
Maligní tumor z pochvy periferního nervu v oblasti cervikálního plexu – kazuistika
Malignant Peripheral Nerve Sheath Tumour of Cervical Plexus – a Case Report
Malignant peripheral nerve sheath tumours are rare neoplasms, especially in the head and neck. They are often asymptomatic. We present a case of 29-years old patient with neurofibromatosis type 1 with malignant tumour of the cervical plexus. A small resistance in the upper mediastinum was diagnosed (ganglioneurinoma) in 2005 and treated surgically (total exstirpation). A small infiltration in the right supraclavicular area occurred five years later and was also managed surgically (debulking) – histologically MPNST. There was a relapse of this tumour in the same area in 2011; this was treated by en bloc resection, followed by chemotheraphy. At present, the patient is in a good clinical status, with no neurological deficit.
Key words:
malignant peripheral nerve sheath tumour – cervical plexus – malignant schwannoma – neurofibromatosis type 1
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
Autoři:
Z. Kadaňka Jr 1; J. Hanák 2; B. Gál 2
![]()
Působiště autorů:
Neurologická klinika LF MU a FN Brno
1; ORL klinika LF MU a FN u sv. Anny v Brně
2
Vyšlo v časopise:
Cesk Slov Neurol N 2013; 76/109(6): 751-755
Kategorie:
Kazuistika
Souhrn
Maligní tumory z pochvy periferního nervu jsou vzácná neoplazmata, obzvláště v oblasti hlavy a krku. Bývají často asymptomatická. Popisujeme případ 29letého pacienta s neurofibromatózou 1. typu s maligním tumorem v oblasti cervikálního plexu. V roce 2005 u něj bylo diagnostikováno ložisko v oblasti horního mediastina, které bylo následně chirurgicky odstraněno a diagnostikováno jako ganglioneurinom. Po pěti letech se objevila rezistence v pravém nadklíčku, dle CT a CT angiografie byla prokázána infiltrace v horním hrudním mediastinu vpravo, jež byla chirurgicky řešena (debulking). Histologicky byl verifikován maligní tumor z pochvy periferního nervu. Roku 2011 došlo k recidivě tumoru v této lokalizaci, nádor byl v bloku resekován a podána chemoterapie. V roce 2013 je pacient v dobrém klinickém stavu, bez neurologického deficitu.
Klíčová slova:
maligní tumor z pochvy periferního nervu – cervikální plexus – maligní schwannom – neurofibromatóza 1. typu
Úvod
Maligní tumory z pochvy periferního nervu (MPNST) jsou vzácná neoplazmata.
Nejčastěji bývají lokalizovány v oblasti trupu a končetin (cca v 80 % případů), jak již bylo popsáno i českými autory [1,2]. V oblasti hlavy a krku však bývají tyto malignity většinou jen ojediněle popisovany v literatuře. Jedná se ale o jedny z nejzhoubnějších nádorů, které často recidivují a metastazují [3]. Lokalizace v cervikálním plexu je vzácná a mnohdy bývá spojena s absencí senzitivních či motorických příznaků [4]. MPNST se vyskytují buď sporadicky, či jsou spojeny s neurofibromatózou, zejména 1. typu. U části pacientů bývá popisována v anamnéze radiační expozice [4]. Uvádíme případ maligního tumoru z pochvy periferního nervu v oblasti cervikálního plexu u 29letého pacienta s neurofibromatózou 1. typu. Jedná se pravděpodobně o první případ MPNST v této lokalizaci publikovaný u nás.
Kazuistika
Pacient (29 let) s diagnostikovanou neurofibromatózou 1. typu (morbus von Recklinghausen) a typickým klinickým nálezem mnohočetných kutánních a subkutánních tumorózních uzlíků. Onemocnění zůstávalo dlouhodobě stacionární.
V roce 2005 bylo provedeno v rámci vstupní prohlídky do zaměstnání RTG plic a bylo popsáno ložisko v oblasti horního mediastina. Na MR hrudníku byla zjištěna postkontrastně se sytící tumorózní formace vysoko paramediastinálně a paravertebrálně velikosti 67 × 40 × 5,5 mm, zasahující až do hrotu hemithoraxu. Dle FNAB (aspirační biopsie tenkou jehlou) byl histologicky verifikován ganglioneurinom. Následně bylo indikováno radikální odstranění tumoru, které histologicky diagnózu potvrdilo.
Po pěti letech byla zjištěna rezistence v oblasti pravého nadklíčku. Podle CT a CT angiografie v květnu 2010 byl prokázán infiltrát v oblasti horní hrudní apertury vpravo velikosti 61 × 64 × 98 mm, přerůstající do měkkých tkání supraklavikulárně. Do něj byla zavzata i pravá vertebrální tepna a a. subclavia. Infiltrát dosahoval až k laterálnímu okraji trachey a k pravému laloku štítné žlázy. Hrudním multidisciplinárním chirurgickým týmem byla provedena operační revize, při níž byl nalezen multicentrický tumor v oblasti pravého nadklíčku vycházející z nervových kmenů plexus cervicalis. Byla provedena subtotální resekce nádoru a histologicky verifikován maligní tumor periferní nervové pochvy G2 (grading FNCLCC, Fédération Nationale des Centres de Lutte Contre le Cancer), proliferační frakce (Ki ‑ 67/ MIB ‑ 1) – 23 %. Pooperační průběh byl komplikován zánětlivým procesem v supraklavikulární krajině, avšak byl úspěšně zvládnut antibiotickou léčbou.
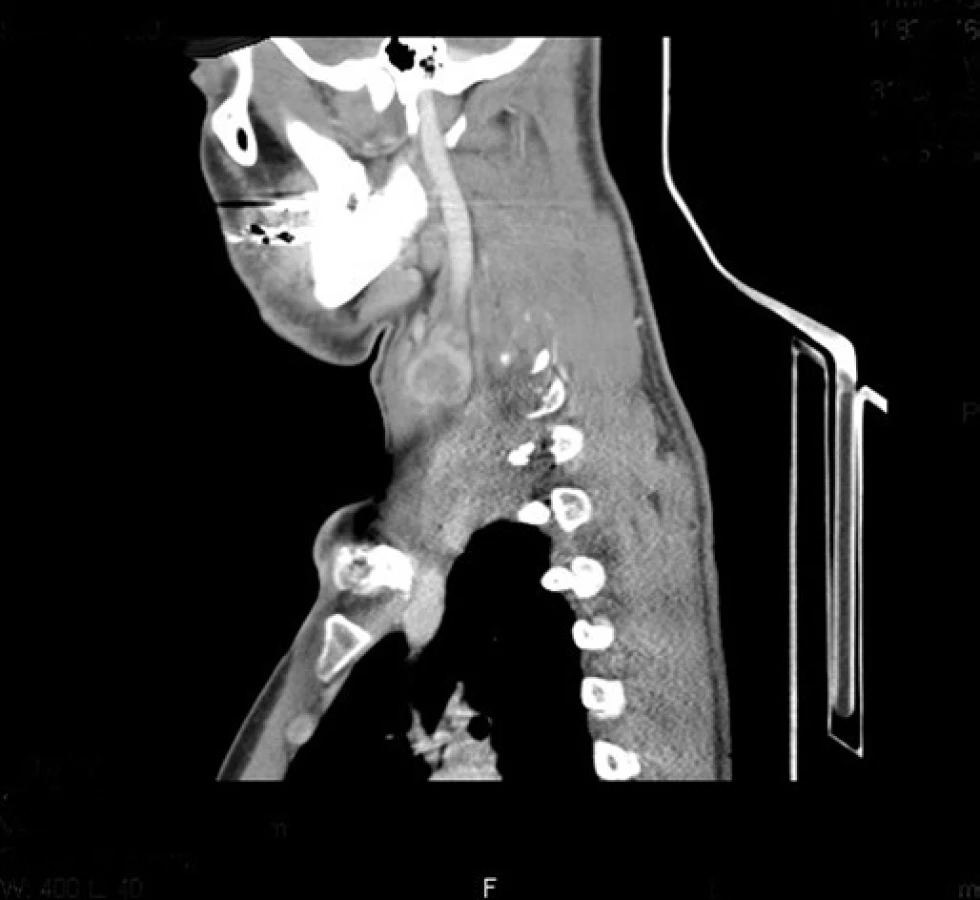
V dubnu 2011 byla s pomocí zobrazovacích vyšetření (MR, CT angiografie) zjištěna recidiva tumoru a indikována chirurgická revize (obr. 1, 2). Peroperačně byl ověřen multicentrický tumor formovaný z kuželovitě ztluštělých nervových kmenů cervikální pleteně a n. vagus (obr. 3). Hlavní masa tumoru vyplňovala supraklavikulární oblast, uzavírala vnitřní jugulární žílu, obkružovala a.carotis communis, distálně naléhala k a. subclavia a dorzálně se zanořovala do skalenických svalů. Po subadventiciální preparaci a uvolnění společné krkavice byl tumor v bloku s vnitřní jugulární žílou a postiženými nervovými kmeny resekován (obr. 4). Histologicky byla opět potvrzena recidiva maligního tumoru periferní nervové pochvy s invazí do stěny vnitřní jugulární žíly a pozitivní distální okrajovou excizí v oblasti horního mediastina (obr. 5, 6). Pooperační CT angiografie je na obr. 7 a 8.







S výsledkem histologického vyšetření byl stav zhodnocen na indikační mezioborové onkologické komisi pro léčbu tumorů hlavy a krku a indikována pooperační chemoterapie. Ta byla aplikována v měsíčních intervalech, doposud 15 sérií, režim: ifosfamid v kombinaci s doxorubicinem (od 8. série monoterapie ifosfamidem). Stav je od operace stabilizovaný. Po celou dobu je objektivní neurologický nález pacienta bez ložiskových projevů, je přítomna pouze lehčí hypestezie na pravém rameni.
Diskuze
Maligní tumor z pochvy periferního nervu (MPNST) byl poprvé popsán A. P. Stoutem v roce 1935 jako „maligní neurilemmom“ [5]. Tento typ nádoru byl následně v literatuře nazýván různými popisnými názvy jako neurogenní sarkom, maligní schwannom, neurofibrosarkom. Z těchto důvodů byl Světovou zdravotnickou organizací zaveden termín „maligní tumor z pochvy periferního nervu“ (MPNST), aby nahradil výše uvedené zavádějící názvy [6].
MPNST jsou sarkomy, které pocházejí z periferních nervů nebo z buněk souvisejících s pochvou periferního nervu (Schwannovy buňky, perineurální buňky nebo fibroblasty). Představují asi 10 % všech sarkomů měkkých tkání [7]. Vyskytují se buď sporadicky, nebo ve spojení s neurofibromatózou 1. typu, a to až v 50 % případů [6].
Neurofibromatóza typu 1 je dědičný syndrom spojený s dysregulací v systému proteinů Ras [8]. Predispozice ke vzniku nádoru se vytváří mutací genu pro neurofibromin (280 kDa protein lokalizovaný na 17. chromozomu), který působí jako negativní regulátor systému proteinů Ras. NF1 deficientní buňky jsou hypersenzitivní na některé růstové faktory, jež vedou signál systémem Ras [9]. Ačkoliv byl neurofibromin typu 1 izolován před více než 20 lety, naše znalosti, jak ovlivňuje růst, metabolizmus a apoptózu buněk jako klíčový supresorový gen, zůstávají neúplné. V poslední době se zdá, že klíčovou roli v tumorigenezi hraje aktivace Heat Shock Proteinu 1 (HSP1), který byl ve velkém množství prokázán u pacientů s MTPPN. Neurofibromin typu 1 působí jako silný regulátor hladiny HSP1 a jeho aktivity [10]. U pacientů s MTPPN bývají též často popisovány mutace supresorového genu p53 [11] a dále v lokusu 9p21 [12], což vede k předpokladu, že k maligní transformaci je zapotřebí ještě druhá genová mutace.
MPNST se vyskytují zhruba ve stejném procentu u obou pohlaví. Jsou lokalizovány zejména na končetinách a trupu, cca v 80 % případů, v oblasti hlavy a krku – dle různých autorů v 10 – 20 % [3,4,13]. Typicky se objevují v sedmé dekádě života, u pacientů s neurofibromatózou však podstatně dříve – nejčastěji ve třetím až čtvrtém deceniu [6]. U pacientů s NF1 je riziko rozvoje MPNST 8 – 13% [7].
Příčina genové mutace není známa. Byla však popsána vyšší incidence tohoto nádoru u pacientů s anamnézou radiační expozice [14], u našeho pacienta ale přítomna nebyla.
Diagnostika těchto tumorů bývá často nesnadná a zavádějící, neboť se i přes značnou agresivitu tumoru často jedná o léze, které nepůsobí větší klinické obtíže, a bývají proto považovány za benigní.
Nejčastějším příznakem MPNST u dospělých je rychle se zvětšující útvar v příslušné oblasti, který bývá obvykle asymptomatický [15]. Nejinak tomu bylo i u našeho pacienta. Mnohem vzácněji se může MPNST manifestovat celou škálou neurologických příznaků zahrnujících radikulární bolesti, parestezie a eventuálně i parézy – vše v závislosti na lokalizaci léze.
Radiologicky a ultrasonograficky se MPNST u dospělých manifestuje jako kulovitý útvar, který většinou přesahuje 5 cm v průměru [16], zejména u pacientů s neurofibromatózou [4].
Magnetická rezonance bývá u MPNST metodou volby a je považována za značně senzitivní a specifické vyšetření k odlišení benigních a maligních tumorů, se specificitou až 81 % pro benigní tumory [17]. Typický je zejména hyperintenzní okraj s centrálním poklesem intenzity na T2 vážených sekvencích, homogenně izodenzní nebo mírně hyperintenzní ke svalu na T1 vážených sekvencích. Zejména rozsah tumoru (nad 5 cm), invaze do tukových tkání, heterogenita, neostré okraje a perifokální edém budí podezření na maligní typ nádoru [18].
FDG (fludeoxyglukóza) PET se ukazuje jako přínosné v detekci metastáz či relapsu choroby [19], ačkoliv jeho role v odlišení benigního a maligního nádoru se původně považovala za spornou [20], nicméně dle recentních studií je velmi přínosná [21].
Histopatologické zhodnocení je pro stanovení definitivní diagnózy zásadní. Typicky bývají popisovány vřetenovité buňky uspořádané do fasciklů, kdy histologický vzorec je podobný fibrosarkomu. Jsou přítomny četné mitózy, celulární atypie, zvýšená celularita a infiltrované, špatně definované buněčné okraje. Imunohistochemické stanovení pozitivity S100 proteinu ukazuje na neuronální původ nádoru, negativita však MPNST nevylučuje. Přítomnost proteinu S ale může být negativní až v 10 – 50 % MPNST [21].
CT hrudníku bývá preferované pomocné vyšetření při pátrání po vzdálených metastázách, scintigrafie skeletu odhalí eventuální kostní postižení.
Resekce nádoru je univerzálně přijímána jako základní metoda léčby [19,22,23]. Cílem je kompletní odstranění tumoru se širokými (histologicky negativními) okraji, bez vynětí regionálních mízních uzlin. Ty bývají postiženy jen extrémně vzácně [4]. Kompletní resekce tumoru však není často možná – zejména v oblasti trupu a hlavy (na rozdíl od končetin), což samozřejmě výrazně zhoršuje další prognózu.
Role přídatné radio ‑ a chemoterapie zůstává nejasná. D’Agostino předpokládá, že chirurgické řešení je jediné vhodné pro pacienta, další autoři rovněž zdůrazňují, že přídatná radio ‑ a chemoterapie nezmění výrazně prognózu onemocnění [5,24]. Léčba sarkomů měkkých tkání adjuvantní radioterapií totiž sice vedla ke statisticky nižšímu počtu lokálních recidiv, nevedla však k poklesu počtu metastáz nebo k prodloužení mediánu dlouhodobého přežití [25]. Někteří autoři se naopak domnívají, že adjuvantní radioterapie by měla být vždy použita spolu s co možná nejradikálnější resekcí tumoru [3]. Přídatná chemoterapie zůstává ještě kontroverznější. Dle některých autorů nemá vůbec smysl [3,4], jiní ji doporučují pouze u nádorů s vysokým stupněm malignity, u nichž je pravděpodobný rozvoj metastáz [8]. Onkologickou komisí pro tumory hlavy a krku byl u našeho pacienta při recidivě tumoru zvolen tento způsob léčby, prozatím s uspokojivým efektem. Radioterapie indikována nebyla.
Prognóza MPNST je velmi závažná, zejména v oblasti hlavy a krku a u pacientů s von Recklinghausenovou nemocí [4]. Většina MPNST jsou sarkomy vysokého stupně malignity, s velkou tendencí k lokální recidivě i k tvorbě metastáz. Ve velkých sériích byla lokální recidiva popsána mezi 40 a 65 % a metastázy v 40 – 68 % [19]. Nádory metastazují nejčastěji do plic, následně pak do kostí a pleury. Pětiletá doba přežití je uváděna od cca 15 do 20 % u pacientů s NF1 a 50 – 55 % u pacientů bez této nemoci [4,15,19,23]. Delší doba přežití je dokumentována u pacientů s totální excizí tumoru, menší velikostí tumoru (pod 5 cm) a u tumorů nižšího stupně [19].
Závěr
Maligní tumor z pochvy periferního nervu v oblasti cervikálního plexu je vzácný. Stejně jako ostatní MPNST je velmi zhoubný a má špatnou prognózu. Léčba spočívá především v subtotální až totální resekci, přičemž role přídatné aktino ‑ a chemoterapie zůstává kontroverzní. Mělo by se na něj myslet u rychle rostoucí kulovité podkožní expanze bez neurologických příznaků, a to především u nemocných s neurofibromatózou 1. typu.
Podpořeno MZ ČR – RVO (FNBr, 65269705).
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
MUDr. Zdeněk Kadaňka jr
Neurologická klinika LF MU a FN Brno
Jihlavská 20
625 00 Brno
e-mail: zdenek.kadanka@fnbrno.cz
Přijato k recenzi: 7.1. 2013
Přijato do tisku: 12. 6. 2013
Zdroje
1. Beneš V III, Kramář F, Hrabal P, Kaiser M, Buchvald P. Maligní tumor z pochvy periferního nervu – dvě kazuistiky. Cesk Slov Neurol N 2009; 72/ 105(2): 163 – 167.
2. Bludovsky D, Zidek S, Hes O, Kazakov D. Melanotic Schwannoma of the Th9Nerve Root. Case Report. World Spinal Column Journal, 2010 : 1(2). [on-line]. Available from:http://www.wscj.org.
3. Minovi A, Basten O, Hunter B, Draf W, Bockmühl U.Malignant peripherical nerve sheath tumors of the head and neck: management of 10 cases and literature review. Head Neck 2007; 29(5): 439 – 445.
4. Ducatman BS, Scheithauer BW, Piepgras DG, Reimann HM, Ilstrup DM. Malignant peripheral nerve sheat tumors: a clinicopathologic study of 120 cases. Cancer 1986; 57(10): 2006 – 2021.
5. D‘Agostino AN, Soule EH, Miller RH. Primary malignant neoplasms of nerves (Malignant neurilemomas) in patients without manifestations of multiple neurofibromatosis (von Recklinghausen‘s disease). Cancer 1963 : 16(8): 1003 – 1014.
6. Gupta G, Mammis A, Maniker A. Malignant peripheral nerve sheath tumours. Neurosurg Clin N Am 2008; 19(4): 533 – 543.
7. Rawal A, Yin Q, Roebuck M, Sinopidis C, Kalogrianitis S, Helliwell TR et al. Atypical and malignant peripheral nerve ‑ sheath tumors of the brachial plexus: report of three cases and review of the literature. Microsurgery 2006; 26(2): 80 – 86.
8. Bollag G, Clapp DW, Shih S, Adler F, Zhang YY, Thompson P et al. Loss of NF1 results in activation of the Ras signaling pathway and leads to aberrant growth in haematopoietic cells. Nat Genet 1996; 12(2): 144 – 148.
9. Largaespada DA, Brannan CI, Jenkins NA, Copeland NG. NF1 deficiency causes Ras ‑ mediated granulocyte/ macrophage colony stimulating factor hypersensitivity and chronic myeloid leukaemia. Nat Genet 1996; 12(2): 137 – 143.
10. Dai Ch, Santagata, S Tang Z, Shi J, Cao J, Kwon H et al. Loss of tumor suppressor NF1 activates HSF1 to promote carcinogenesis J Clin Invest 2012; 122(10): 3742 – 3754.
11. Kindblom LG, Ahldén M, Meis ‑ Kindblom JM, Stenman G, Virchows. Immunohistochemical and molecular analysis of p53, MDM2, proliferating cell nuclear antigen and Ki67 in benign and malignant peripheral nerve sheath tumours. Virchows Arch 1995; 427(1): 19 – 26.
12. Berner JM, Sørlie T, Mertens F, Henriksen J, Saeter G,Mandahl N et al. Chromosome band 9p21 is frequently altered in malignant peripheral nerve sheath tumors: studies of CDKN2A and other genes of the pRB pathway. Genes Chromosomes Cancer 1999; 26(2): 151 – 160.
13. Stark AM, Buhl R, Hugo HH, Mehdorn HM. Malignant peripheral nerve sheath tumours ‑ report of 8 cases and review of the literature. Acta Neurochir (Wien) 2001; 143(4): 357 – 363.
14. Evans DG, Baser ME, McGaughran J, Sharif S, Howard E, Moran A. Malignant peripheral nerve sheath tumours in neurofibromatosis 1. J Med Genet 2002; 39(5): 311 – 314.
15. Adamson DC, Cummings.TJ, Friedman AH. Malignant peripheral nerve sheath tumor of the spine after radiation terapie for Hodgkinʼs lymphoma. Clin Neuropathol 2004; 23(5): 245 – 255.
16. Baehring JM, Betensky RA, Batchelor TT. Malignant peripheral nerve sheath tumor: the clinical spectrum and outcome of treatment. Neurology 2003; 61(5): 696 – 698.
17. Ghosh BC, Ghosh L, Huvos AG, Fortner JG. Malignant schwannoma. A clinicopathologic study. Cancer 1973; 31(1): 184 – 190.
18. Furniss D, Stan MC, Morritt D, Lim J, Khanna T, Way BL et al. A 10‑year review of benign and malignant peripheral nerve sheath tumors in a single center: clinical and radiographic features can help to differentiate benign from malignant lesions. Plast Reconstr Surg 2008; 121(2): 529 – 533.
19. Friedrich RE, Kluwe L, Funsterer C, Mautner VF. Malignant peripheral nerve sheath tumors (MPNST) in neurofibromatosis type 1 (NF1) diagnostic findings on magnetic resonance images and mutational analysis of the NF1 gene. Anticancer Res 2005; 25(3A): 1699 – 1702.
20. Hruban RH, Shiu MH, Senie RT, Woodruff JM. Malignant peripheral nerve sheath tumor of the buttock and lower extremity. A study of 43 cases. Cancer 1990; 66(6): 1253 – 1265.
21. Ferner RE, Lucas JD, O‘Doherty MJ, Hughes RAC, Smith MA, Cronin B et al. Evaluation of 18 fluorodeoxyglucose positron emission tomography in the detection of malignant peripheral nerve sheath tumors in neurofibromatosis 1. J Neurol Neurosurg Psychiatry 2000; 68(3): 353 – 357.
22. Perry A, Roth KA, Banerjee R, Fuller CE, Guttmann DH.NF1 deletions in S ‑ 100 protein‑positive and negative cells of sporadic and neurofibromatosis 1 (NF1) – associated plexiform neurofibromatosis and malignant peripheral nerve sheath tumors. AM J Pathol 2001; 159(1): 57 – 61.
23. Wong WW, Hirose T, Scheithauer BW, Schild SE, Gunderson LL. Malignant peripheral nerve sheath tumor: analysis of treatment outcome. Int J Radiat Oncol Biol Phys 1998; 42(2): 351 – 360.
24. Wanebo JE, Malik JM, VandenBerg SR, Wanebo HJ,Driesen N, Persing JA. Malignant peripheral nerve sheath tumors. A clinicopathologic study of 28 cases. Cancer 1993; 71(4): 1247 – 1253.
25. Vraa S, Keller J, Nielsen OS, Sneppen O, Jurik AG, Jensen OM. Prognostic factors in soft tissue sarcomas: the Aarhus experience. Eur J Cancer 1998; 34(12): 1876 – 1882.
Štítky
Dětská neurologie Neurochirurgie NeurologieČlánek vyšel v časopise
Česká a slovenská neurologie a neurochirurgie

2013 Číslo 6
- Přibývající důkazy o přínosu léčebného konopí u pacientů s chronickou bolestí
- Nízká hladina vitamínu D v prenatálním období může zvýšit riziko roztroušené sklerózy
- Pacientům s roztroušenou sklerózou pomáhá intermitentní pohybová aktivita
- Zahájení léčby intramuskulárním interferonem beta-1a během první epizody demyelinizace u RS
- Časné klinické prediktory a progrese ireverzibilní invalidity u RS
Nejčtenější v tomto čísle
- Frontotemporálna lobárna degenerácia z pohľadu nových klinicko‑patologických korelácií
- Tuberózní skleróza u dětí sledovaných od novorozeneckého věku pro prenatální nález rhabdomyomů srdce – dvě kazuistiky
- Expanze pineální krajiny
- Komorbidita migrény a deprese – metaanalytická studie
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy