most patients. Blood 2009; 113 : 5418-5422.
22. Bladé J. Multiple myeloma: diagnosis, staging and criteria of response. Hematology, Education program EHA 2007; 1 : 102-107.
23. Baldini L, Goldaniga M, Guffanti A, et al. Immunoglobulin M monoclonal gammopathies of undetermined significance and indolent Waldenströmęs makroglobulinemia recognize the same determinants of evolution into symptomatic lymphoid disorders: proposal for a common prognostic scoring system. J Clin Oncol 2005; 23 : 4662-4668.
24. Sakalová A, Ilenčíková D, Weiglová K, et al. Včasná diagnostika mnohopočetného myelómu – jeho latentných a variantných foriem. Interná Med 2008; 8 : 91-97.
25. Chiecchio L, Dagrada GP, Protheroe RK, et al. Loss of 1p and rearrangement of MYC are associated with progression of smouldering myeloma to myeloma: sequential analysis of a single case. Haematologica 2009; 94 : 1024-1028.
26. Blotta S, Tassone P, Prabhala RH, et al. Identification of novel antigens with induced immune response in monoclonal gammopathy of undetermined significance. Blood 2009; 114 : 3276-3284.
27. Kristinsson S, Björkholm M, Goldin LR, et al. Risk of lymphoproliferative disorders among first-degree relatives of lymphoplasmacytic lymphoma/Waldenström macroglobulinemia patients: a population-based study in Sweden. Blood 2008; 112 : 3052-3056.
28. Durie BGM, Harousseau J-L, Miguel JS, et al. International uniform response criteria for multiple myeloma. Leukemia 2006; 20 : 1467-1473.
29. Bladé J. Diagnostic criteria for MGUS, multiple myeloma and other conditions. N Engl J Med 2006; 355 : 2765-2771.
30. Cesana C, Miqueleiz S, Bernuzzi P, et al. Smouldering Waldenströmęs macroglobulinemia: Factors predicting evolution to symptomatic disease. Semin Oncol 2003; 30 : 231-235.
31. UK Myeloma Forum: Guidelines on the diagnosis and management of AL amyloidosis. Br J Haematol 2004; 125 : 681-700.
32. Gertz MA, Comenzo R, Falk RH, et al. Definition of organ involvement and treatment response in immunoglobulin light chain amyloidosis (AL): A consensus opinion from the 10th International Symposium on Amyloid and Amyloidosis. Am J Hematol 2005; 79 : 319-328.
33. Comenzo RA. How I treat amyloidosis. Blood 2009; 114 : 3147-3157.
34. Sakalová A, Plank L, Škultétyová D, et al. Primárna amyloidóza obličiek a asymptomatický (smoldering) mnohopočetný myelóm. Interná Med 2009; 9 : 338-342.
35. Gameren I, Rijswijk M, Bijzet J, et al. Histological regression of amyloid in AL amyloidosis is exclusively seen after normalisation of serum free light chain. Haematologica 2009; 94 : 1094-1100.
36. Sakalová A, Plank L, Škultétyová D, et al. Súčasný pohľad na systémovú AL amyloidózu z hľadiska včasnej diagnózy. Lek Obz 2009; 58 : 425-430.
37. Čapek J, Pěnička M, Kment M. Srdeční AL-amyloidóza s relativně benigním průběhem. Prakt Lék 2009; 89 : 320-323.
38. Bradwell AR, Carr-Smith HD, Mead GP, et al. Free light chains in patients with Bence Jones myeloma. Lancet 2003; 361 : 489-495.
39. Katzmann JA, Abraham RS, Dispenzieri A, et al. Diagnostic performance of quantitative serum free light chain assays in clinical practice. Clin Chem 2005; 51 : 878-881.
40. Nowrousian MR, Brandhorst D. Free light chain assays are more sensitive than urine IFE in patients with multiple myeloma. Clin Cancer Res 2005; 11 : 8706-8714.
41. Gertz M, Merlini G, Treon S. Amyloidosis and Waldenströmęs macroglobulinemia Hematology 2004; 257 : 1-34.
42. Merlini G, Palladini G. Advances in AL amyloidosis. Hematology Education 2008; 2 : 287-293.
43. Rossi F, Petrucci M, Guffanti A, et al. Proposal and validation of prognostic scoring systems for IgG and IgA monoclonal gammopathies of undetermined significance. Clin Cancer Res 2009; 15 : 4439-4445.
44. Koshiol J, Gridley G, Engels E, McMaster M, Landgren O. Chronic immune stimulation and subsequent Waldenström macroglobulinemia. Arch Intern Med 2008; 168 : 1903-1909.
45. Wechalekar A, Lachmann H, Goodman H, Bradwell A, Hawkins P, Gillmore J. AL amyloidosis associated with IgM paraproteinemia: clinical profile and treatment outcome. Blood 2008; 112 : 4009-4016.
46. Bojtárová E. Transplantácia krvotvorných buniek. Interná Med 2009; 9 : 533-538.
47. Brenner H, Gondos A, Pulte D. Expected long-term survival of patients diagnosed with multiple myeloma in 2006-2010. Haematologica 2009; 94 : 270-275.
48. Palumbo A, Sezer O, Kyle R, et al. International myeloma working group guidelines for the management of multiple myeloma patients ineligible for standard high-dose chemotherapy with autologous stem cell transplantation. Leukemia 2009; 23 : 1716-1730.
49. Paiva B, Vidriales M, Cerveró J, et al. Multiparameter flow cytometric remission is the most relevant prognostic factor for multiple myeloma patients who undergo autologous stem cell transplantation. Blood 2008; 112 : 4017-4023.
50. Sakalová A, Mistrík M, Hrubiško M, et al. Súčasná liečba mnohopočetného myelómu a Waldenströmovej makroglobulinémie. Lek Obzor 2004; 53 : 280-284.
51. Hájek R, Adam Z, pro CMG a ČHS. Léčba mnohočetného myelomu: Indikace k léčbě novými léky dle guidelines 2008 České myelomové skupiny 2008 a ČHS. Transfuze Hematol dnes 2008; 14 (Suppl. 2): 37-38.
52. Sakalová A, Dedík L, Škultétyová D, et al. Problém kurability mnohopočetného myelómu. Acta Chemoterapeutica 2006; 16 : 32-39.
53. Sakalová A, Chabroňová I, Gažová S, et al. Retrolective cohort study of an additive therapy with an oral enzyme preparation in patients with multiple myeloma. Cancer Chemother Pharmacol 2001; 47 : 38-44.
54. Škultéty J., Žiak M., Durdík Š. Zytokinaktivität bei Patienten mit gastrointestinalen Karzinomen. MMW-Taschenbuch: Systemische Enzymtherapie, München, Mediz. Verlag, 2995, s. 251-253.
Štítky
Hematologie a transfuzní lékařství Interní lékařství OnkologieČlánek vyšel v časopise
Transfuze a hematologie dnes

2010 Číslo 4
- Diosmin a hesperidin: Co ukazuje farmakokinetika?
- Evaluace skóre m7-FLIPI u pacientů s folikulárním lymfomem odhalila slibný prediktivní biomarker k určení vhodné chemoterapie
- Neuropatie výrazně zpomaluje proces hojení u povrchových ran
- Diagnostika von Willebrandovy choroby krok za krokem
- Těhotenství a porod u ženy s VWD − kazuistika
Nejčtenější v tomto čísle
- Súčasná klasifikácia, diagnostika a prognóza primárnych monoklonových gamapatií (paraproteinémií)
- Vliv dárcovství krve na zásoby železa u dárců: porovnání dvojité erytrocytaferézy a dárcovství plné krve
- Česká asociace zdravotních laborantů - co je a proč vznikla
-
Radioterapie u folikulárního lymfomu.
Stará historie v nové perspektivě?
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy