
Akutní zadní multifokální plakoidní pigmentová epiteliopatie
Acute Posterior Multifocal Placoid Pigment Epitheliopathy – Case Report
Acute posterior multifocal placoid pigment epitheliopathy (APMPPE) affects individuals between 20 to 30 years of age and ocular manifestations are often preceded by flu-like illness. Symptoms of the disease include acute vision decrease associated with central and paracentral scotoma. Impairment of vision is usually bilateral, but may be asymmetric. We report the case of a man at the age of twenty-one, who was treated for APMPPE. Due to the involvement of the macula, low visual acuity and related immunogenetic predisposition (HLA-B27 positivity) was recommended corticosteroid therapy with a good therapeutic effect.
Key words:
acute posterior multifocal placoid pigment epitheliopathy (APMPPE), white dot syndromes, macular edema
Autoři:
A. Stěpanov; A. Feuermannová; J. Studnička; L. Hejsek; M. Burova; N. Jirásková; P. Rozsíval
Působiště autorů:
Oční klinika, Fakultní nemocnice, Hradec Králové, přednosta prof. MUDr. Pavel Rozsíval, CSc., FEBO
Vyšlo v časopise:
Čes. a slov. Oftal., 70, 2014, No. 2, p. 72-76
Kategorie:
Kazuistika
Souhrn
Akutní zadní multifokální plakoidní pigmentová epiteliopatie (APMPPE) postihuje jedince mezi 20.–30. rokem věku. Očním projevům často předchází chřipkové onemocnění. K příznakům onemocnění patří akutní zhoršení zraku, spojené s centrálními a paracentrálními skotomy. Zhoršení vidění je obvykle oboustranné, ale může být asymetrické. Referujeme kazuistické sdělení případu muže ve věku 21 let, léčeného pro APMPPE. Vzhledem k postižení centrální krajiny sítnice, nízké vstupní zrakové ostrosti a související imunogenetické predispozici (pozitivita HLA-B 27) byla doporučena celková kortikosteroidní léčba s dobrým terapeutickým efektem.
Klíčová slova:
akutní zadní multifokální plakoidní pigmentová epiteliopatie (APMPPE), syndrom bílých teček, edém makuly
ÚVOD
Akutní zadní multifokální plakoidní pigmentová epiteliopatie (APMPPE) je vzácné onemocnění, které postihuje hlavně mladší jedince (mezi 20. a 30. rokem věku) bez ohledu na pohlaví. Poprvé bylo popsáno Gassem v roce 1968 [6]. V 30–50 % případů očním projevům předchází chřipkové onemocnění [5, 7, 8].
K příznakům APMPPE patří akutní zhoršení zraku, spojené s centrálními a paracentrálními skotomy. Zhoršení vidění je obvykle oboustranné, ale může být asymetrické. Druhé oko bývá postiženo následně během několika dní až týdnů.
Ve většině případů se zraková ostrost vrátí k původním hodnotám během 3–6 týdnů, ale zlepšení může trvat i delší dobu, až 6 měsíců [7]. Recidivy jsou vzácné a obvykle se vyskytují v průběhu 6 měsíců od prvního záchytu. Vzhledem k existující souvislosti mezi tímto onemocněním a HLA-B7 a HLA-DR2 pozitivitou je možné předpokládat, že existuje genetická predispozice. Akutní zadní multifokální plakoidní pigmentová epiteliopatie může být spojená s celkovými infekcemi (například plicní tuberkulóza, skupina streptokokových infekcí a borelióza) i s neinfekčními nemocemi, včetně Wegenerovy granulomatózy, erythema nodosum, polyarteritis nodosa, tyreoiditidy, ulcerózní kolitidy, vaskulitidy mozkových cév, skleritidy a episkleritidy [12].
KAZUISTIKA
Muž ve věku 21 let byl přijat na Oční kliniku Fakultní nemocnice Hradec Králové pro zhoršení vidění pravého oka. Z anamnézy vyplývá, že před 4 týdny prodělal chřipku. Celkově se s ničím neléčil, pravidelně žádné léky neužíval. Předchozí oční anamnéza byla bez pozoruhodností.
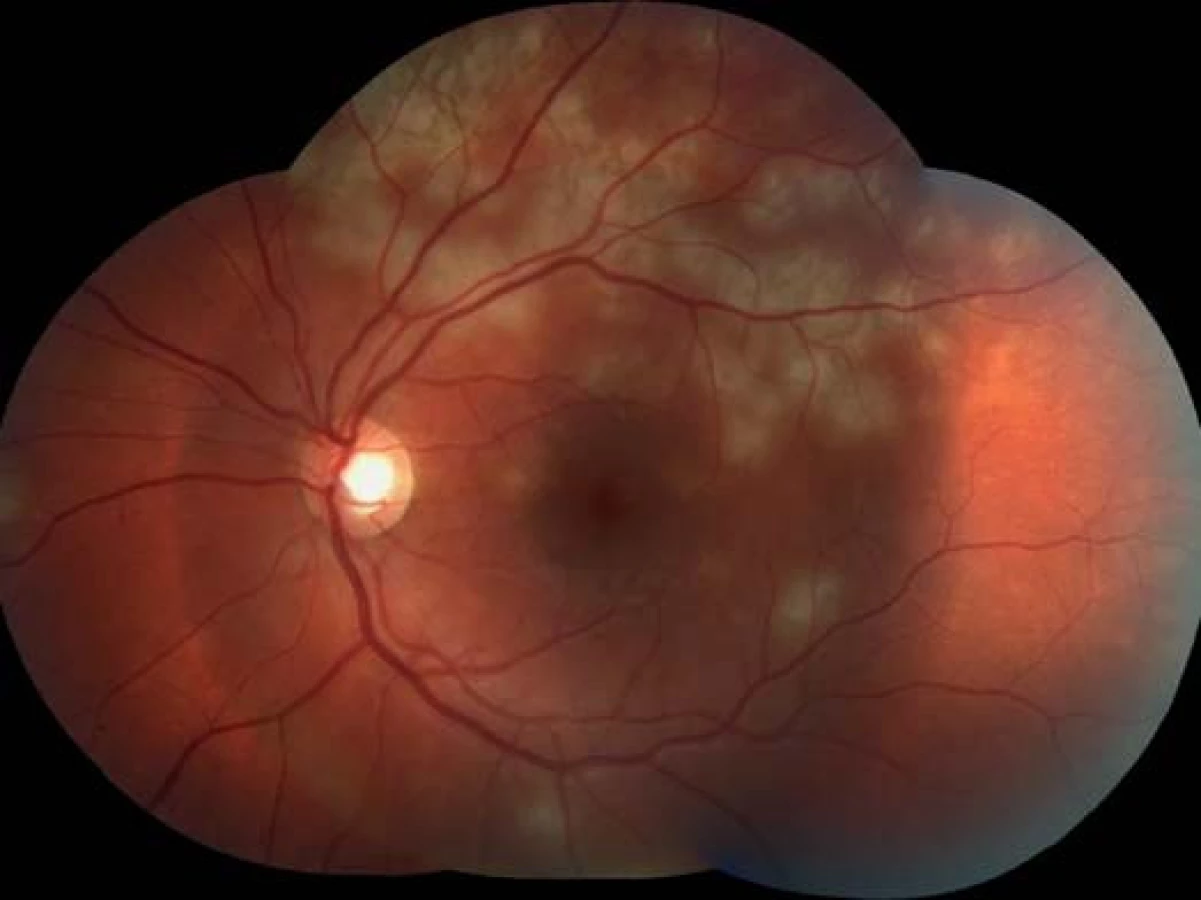
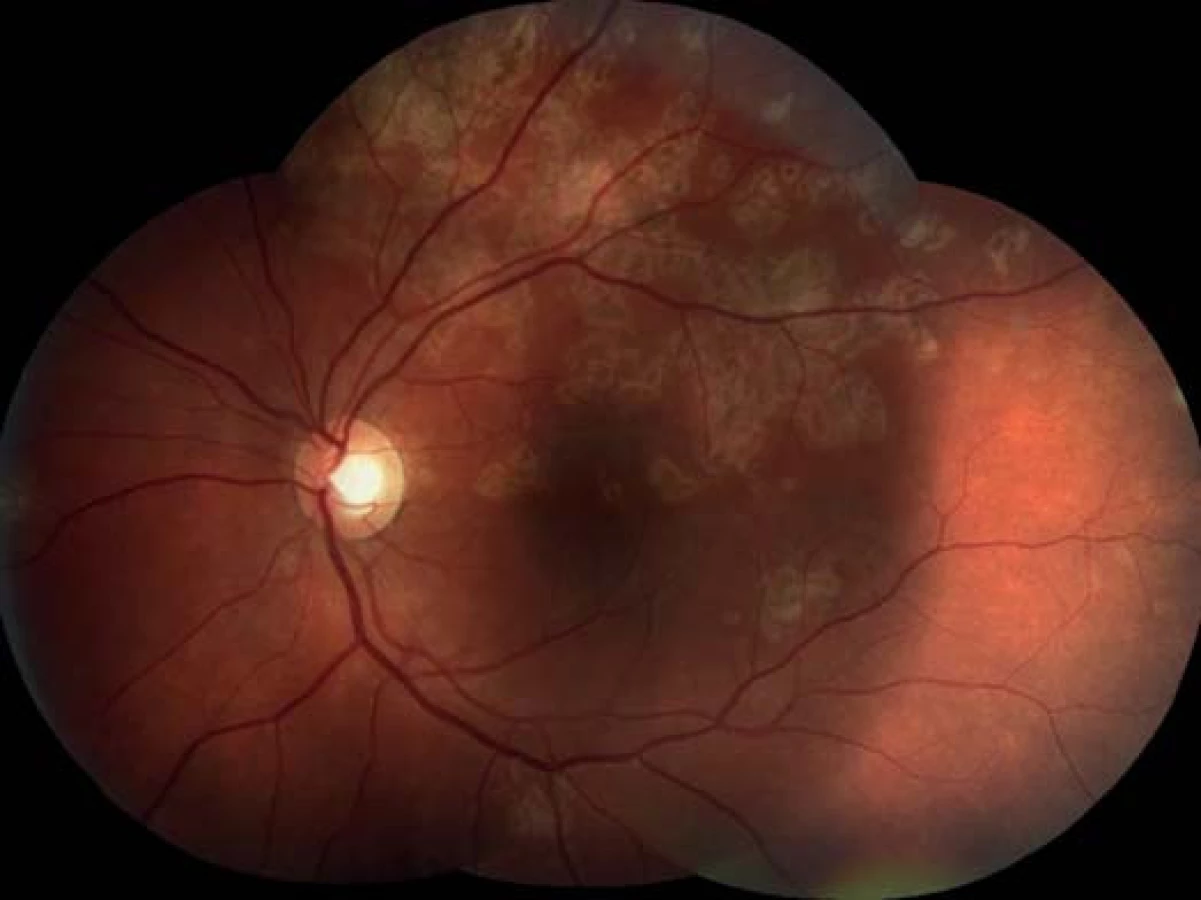
Oční obtíže pozoroval tři dny, vnímal postupně se zvětšující černou skvrnu v centru zorného pole pravého oka. Vstupní zraková ostrost pravého oka byla 1,5/50, korekce nelepšila. Zraková ostrost levého oka byla 6/6 naturálně. Na endotelu rohovky vpravo jsme pozorovali ojedinělé precipitáty, v přední komoře tyndalizaci 1+ a ve sklivci četné práškovité zákalky. Terč zrakového nervu byl ohraničený, růžové barvy, na sítnici podél cévních arkád bylo množství žlutobělavých ložisek, některá s pigmentovými shluky, v centrální krajině byl přítomen výrazný edém (obr. 1). Na levém oku byl nález na předním segmentu přiměřený věku, na očním pozadí dominovala žlutobělavá ložiska podél cévních arkád, v centrální krajině tři bělavá ložiska v horní části makuly (obr. 2). Optická koherenční tomografie (OCT) (Zeiss Cirrus) centrální krajiny oka pravého ukázala serózní ablaci neuroretiny, nález volné tekutiny mezi neuroretinou a retinálním pigmentovým epitelem (RPE), hyperrefletivní zánětlivé ložisko ve vrstvě zevních a vnitřních fotoreceptorů (IS/OS) a edém neuroretiny (obr. 3). Centrální retinální tloušťka (CRT) byla 648 μm. Na levém oku jsme na OCT zjistili 3 hyperrefletivní ložiska ve vrstvě IS/OS fotoreceptorů. Centrální retinální tloušťka byla 230 μm (obr. 4).



Již na začátku první minuty fluorescenční angiografie (FAG) jsme pozorovali hypofluorescenci v místě ložisek (obr. 5), která postupně přecházela ve splývající hyperfluorescenci. Pozánětlivá ložiska měla charakter negradující hyperfluorescence „window“ defektů RPE (obr. 6).


Na základě objektivního nálezu a anamnézy pacienta byla stanovena diagnóza APMPPE oboustranně s převahou vpravo. Byly provedeny základní krevní a cílené revmatologické odběry včetně HLA typizace. Zjistili jsme vyšší hodnoty CRP 73,5 mg/l a HLA B-27 pozitivitu, ostatní hodnoty byly v normě. Vzhledem k postižení centrální krajiny, nízké zrakové ostrosti a související imunogenetické predispozici (HLA-B 27 pozitivita) byla doporučena kortikosteroidní léčba – celkem 3 intravenózní pulsy Methylprednisolonu 500 mg po dobu tří dnů, dále bylo pokračováno v perorálním užívání Prednisonu 60 mg/den s postupným snižováním dávky po dobu 7 dní. Při nastavené terapii došlo k subjektivnímu i objektivnímu zlepšení nálezu na obou očích. Zraková ostrost pravého oka se zlepšila na 6/24, u levého oka zůstala zachována 6/6. Na očním pozadí oboustranně po 6 dnech od nasazení léčby se původně aktivní ložiska postupně ohraničují a částečně pigmentují (obr. 7, 8). Optická koherenční tomografie centrální krajiny pravého oka (obr. 9) ukazuje zlepšení stavu, edém neuroretiny v centrální krajině vymizel, foveolární deprese se vytvořila, CRT 164 μm. Na auto-fluorescenčním snímku sítnice jsou oboustranně patrná ložiska atrofie RPE a hrubých pigmentových změn v postiženích oblastech (obr. 10, 11). Další sledování vývoje onemocnění nebylo možné, protože pacient se nedostavil na plánovanou kontrolu.




DISKUSE
Typickým nálezem APMPPE na očním pozadí jsou mnohočetná, nažloutlá ložiska velikosti přibližně 1500–3000 μm, která se táhnou od zadního pólu k ekvátoru oka a jsou lokalizována v úrovni RPE. V průběhu jednoho až dvou týdnů se akutní ložiska postupně ohraničují a objevují se přesuny pigmentu různého stupně s atrofií RPE. V našem případě jsme kromě typických známek APMPPE ještě zaznamenali méně častý nález iridocyklitidy a také přítomnost zánětlivých buněk ve sklivci. Jinými méně častými nálezy jsou periflebitida, centrální okluze retinální žíly, neovaskularizace terče zrakového nervu, serózní odchlípení sítnice, retrohyaloidní krvácení, episkleritida, zánět a otok terče zrakového nervu, rozšíření a tortuozita cév sítnice [1, 2, 7, 8, 9, 12].
Etiopatogeneze onemocnění zůstává nejasná. V literatuře je popsáno několik teorií. Gass předpokládá, že příčinou onemocnění je přechodná porucha struktury a funkce RPE na podkladě potenciální virové infekce (edém RPE buněk, který blokuje choroidální fluorescenci). Dobrá prognóza tohoto onemocnění a rychlé zlepšení zrakové ostrosti tuto teorii podporují. Další autoři uvádějí jako primární příčinu choriokapilární hypoperfuzi se sekundárními změnami RPE [4]. Příčinou této hypoperfuze mohou být vaskulární změny imunitního původu, což by korelovalo s HLA-B7 a HLA-DR2 pozitivitou a asociací se systémovými vaskulitidami u většiny pacientů s APMPPE [5]. V našem případě v akutní fázi onemocnění přítomnost hyperreflektivního zánětlivého ložiska ve vrstvě IS/OS fotoreceptorů na OCT a nález volné tekutiny mezi neuroretinou a RPE, podporují spíše tuto teorii. Choriokapilární hypo - až nonperfuze vede k ischémii choroidey, pak následně k změnám RPE, defektům vrstvy junkce IS/OS fotoreceptorů a zvýšené permeabilitě cév. Objektivně jsme na FAG pozorovali projev těchto procesů na sítnici jako hypofluorescenci v místě aktivních ložisek na začátku FAG, která postupně přecházela ve splývající hyperfluorescenci.
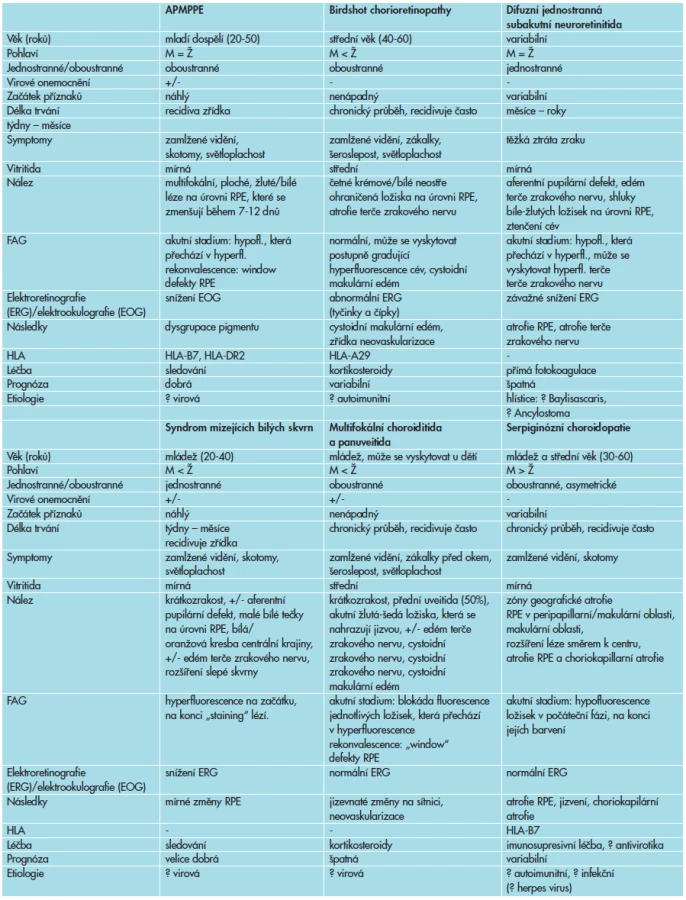
Akutní zadní multifokální plakoidní pigmentová epiteliopatie patří do heterogenní skupiny onemocnění, zvaných syndrom bílých teček (white dot syndromes), jejichž klinické nálezy jsme v rámci diferenciální diagnostiky porovnávali s nálezem u našeho pacienta (tab. 1) [11].
Většina autorů nedoporučuje zahajovat léčbu APMPPE, ale v případě přítomnosti negativních prognostických faktorů, kterými jsou postižení makuly, nízká zraková ostrost, související imunogenetická predispozice, věk vyšší než 60 roků při první atace, jednostranný projev onemocnění a interval delší než 6 měsíců před postižením druhého oka, se užívání kortikosteroidů doporučuje [3, 10]. V našem případě vzhledem k postižení centrální krajiny, nízké zrakové ostrosti oka pravého a související imunogenetické predispozici (HLA-B 27 pozitivita) byla doporučena celková kortikosteroidní léčba s dobrým terapeutickým efektem.
ZÁVĚR
Akutní zadní multifokální plakoidní pigmentová epiteliopatie je získané zánětlivé onemocnění, které patří do heterogenní skupiny syndromu bílých teček. V našem případě jsme na základě klinického obrazu a anamnézy diagnostikovali méně častou formu onemocnění s postižením centrální krajiny a výrazným poklesem zrakové ostrosti na více postiženém oku s doprovodnou zánětlivou reakcí na předním segmentu oka a ve sklivcovém prostoru. Při nasazené intenzivní imunosupresivní léčbě došlo k ústupu zánětlivé reakce v celém rozsahu a zlepšení zrakové ostrosti. Neúplný návrat zrakové ostrosti na více postiženém oku souhlasí s trvalými změnami RPE v makule.
Do redakce doručeno dne 27. 3. 2014
Do tisku přijato dne 22. 4. 2014
MUDr. Alexandr Stepanov
Oční klinika
Fakultní nemocnice Hradec Králové
Sokolská 581
500 05 Hradec Králové
e-mail: stepanov.doctor@gmail.com
Zdroje
1. Abu El-Asrar, AM., Aljazairy, AH.: Acute posterior multifocal placoid pigment epitheliopathy with retinal vasculitis and papillitis, Eye, 2002; 16 : 642–644.
2. Allee, SD., Marks, SJ.: Acute posterior multifocal placoid pigment epitheliopathy with bilateral central retinal vein occlusion, Am J Ophthalmol, 1998; 126 : 309–312.
3. Burés-Jelstrup, A., Adán, A., Casaroli-Marano, R.: Epiteliopatía pigmentaria placoide posterior multifocal aguda. Estudio de 16 casos., Arch Soc Esp Oftalmol, 2007; 82 : 291-298
4. De Souza, S., Aslanides, IM., Altomare, F.: Acute posterior multifocal placoid pigment epitheliopathy associated with retinal vasculitis, neovascularization and subhyaloid hemorrhage., Can J Ophthalmol, 1999; 34 : 343–345.
5. Ducos de Lahitte, G., Fajnkuchen, F., Giraud, C., Chabne, G.: Épithéliopathie en plaque et mauvais pronostic visuel: ą propos d’un cas., J Fr Ophtalmol., 2004, 27 : 617-622.
6. Gass, JDM.: Acute posterior multifocal pigment epitheliopathy., Arch Ophthalmol., 1968; 80 : 177–185.
7. Gass, JDM.: Stereoscopic atlas of macular diseases: diagnosis and treatment., 3rd ed. St Louis: CV Mosby, 1987; pp. 504 –506.
8. Holt, WS., Regan, CDJ., Trempe, C.: Acute posterior multifocal placoid pigment epitheliopathy. Am J Ophthalmol, 1976; 81 : 403–412.
9. Isashiki, M., Koide, H., Yamashita, T., Ohba, N.: Acute posterior multifocal placoid pigment epitheliopathy associated with diffuse retinal vasculitis and late haemorrhagic macular detachment., Br J Ophthalmol, 1986; 70 : 255–259.
10. Pagliarini, S., Piruet, B., Ffytche, TJ., Bird, AC.: Foveal involvement and lack of visual recovery in APMPPE associated with uncommon features., Eye, 1995, 9 : 42–47.
11. Quillen, D., Davis, J., Gottlieb, J. et al.: The White Dot Syndromes., Am J Ophthalmol, 2004; 137 : 5 38–50
12. Rose, SJ., Lou, PL.: Acute posterior multifocal placoid pigment epitheliopathy., in Albert, DM., Miller, JW.,: Principles and practice of ophtalmology II. Philadelphia, Elsevier; 2008., pp. 2089–2095.
Štítky
OftalmologieČlánek vyšel v časopise
Česká a slovenská oftalmologie

2014 Číslo 2
- Patofyziologie glaukomu
- Současná oční operativa – bezpečná investice do výborného zraku
- Selektivní laserová trabekuloplastika nesnižuje nitroční tlak více než argonová laserová trabekuloplastika
- Progresi glaukomu je třeba hodnotit strukturálními i funkčními parametry
- Ztráta centrálního vidění po filtrujících operacích glaukomu
Nejčtenější v tomto čísle
- Postižení okohybných svalů u pacientů s endokrinní orbitopatií
- Retinopatia prematúrnych detí I. časť
- Akutní zadní multifokální plakoidní pigmentová epiteliopatie
- Retinopatia prematúrnych detí – terapia II. časť
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy