
Klinický význam analýz genů středního rizika pro hodnocení rizika vzniku karcinomu prsu a dalších nádorů v České republice
The Clinical Importance of a Genetic Analysis of Moderate-Risk Cancer Susceptibility Genes in Breast and Other Cancer Patients from the Czech Republic
Background:
Analysis of the major breast cancer (BC) predisposition genes BRCA1 and BRCA2 enables identification of high-risk individuals. Specialized programs enrolling the carriers of BRCA1/2 mutations facilitate an improvement in prevention and early diagnostics in asymptomatic individuals and rationalize the selection of individualized treatment in case of a BC onset. However, the carriers of mutations in the major predisposition genes represent only approximately 25% of cases among high-risk BC patients. Numerous candidate predisposing genes for breast and other cancers have recently been identified. The risk of cancer development associated with alterations in these genes is lower, and there is a considerable population variability in different regions worldwide.
Aim:
We have performed mutation analyses of moderate-risk cancer susceptibility genes to evaluate their clinical importance for genetic counseling in high-risk patients suffering from breast and other cancers in the Czech population.
Results:
Czech oncological patients were analysed for mutation in ATM, CHEK2, NBS1 (NBN) and PALB2 genes. The majority of analyzed individuals represent the population of high-risk BRCA1/2-negative BC patients.
Conclusions:
Based on results of this study, we recommend an analysis of recurrent truncating mutations in the CHEK2 gene (the c.1100delC mutation and a large deletion affecting exons 9–10) in BRCA1/2-negative patients from high-risk BC families. A clinical assessment of missense variants in CHEK2 is not suitable. A routine mutation analysis of the ATM and NBS1 (NBN) genes is not recommended in BC patients due to the low frequency of alterations in these genes in the Czech Republic. An identification of truncating mutations in the PALB2 gene is important in BRCA1/2-negative BC patients from families with a strong history of BC (HBC families). The frequency of PALB2 mutations may be comparable to the frequency of mutations in the BRCA2 gene in Czech HBC families.
Key words:
hereditary cancer – genetic testing – ATM – CHEK2 – NBS1 (NBN) – PALB2
This study was supported by IGa MZ ČR NT/13343 grant and by the European Regional Development Fund and by the National budget of the Czech republic (OP VaVpI - RECaMO, CZ.1.05/2.1.00/03.0101).
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
Submitted:
15. 5. 2012
Accepted:
26. 5. 2012
Autoři:
P. Pohlreich 1; Z. Kleibl 1; P. Kleiblová 1; M. Janatová 1; J. Soukupová 1; E. Macháčková 2; J. Hazova 2; P. Vašíčková 2; E. Sťahlová Hrabincová 2; M. Navratilova 2; M. Svoboda 2,3
![]() ; L. Foretová 2
; L. Foretová 2
Působiště autorů:
Ústav biochemie a experimentální onkologie, 1. LF UK a VFN v Praze
1; Oddělení epidemiologie a genetiky nádorů, Masarykův onkologický ústav, Brno
2; Klinika komplexní onkologické péče, Masarykův onkologický ústav, Brno
3
Vyšlo v časopise:
Klin Onkol 2012; 25(Supplementum): 59-66
Souhrn
Východiska:
Analýza hlavních predispozičních genů BRCA1 a BRCA2 podmiňujících vznik dědičných forem karcinomu prsu umožňuje identifikaci vysoce rizikových osob. Kromě zpřesnění odhadu rizika onemocnění lze nosiče patogenních mutací zařadit do programů pro časnou diagnostiku a prevenci onemocnění a v případě jeho rozvoje i specifickou léčbu. Nosiči mutací v hlavních predispozičních genech však tvoří přibližně jen 25 % populace vysoce rizikových pacientů. V nedávné době byla identifikována řada dalších genů, jejichž alterace zvyšují riziko karcinomu prsu a některých dalších nádorů. Tyto geny se však vyznačují nižším rizikem vzniku onemocnění a výraznou populační variabilitou ve světě.
Cíl:
Provedli jsme shrnutí studií analyzujících dědičné mutace v genech středního rizika v populaci pacientů s karcinomem prsu a případně dalšími nádorovými onemocněními v ČR s cílem vyhodnotit současné možnosti uplatnění analýz těchto genů v klinické praxi.
Výsledky:
V populaci onkologických pacientů v ČR byly provedeny analýzy genů ATM, CHEK2, NBS1 (NBN) a PALB2. Vyšetřované populace pacientů zahrnovaly především soubory vysoce rizikových pacientek s karcinomem prsu bez nálezu alterací v hlavních predispozičních genech BRCA1/2.
Závěry:
Na základě výsledků provedených studií lze u BRCA1/2-negativních pacientek z rizikových rodin s karcinomem prsu pro klinické testování v ČR doporučit analýzu patogenních variant genu CHEK2 (především mutace c.1100delC a rozsáhlé delece postihující exony 9–10). Missense varianty genu CHEK2 nelze zatím jednoznačně klinicky hodnotit. Analýzy mutací genů ATM a NBS1 (NBN) nejsou s ohledem na malou frekvenci jejich výskytu vhodné pro klinické hodnocení. Nadějná je analýza patogenních variant genu PALB2u BRCA1/2-negativních pacientek z rodin s mnohočetným výskytem karcinomu prsu, jejíž význam v této skupině může být v naší populaci srovnatelný s významem mutací v genu BRCA2.
Klíčová slova:
dědičné nádory – genetické testování – ATM – CHEK2 – NBS1 (NBN) – PALB2
Úvod
Karcinom prsu se vyskytuje ve dvou patogeneticky rozdílných formách. U většiny žen se karcinom prsu vyvíjí ve formě sporadického onemocnění, které vzniká v důsledku akumulace somatických mutací v buňkách prsní žlázy. Nádorová transformace mamárních epitelií se dotýká deregulace kritických signálně-transdukčních cest (buněčného dělení, apoptózy a reparace genomové DNA) způsobených aktivací protoonkogenů a inaktivací tumor supresorových genů na základě genetických inzultů doprovázených epigenetickými změnami. Výsledkem těchto selhání je vznik maligně transformované buňky in situ, která může na základě genomové nestability způsobené poruchami DNA reparačních pochodů vytvářet řadu geneticky nestabilních dceřiných buněk tolerujících genomové defekty postihující další regulační mechanizmy.
U dědičných forem karcinomu prsu tvořících 5–10 % mamárních tumorů dochází k obdobnému vývoji jako v případě nádorů sporadických. Proces nádorové transformace je však výrazně pravděpodobnější kvůli existenci patogenní mutace v některém z predispozičních genů kódujících protein zúčastněný v (pravděpodobně tkáňově specifickém) procesu nádorové supresivity. Pro nosičky mutací v genech predisponujících ke vzniku hereditární formy karcinomu prsu je tak typické, že riziko vzniku karcinomu prsu je oproti populačnímu riziku významně zvýšeno, k onemocnění dochází v nižším věku, postižení je často bilaterální, a kromě samotného karcinomu prsu jsou nosiči onemocnění ohroženi i zvýšeným rizikem vzniku nádorů v dalších lokalizacích. Do současné doby bylo charakterizováno několik predispozičních genů, jejichž patogenní alterace významně zvyšují riziko vzniku karcinomu prsu (BRCA1 a BRCA2, TP53, PTEN, LKB1/STK11) [1]. Další geny ovlivňující riziko vzniku karcinomu prsu se vyskytují v podstatně menší míře (geny s nízkou a střední penetrancí), avšak tvoří rozrůstající se pestrou skupinu genů (ATM, CHEK2, NBS1 (NBN), PALB2, TGFβ1, CASP-8 a další).
Až nápadným, ve výčtu genů podílejících se na vzniku hereditárních forem karcinomu prsu, je jejich blízký vztah k pochodům přímo zprostředkujícím nebo regulujícím kontrolní body (checkpoints) buněčného cyklu a reparaci genomové DNA (obr. 1) [2].

Jak již bylo řečeno, predispoziční geny modifikují riziko vzniku karcinomu prsu (a dalších nádorů) nestejnoměrně. Zatímco u některých vysoce penetrantních genů (BRCA1/2) je nosičství patogenních alterací spojené s rizikem vysokým, u jiných je zvýšení rizika pouze střední či malé (tab. 1) a lze předpokládat, že některé z variant riziko vzniku nádorů snižují – jejich význam je tak protektivní.
![Relativní riziko (RR) vzniku karcinomu prsu u nosičů mutací v jedné z alel uvedených genů (podle [46]).](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image/44e73e5917ba305bc1550da6dde7c874.png)
Po objevu BRCA1 a BRCA2 v polovině 90. let bylo věnováno enormní úsilí charakterizaci dalších predispozičních genů (BRCA3/BRCAX). Přes jeho neúspěch byla na základě asociačních studií a vazebných analýz vytipována řada genů ovlivňujících riziko vzniku karcinomu prsu, avšak na rozdíl od hlavních predispozičních genů je pravděpodobnost vývoje nádoru u nosičů mutací jen mírně či středně zvýšena ve srovnání s rizikem v obecné populaci. Tyto geny byly nazvány jako geny se střední až nízkou penetrancí. Zatímco alterace vysoce penetrantních genů se v populaci vyskytují s malou četností (~0,1–0,01 %), alterace v genech s nízkou penetrancí (resp. nízkopenetrantní alely) jsou relativně časté (1–30 %) [3]. Přestože se geny se střední a nízkou penetrancí vyznačují klinicky méně závažným ovlivněním rizika onemocnění, vysoká alelická frekvence umožňuje kooperativní účinek nízkopenetrantních alel, které tak mohou být příčinou zvýšeného rodinného výskytu nádorového onemocnění u osob bez alterací v hlavních predispozičních genech. Za arbitrárně stanovenou hranici odlišující geny s nízkou penetrancí od vysoce penetrantních genů se považuje relativní riziko (RR) ~2. S ohledem na skutečnost, že penetrance je spojitou veličinou a skupina kandidátních genů se významně zvyšuje, setkáváme se v poslední době ještě s termínem „geny se střední penetrancí“ (geny středního rizika), u kterých se RR vyskytuje v intervalu 2–5.
V předkládané práci uvádíme přehled vybraných genů se střední penetrancí (ATM, CHEK2, NBS1(NBN) a PALB2) zvyšujících riziko vzniku karcinomu prsu, u kterých byly provedeny alespoň pilotní studie v populaci pacientů v ČR. Zaměřujeme se na zhodnocení jejich klinického významu v onkogenetice.
1. ATM (Ataxia Telangiectasia Mutated)
Hereditární mutace obou alel genu ATM (OMIM 607585) zapříčiňují vznik vzácného autozomálně recesivního onemocnění ataxia telangiectasia (AT) charakterizovaného mozečkovou ataxií, okulokutánními telangiektáziemi, defekty imunitního systému, zvýšenou citlivostí na účinky ionizujícího záření a výrazným sklonem ke vzniku nádorů, především lymfomů a leukemií [4]. Vznik karcinomu prsu u pacientů s AT je poměrně raritní, na čemž se podílí především významné zkrácení života nosičů bialelických mutací. Vztah mutací ATM ke karcinomu prsu tak vychází z epidemiologických analýz prokazujících mírné, avšak signifikantně zvýšené celoživotní riziko karcinomu prsu u heterozygotů s mutací genu ATM.
Protein ATM je iniciální kinázou regulující buněčnou odpověď na přítomnost dvouřetězcových zlomů genomové DNA. Tyto alterace znamenají závažné poruchy integrity genomu s výraznými patologickými důsledky pro postižené buňky. Jejich příčinou jsou exogenní vlivy (ionizující záření, radiomimetika) i endogenní procesy (poruchy replikace, radikálové ataky, meiotické rekombinace). V závislosti na proliferačním stavu buňky jsou dvouřetězcové zlomy reparovány v G0, G1–S fázi buněčného cyklu pomocí nehomologního spojení konců DNA (NHEJ) nebo přesnějším způsobem reparace docíleným homologní rekombinací v postreplikačních fázích buněčného cyklu. ATM kináza primárně iniciuje reparaci procesem homologní rekombinace, čímž se významně podílí na tvorbě bariéry zabraňující vzniku maligně transformovaných buněk in vivo [5]. Aktivovaná kináza ATM následně fosforyluje řadu substrátů, které se účastní reparace DNA, ale i regulace buněčného cyklu a apoptózy [6]. Gen ATM, zahrnující oblast 150 kb (11q22–23), kóduje v 66 exonech protein o velikosti 350 kDa (62 kódujících exonů).
Mutační analýza ATM u pacientek s karcinomem prsu v ČR
Soukupová et al [7] analyzovali gen ATM u 161 osob s karcinomem prsu z vysoce rizikových rodin, u kterých nebyla zaznamenána přítomnost patogenních alterací v genech BRCA1/2 a 183 vzorků kontrolní nenádorové populace. Analýza prokázala přítomnost alterací vedoucích ke zkrácení proteinového produktu u 4 (2,5 %) ze 161 vyšetřených pacientů (tab. 2). S výjimkou alterace postihující sestřih exonu 11 (c.1066–6 T>G), která byla detekována u dvou ze 183 souběžně vyšetřených vzorků kontrolní skupiny a jejíž patogenita je sporná, nebyly u kontrol zachyceny žádné další patogenní varianty genu ATM.

Klinický význam analýzy ATM v ČR
Z výsledků jediné doposud provedené studie vyplývá, že četnost hereditárních alterací v genu ATM v naší populaci je pravděpodobně nízká (< 2 % u osob s karcinomem prsu z vysoce rizikových rodin). Míra zvýšení rizika vzniku karcinomu prsu u vysoce rizikových osob je RR = 2,37 (95% CI = 1,51–3,78) [8]. Provádění mutační analýzy genu ATM (i s ohledem na velikost genu) se, kromě podezření na onemocnění ataxií telangiektázií, u rizikových pacientek s karcinomem prsu zatím nedoporučuje. Diskutabilní otázkou zůstává vztah alterací genu ATM k radiosenzitivitě.
2. CHEK2 (Checkpoint kinase 2)
Gen CHEK2 (OMIM 604373) je lokalizován na chromozomu 22q12.1. Výsledný protein, označovaný jako CHK2, má velikost přibližně 60 kD a tvoří jej 546 aminokyselinových zbytků [9]. CHK2 je jaderný fosfoprotein, který se účastní přenosu a amplifikace signálů od senzorických proteinů rozpoznávajících přítomnost dvouřetězcových zlomů v DNA na efektorové proteiny fosforylované CHK2 kinázou, které následně regulují intracelulární pochody v odpovědi na poškození genomu (obr. 1).
Mutace v genu CHEK2 byly na přelomu tisíciletí popsány v souvislosti se vznikem Li-Fraumeni syndromu u nemocných bez přítomnosti mutace v genu TP53 [10]. Třebaže tato asociace byla v dalších studiích značně zpochybněna, provedené analýzy naznačily, že alterace CHEK2 genu se podílejí na zvýšení rizika vzniku řady nádorových onemocnění čítajících nejen karcinomy prsu, ale i karcinomy kolorekta, prostaty, ovaria, štítné žlázy a ledviny [11–15]. Mutace c.1100delC, vedoucí k posunu čtecího rámce a translaci zkráceného proteinového produktu s nefunkční kinázovou aktivitou, je nejvíce studovanou alterací CHEK2 genu. V současné době známe výsledky řady rozsáhlých studií provedených především na populacích pacientů s hereditárními a sporadickými karcinomy prsu, které ukazují, že frekvence této alterace oplývá výraznou geografickou odlišností, s vysokou četností v severní a západní Evropě a Rusku, nižším výskytem ve střední a východní Evropě, USA a Austrálii a zanedbatelným výskytem v jižní Evropě, Jižní Americe a Asii [16].
Mezi další často sledované alterace CHEK2 patří mutace c.470C>A (I157T) postihující funkci FHA domény (vysoce konzervativní oblasti genu CHEK2), alterace donorového místa sestřihu c.444+1G>A (známé jako IVS2+1G>A) způsobující aberantní sestřih vedoucí k posunu čtecího rámce a vzniku zkráceného translačního produktu a rozsáhlá delece c.909-2028_1095+330del5395 (p.Met304Leufs15X) postihující dva exony v centrální části genu, která se endemicky objevuje v regionu zahrnujícím ČR [17]. Kromě těchto patogenních alterací genu CHEK2 však bylo do současné doby popsáno více než 80 dalších genetických změn, mezi kterými jasně dominují missense alterace, jejichž význam je ve většině případů nejasný.
Analýza genu CHEK2 v ČR
Analýza posunových mutací
Výskyt patogenní mutace c.1100delC vedoucí k syntéze zkráceného proteinového produktu (p. T367MfsX15) byl analyzován u 358 nemocných s karcinomem prsu z vysoce rizikových rodin a 688 neselektovaných pacientek se sporadickým karcinomem prsu [18]. Výsledky analýzy ukázaly, že četnost této alterace ve vyšetřované populaci byla nízká (1/358 (0,28 %) a 3/688 (0,44 %)). Vyšetření kontrolního souboru odhalilo dva nosiče této alterace v 730 vyšetřených vzorcích (0,27 %). Od uvedení do praxe v roce 2006 je screening mutace c.1100delC zahrnut do vyšetření rozsáhlých delecí BRCA2 pomocí standardně prováděné MLPA. Na základě souhrnných výsledků analýz provedených na pracovištích Oddělení epidemiologie a genetiky nádorů MOÚ Brno a Ústavu biochemie a experimentální onkologie (ÚBEO) 1. LF UK v Praze se ukazuje, že četnost mutace c.1100delC je v ČR vyšší než prvotní zjištění. Mutace byla identifikována u 17 z 1 866 vysoce rizikových BRCA1/2-negativních pacientů (0,91 %). U 18 pacientů z tohoto souboru (0,96 %) byla nalezena rozsáhlá delece 5395bp. V současné době dokončujeme analýzu CHEK2 v souboru vysoce rizikových BRCA1/2-negativních pacientů, která kromě shora uvedených alterací odhalila i přítomnost čtyř dalších posunových mutací u 6 z 616 vyšetřovaných pacientů.
Význam posunových mutací v genu CHEK2 byl rovněž studován jako rizikový faktor vzniku maligního onemocnění i u dalších onkologických pacientů v ČR. Četnost výskytu nosičů c.1100delC ve skupině nemocných s karcinomem kolorekta (4/631, 0,63 %) se nelišila od výskytu v kontrolní populaci (2/730, 0,27 %, p = 0,4) [19]. Ve vzorcích 113 osob s adenomatózní familiární polypózou (FAP) a její atenuovanou formou (AFAP) nebyl nalezen žádný nosič mutace c.1100delC [20]. Rovněž jsme nenalezli žádného nosiče této mutace ve skupině 270 nemocných s karcinomem pankreatu [21].
Rozsáhlá delece genu CHEK2 postihující exony 9 a 10 nebyla nalezena u nemocných s kolorektálním karcinomem či sporadickým karcinomem pankreatu, a je tedy pravděpodobně asociována pouze se zvýšeným rizikem vzniku karcinomu prsu.
Analýza dalších alterací genu CHEK2
V rámci výzkumných programů jsme se zabývali i studiem výskytu missense variant (mutací vedoucích k záměně jedné aminokyseliny za jinou) ve vybraných oblastech genu CHEK2 u pacientek s karcinomem prsu a dalšími nádory dospělého věku v české populaci (tab. 3). Analýza byla zaměřena na oblast tzv. FHA domény, kde se vyskytuje missense varianta genu CHEK2 c.470T>C (p.I157T). Tato varianta byla nejčetnější změnou v kódující oblasti genu CHEK2 ve vyšetřovaných souborech. Její výskyt byl ve srovnání s kontrolní populací však významně vyšší pouze u pacientů s karcinomem kolorekta (2,5 vs 4,8 %, p = 0,03), ale nelišil se u nemocných se sporadickým karcinomem prsu (2,5 vs 2,8 %, p = 0,7). Výskyt alterace I157T vedl ke zvýšení rizika vzniku sporadického karcinomu kolorekta (OR = 2,0), ale ne karcinomu prsu. Ve srovnání s výsledky studií v Polsku nebo Finsku je výskyt alterace I157T v ČR nižší [24–26].
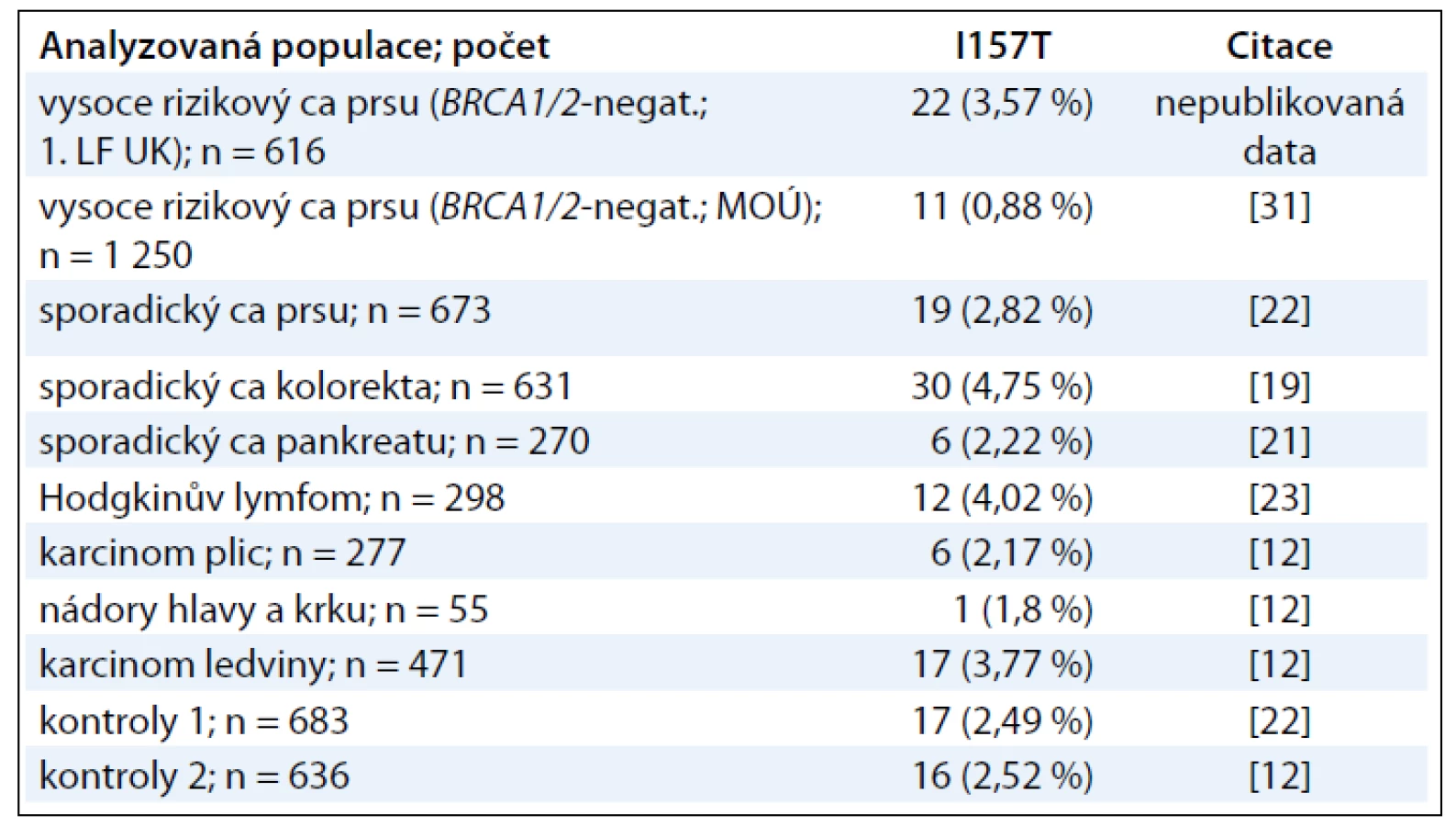
V souborech pacientů se sporadickým karcinomem prsu i kolorekta jsme analyzovali vztah nalezených alterací ve FHA doméně k familiárnímu výskytu nádorových onemocnění, který však nebyl nalezen ani v populaci karcinomu prsu ani v případě karcinomu kolorekta, kde přítomnost mutace I157T prokazatelně zvyšovala riziko vzniku „sporadického“ onemocnění. Hraničního významu dosahovala korelace mezi současným výskytem karcinomu kolorekta a karcinomu plic u nosičů alterace v oblasti FHA domény (p = 0,051). Tento nesignifikantní výsledek je potenciálně zajímavým zjištěním, neboť byla na dvou nezávislých studiích prokázána negativní asociace výskytu mutace I157T a karcinomu plic (RR = 0,44, 95% CI 0,31–0,63) [12,27]. Zjištění, že I157T může vykazovat protektivní účinek na vznik karcinomu plic a rizikový účinek na vznik dalších nádorů, včetně karcinomu prsu a kolorekta, oživil zájem o místně specifický charakter alterací CHEK2, který naznačuje, že v některých tkáních (prs, kolorektum) vedou alterace CHK2 k „vyřazení“ DNA reparačních mechanizmů umožňujících destabilizaci genomu a vznik následných genetických inzultů zodpovědných za nádorovou transformaci, zatímco v jiných tkáních (plíce) iniciuje porucha této signalizace netolerovatelné selhání genomové integrity iniciující zánik takto poškozených buněk [28]. Této spekulaci nahrává i pozorovaný akcentovaný protektivní účinek (snížení RR přibližně o polovinu) mutace I157T na vznik karcinomu plic u kuřáků, kde se předpokládá, že haploinsuficientní stav CHK2 vede k akcentované apoptóze buněk získávajících rozsáhlá genotoxická poškození v důsledku kouření. Odpověď na tuto zatím nevyjasněnou otázku je důležitým předpokladem pro racionální použití specifické terapie založené na ovlivnění signálních cest regulovaných CHK2 kinázou ve smyslu použití jejích inhibitorů na straně jedné, nebo reaktivace její funkce na straně druhé.
Klinický význam analýzy CHEK2 v ČR
Z doposud provedených studií a výsledků genetických analýz vyplývá, že četnost nosičů s vysokou pravděpodobností prokazatelně patogenních alterací (posunových mutací) v genu CHEK2 v české populaci bude vyšší než 2 %.
V roce 2008 Weischer et al [29] publikovali rozsáhlou metaanalýzu (26 000 nemocných a 27 000 kontrol) mutace c.1100delC u pacientů s karcinomem prsu. Její výsledky odhalily větší význam mutace c.1100delC, než vyplývalo z předchozích dílčích výsledků jednotlivých studií (OR = 2,7; 95% CI 2,1–3,4 u pacientek se sporadickým karcinomem prsu, s kumulativním rizikem 37 % do 70 let; 95% CI 26–57 %). Ještě podstatně vyšší riziko bylo nalezeno pro pacientky s familiárním karcinomem prsu (OR = 4,8; 95% CI 3,3–7,2). Autoři doporučují zařazení nosičů mutace c.1100delC do intenzivních screeningových programů pro nosiče mutací v hlavních predispozičních genech zodpovědných za vznik hereditárních karcinomů prsu a/nebo ovaria. S tímto závěrem se ztotožňuje i shrnující práce Naroda [30], ve které je doporučen screening nosiček mutace c.1100delC pomocí MR a chemoprevence tamoxifenem. Je pravděpodobné, že ostatní posunové mutace v genu CHEK2 se budou vyznačovat obdobným ovlivněním rizika, jako je tomu v případě c.1100delC.
Testování dvou prokazatelně patogenních a rekurentních mutací v genu CHEK2 (c.1100delC a rozsáhlé delece postihující dva exony centrální oblasti) je možné v ČR doporučit v rodinách s familiárním nebo sporadickým výskytem nádoru prsu, kde je indikováno vyšetření i BRCA1/2 genů pro možnou dědičnou příčinu. Pokud byla mutace zjištěna v rizikové rodině s nádory prsu, je vhodné provést prediktivní testování u příbuzných. U nosiček mutace c.1100delC přesahuje přepokládané celoživotní riziko nádorů prsu 25 %, pokud má žena příbuznou prvního stupně s nádorem prsu, nicméně může být vyšší v rodinách s mnohočetným výskytem nádorů prsu. Přesto se běžně nenabízí nosičkám CHEK2 mutace profylaktická mastektomie.
V preventivní péči je nutné použití i roční magnetické rezonance prsů. Nádory prsu jsou u většiny pacientek ER a PR pozitivní, objevují se o něco později než u nosiček BRCA1/2 mutací. V souboru byl průměrný věk onemocnění nádorem prsu u žen s velkou delecí dvou exonů CHEK2 48,1 let (32–71 let), u nosiček c.1100delC CHEK2 43,3 let (24–59 let) [31].
Genetické poradenství v rodinách s CHEK2 mutací je na základě neúplné penetrance patogenních alterací problematické, jelikož riziko nádorů nemusí záviset pouze na samotné mutaci, ale i na významnosti rodinné anamnézy. Nelze zcela vyloučit, že mutace v CHEK2 genu může být modifikačním faktorem, který moduluje riziko dalšího predispozičního genu.
U žen, které nejsou nosičkami familiární CHEK2 mutace, je nutné opatrně posuzovat nutnost preventivní péče. Vzhledem k tomu, že je možná polygenní etiologie rizika nádoru prsu v rodině, je vhodné navrhnout preventivní péči dle empirického rizika rodinné anamnézy.
CHEK2mutace se pouze minimálně vyskytuje v rodinách s BRCA1/2 mutací. V souboru 1 250 pacientek analyzovaných v MOÚ byly nalezeny obě mutace BRCA1 a CHEK2 pouze v jedné rodině. V běžném testování vyšetřování končí nálezem patogenní BRCA1/2mutace a mutace v CHEK2 genu již není zjišťována. Zachycení obou mutací je proto možné pouze v rámci studií.
Metaanalýza mutace c.1100delC u kolorektálního karcinomu (4 194 pacientů a 10 010 kontrol) prokázala zvýšení rizika vzniku onemocnění u neselektovaných nemocných s kolorektálním karcinomem (OR = 2,11; 95% CI 1,41–3,16), u nemocných s familiárním onemocněním (OR = 2,80; 95% CI 1,74–4,51), ale ne se sporadickou formou onemocnění (OR = 1,45; 95% CI 0,49–4,30). S ohledem na malou četnost této varianty a nejasná indikační kritéria nelze zatím vyšetření v klinické praxi doporučit.
Výsledky nedávné rozsáhlé studie mutace c.1100delC u pacientů s maligním melanomem z Německa a Dánska rovněž ukazují, že tato alterace se podílí na dvojnásobném zvýšení rizika onemocnění (OR = 2,01; 95% CI 1,03–3,91). Analýza genu CHEK2 u nemocných s melanomem v ČR zatím neproběhla.
Analýza mutací v genu CHEK2 u ostatních nádorů, u kterých byla nalezena asociace nosičství mutace v genu CHEK2 (obvykle c.1100delC) se zvýšeným rizikem vzniku onemocnění (karcinom prostaty (OR = 3,0), karcinom štítné žlázy, karcinom ledviny), není zatím pro klinickou praxi doporučena. Výsledky vyplývají z ojedinělých studií a analýza u pacientů v ČR neproběhla vůbec nebo jen na malém souboru nemocných.
Mutace I157T CHEK2 je missense variantou, která by mohla zvyšovat rizika různých nádorů; riziko nádorů prsu asi 1,5násobně, uváděna jsou i vyšší rizika nádorů tlustého střeva a prostaty. Na základě současných výsledků nelze přesně hodnotit její vliv na vznik karcinomu prsu a dalších nádorů a její klinické využití zatím není možné. Pro další missense varianty v CHEK2 genu (Arg180Cys, Thr387Asn, Ala392Val) je v současnosti klinické využití ještě více problematické.
3. NBS1 (Nibrin, NBN)
Gen NBS1 (dnes NBN; OMIM*602667) se nachází na chromozomu 8q21 a kóduje protein skládající se ze 754 aminokyselin nazývaný nibrin [32]. Nibrin je součástí heterotrimerního MRN komplexu (složeného z proteinů MRE11/RAD50/NBS1; obr. 1), který sehrává základní úlohu v reparaci dvouřetězcových zlomů procesem homologní rekombinace. Zatímco MRE11 svou nukleolytickou aktivitou upravuje místo zlomu DNA pro zahájení homologní rekombinace a RAD50 udržuje MRN komplex a přerušené konce DNA, NBS1 působí jako komunikační uzel dodávající celému komplexu možnost interakce s dalšími proteiny odpovědi na vznik dvouřetězcových zlomů (ATM, CtIP), čímž vzniká velmi komplexní struktura reparačního ohniska [33]. Dědičné mutace v genu NBS1způsobují autozomálně recesivní onemocnění Nijmegen Breakage Syndrome (NBS), charakterizovaný mikrocefalií, růstovou retardací, imunodeficiencí, hypersenzitivitou na ionizující záření a zvýšeným rizikem vzniku lymfoidních malignit [34]. U více než 90 % nemocných s NBS se vyskytuje patogenní mutace c.657del5 (p.K219DfsX16) v exonu 6, méně častá je pravděpodobně rovněž patogenní missense varianta c.643C>T (R215W). Řada studií nalezla zvýšené riziko vzniku karcinomu prsu a dalších nádorů (karcinom kolorekta, leukemie) u heterozygotů s c.657del5 [35].
Mutační analýza v genech kódujících dalších dva proteiny MRN komplexu (MRE11 a RAD50) v ČR doposud provedena nebyla. Předpokládá se, že alterace v těchto genech by se mohly podílet na zvýšeném riziku vzniku karcinomu prsu [2].
Mutační analýza NBS1 v ČR
V nedávné době byly publikovány výsledky dvou rozsáhlých analýz mutace c.657del5 u pacientů s kolorektálním karcinomem [36] a BRCA1/2-negativních pacientek z vysoce rizikových rodin s karcinomem prsu a v populaci pacientek s neselektovaným karcinomem prsu [37]. Obě studie ukazují nízkou četnost výskytu studované mutace mezi nemocnými s kolorektálním karcinomem (3/750, 0,40 %), vysoce rizikovým karcinomem prsu (2/600, 0,33 %) i sporadickým karcinomem prsu (2/703, 0,28 %). Výskyt mutace v populacích pacientů c.657del5 se nadto nelišil od výskytu v kontrolních populacích (5/1411, 0,35 % a 2/915, 0,22 %).
Klinický význam analýzy NBS1 (NBN) v ČR
S ohledem na velmi nízký výskyt alterací v genu NBS1 je praktický význam mutační analýzy pro pacientky s karcinomem prsu a nemocné s kolorektálním karcinomem malý.
4. PALB2 (Partner and Localizer of BRCA2)
Gen PALB2 (OMIM 610355) byl objeven v roce 2006 [38]. Je lokalizován na chromozomu 16p12.2 a skládá se ze 13 exonů. Proteinový produkt o 1 186 amino-kyselinách je součástí endogenního BRCA2-komplexu. Předpokládá se, že PALB2 proteiny oligomerizují a zprostředkovávají vstup BRCA2 s RAD51 a BRCA1 proteinů do reparačního ohniska. Tato interakce je zásadní pro spuštění HR.
Krátce po objevu genu PALB2 byly prokázány jeho bialelické patogenní mutace jako příčina nového subtypu Fanconiho anémie (FA-N) [39]. Vzhledem k fenotypové podobnosti subtypu Fanconiho anémie (FA-D1) způsobené bialelickými mutacemi genu BRCA2s typem FA-N a s ohledem na úzké funkční spojení proteinů BRCA2 a PALB2 bylo možné předpokládat, že monoalelické mutace PALB2 genu mohou být – jak je tomu u genu BRCA2 – příčinou dědičné predispozice k nádorovému onemocnění, zejména ke karcinomu prsu. Zvýšení rizika vzniku karcinomu prsu na dvou - až šestinásobek bylo následně u heterozygotních nosičů mutací genu PALB2 skutečně prokázáno v řadě především bělošských populací [40]. Jako patogenní byly klasifikovány nonsense mutace, posunové mutace a sestřihové mutace spojené s posunem čtecího rámce; naopak klinický význam různých zachycených missense variant genu PALB2 zůstává nejistý [41]. Relativní riziko karcinomu prsu u nosiček mutací v genu PALB2 bylo zpočátku odhadnuto na 2,3 (95% CI 1,4–3,9), resp. (3,0 u žen mladších 50. let a 1,9 u starších pacientek). V následujících studiích ale bylo popsáno riziko vyšší (2,3–6,1) [42–44]. Do současné doby byla charakterizována řada populačně-specifických alterací genu PALB2, které jsou často unikátní pro studovaný region.
Mutační analýza PALB2 v ČR
Doposud jsou k dispozici pouze pilotní výsledky mutační analýzy genu PALB2 u vysoce rizikových BRCA1/2-negativních pacientek s karcinomem prsu [45]. Tyto výsledky však naznačují, že prokazatelně patogenní posunové mutace v genu PALB2 se u vysoce rizikových pacientek s karcinomem prsu vyskytují v ČR relativně často (6/190, 3,1 %). Nalezené mutace (tab. 4) nebyly zachyceny v souboru 1 227 vzorků kontrolní populace.

Klinický význam analýzy PALB2 v ČR
O zařazení mutační analýzy bude možné kvalifikovaně rozhodnout po dokončení studie. Z dosavadních výsledků vyplývá, že o mutační analýze genu PALB2 by bylo možné uvažovat u pacientek s karcinomem prsu z rodin s familiárním výskytem karcinomu prsu. Nosičky patogenních alterací v genu PALB2 by měly být zařazeny do sledování podobně jako nosičky mutací v genu BRCA2. Vzhledem k neúplné segregaci variant genu PALB2 s nádorovým fenotypem je indikace pacientek s prokazatelně patogenními mutacemi v PALB2k preventivním chirurgickým výkonům doposud nejasná. Alterace v genu PALB2 neasociují se zvýšeným rizikem vzniku karcinomu ovaria. Jejich zvýšený výskyt u nemocných s karcinomem pankreatu je předmětem dalších analýz.
Závěr
Identifikace nosičů patogenních mutací predisponujících ke vzniku nádorů je důležitým cílem klinické onkogenetiky. Umožňuje zařazení onkologických pacientů do specializovaných programů pro časnou diagnostiku případného vzniku metachronního nádorového onemocnění či aplikaci specializovaných léčebných modalit (např. inhibitory PARP1). U klinicky asymtomatických jedinců v rodinách nosičů mutací umožňuje diferencovat preventivní programy na základě přítomnosti, či nepřítomnosti příčinné alterace. Vedle zřejmého klinického významu (záchyt onemocnění v časném, tedy obvykle lépe kurabilním stadiu, možnosti profylaktických výkonů, aplikace specializovaných terapeutických postupů) má identifikace příčinných mutací podmiňující vznik hereditárních nádorových syndromů i psychologicko-sociální význam pro postižené rodiny a v neposlední řadě prokazatelný ekonomický přínos.
V současné době dochází k výraznému rozšiřování spektra kandidátních predispozičních genů. V případě karcinomu prsu tak vedle klasických a relativně častých hlavních predispozičních genů BRCA1/2 a vysoce rizikových, avšak velmi vzácných genů TP53 a PTEN, byla identifikována další skupina genů zahrnující CHEK2, ATM, NBS1, RAD50, BRIP1, a PALB2 [46]. Je vysoce pravděpodobné, že díky rozvoji next-generation sekvenování budou brzy identifikovány i geny další [47,48]. Na rozdíl od hlavních predispozičních genů však patrně nelze očekávat, že jejich výskyt bude uniformní ve všech populacích (pravděpodobnost objevu BRCA3 je velmi malá). Zdá se, že tyto další predispoziční geny (jejich hereditární mutace) budou u jednotlivých nádorových onemocnění nebo skupiny nádorů charakteristické pro určité populace či regiony, z čehož vyplývá urgentní potřeba studií predispozičních genů na národních úrovních. Kromě toho, pro klinické využití umožňující aplikaci racionálních diagnostických, preventivních či terapeutických postupů je nezbytné, aby byl k dispozici dostatečně rozsáhlý a dobře charakterizovaný soubor nosičů mutací, který umožňuje zpřesnění kvalifikovaného odhadu rizika studovaných alterací. Přes nepochybné pokroky tento požadavek dnes splňuje, s výjimkou hlavních predispozičních genů, pouze mizivé procento alterací v ostatních predispozičních genech.
Závěrem lze tak konstatovat, že pro zařazení do rutinního screeningu u vysoce rizikových pacientek s karcinomem prsu lze v současnosti jednoznačně doporučit screening posunových mutací v genu CHEK2 (především c.1100delC a rozsáhlá delece postihující dva exony). V dohledné době bude patrně možné kvalifikovaně rozhodnout i v otázce analýzy genu PALB2. Naopak, analýza dědičných alterací v genu ATM a NBS1 na základě současných výsledků pro identifikaci rizika karcinomu prsu (a u NBS1 i karcinomu kolorekta) vhodná není.
Poděkování
Autoři práce děkují všem spolupracujícím klinickým genetikům a onkologům za jejich pochopení a pomoc.
Práce byla podpořena grantem IGA MZ ČR NT/13343 a Evropským fondem pro regionální rozvoj a státním rozpočtem České republiky (OP VaVpI - RECAMO, CZ.1.05/2.1.00/03.0101).
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
doc. MUDr. Petr Pohlreich, Ph.D.
Ústav biochemie a experimentální onkologie
1. LF UK a VFN v Praze
U Nemocnice 5
128 53 Praha 2
e-mail: ppohl@lf1.cuni.cz
doc. MUDr. Lenka Foretová, Ph.D.
Oddělení epidemiologie a genetiky nádorů
Masarykův onkologický ústav
Žlutý kopec 7
656 53 Brno
e-mail: foretova@mou.cz
Obdrženo: 15. 5. 2012
Přijato: 26. 5. 2012
Zdroje
1. Oldenburg RA, Meijers-Heijboer H, Cornelisse CJ et al. Genetic susceptibility for breast cancer: how many more genes to be found? Crit Rev Oncol Hematol 2007; 63(2): 125–149.
2. Bartkova J, Tommiska J, Oplustilova L et al. Aberrations of the MRE11-RAD50-NBS1 DNA damage sensor complex in human breast cancer: MRE11 as a candidate familial cancer-predisposing gene. Mol Oncol 2008; 2(4): 296–316.
3. Foulkes WD. Inherited susceptibility to common cancers. N Engl J Med 2008; 359(20): 2143–2153.
4. Soukupova J, Pohlreich P, Seemanova E. Characterisation of ATM mutations in Slavic Ataxia telangiectasia patients. Neuromolecular Med 2011; 13(3): 204–211.
5. Bartek J, Bartkova J, Lukas J. DNA damage signalling guards against activated oncogenes and tumour progression. Oncogene 2007; 26(56): 7773–7779.
6. Prokopcova J, Kleibl Z, Banwell CM et al. The role of ATM in breast cancer development. Breast Cancer Res Treat 2007; 104(2): 121–128.
7. Soukupova J, Dundr P, Kleibl Z et al. Contribution of mutations in ATM to breast cancer development in the Czech population. Oncol Rep 2008; 19(6): 1505–1510.
8. Renwick A, Thompson D, Seal S et al. ATM mutations that cause ataxia-telangiectasia are breast cancer susceptibility alleles. Nat Genet 2006; 38(8): 873–875.
9. Matsuoka S, Huang M, Elledge SJ. Linkage of ATM to cell cycle regulation by the Chk2 protein kinase. Science 1998; 282(5395): 1893–1897.
10. Bell DW, Varley JM, Szydlo TE et al. Heterozygous germ line hCHK2 mutations in Li-Fraumeni syndrome. Science 1999; 286(5449): 2528–2531.
11. Cybulski C, Górski B, Huzarski T et al. CHEK2 is a multiorgan cancer susceptibility gene. Am J Hum Genet 2004; 75(6): 1131–1135.
12. Brennan P, McKay J, Moore L et al. Uncommon CHEK2 mis-sense variant and reduced risk of tobacco-related cancers: case control study. Hum Mol Genet 2007; 16(15): 1794–1801.
13. Seppälä EH, Ikonen T, Mononen N et al. CHEK2 variants associate with hereditary prostate cancer. Br J Cancer 2003; 89(10): 1966–1970.
14. Thompson D, Seal S, Schutte M et al. A multicenter study of cancer incidence in CHEK2 1100delC mutation carriers. Cancer Epidemiol Biomarkers Prev 2006; 15(12): 2542–2545.
15. Nevanlinna H, Bartek J. The CHEK2 gene and inherited breast cancer susceptibility. Oncogene 2006; 25(43): 5912–5919.
16. Kleibl Z, Novotny J, Bezdickova D et al. The CHEK2 c.1100delC germline mutation rarely contributes to breast cancer development in the Czech Republic. Breast Cancer Res Treat 2005; 90(2): 165–167.
17. Walsh T, Casadei S, Coats KH et al. Spectrum of mutations in BRCA1, BRCA2, CHEK2, and TP53 in families at high risk of breast cancer. JAMA 2006; 295(12): 1379–1388.
18. Kleibl Z, Novotny J, Malik R et al. Výskyt a význam mutace CHEK2*1100delC u pacientek s karcinomem prsu a v kontrolní skupině zdravých žen v České republice. Klin Onkol 2005; 18(3): 98–101.
19. Kleibl Z, Havranek O, Hlavata I et al. The CHEK2 gene I157T mutation and other alterations in its proximity increase the risk of sporadic colorectal cancer in the Czech population. Eur J Cancer 2009; 45(4): 618–624.
20. Kleibl Z, Havránek O, Novotny J et al. Analýza mutace c.1100delC genu CHEK2 v populaci pacientů se sporadickým karcinomem kolorekta a familiární adenomatózní polypózou. Klin Onkol 2007; 20(2): 224–226.
21. Mohelnikova-Duchonova B, Havranek O, Hlavata I et al. CHEK2 gene alterations in the forkhead-associated domain, 1100delC and del5395 do not modify the risk of sporadic pancreatic cancer. Cancer Epidemiol 2010; 34(5): 656–658.
22. Kleibl Z, Havranek O, Novotny J et al. Analysis of CHEK2 FHA domain in Czech patients with sporadic breast cancer revealed distinct rare genetic alterations. Breast Cancer Res Treat 2008; 112(1): 159–164.
23. Havranek O, Spacek M, Hubacek P et al. Alterations of CHEK2 forkhead-associated domain increase the risk of Hodgkin lymphoma. Neoplasma 2011; 58(5): 392–395.
24. Cybulski C, Wokołorczyk D, Kładny J et al. Germline CHEK2 mutations and colorectal cancer risk: different effects of a missense and truncating mutations? Eur J Hum Genet 2007; 15(2): 237–241.
25. Kilpivaara O, Vahteristo P, Falck J et al. CHEK2 variant I157T may be associated with increased breast cancer risk. Int J Cancer 2004; 111(4): 543–547.
26. Kilpivaara O, Alhopuro P, Vahteristo P et al. CHEK2 I157T associates with familial and sporadic colorectal cancer. J Med Genet 2006; 43(7): e34.
27. Cybulski C, Masojc B, Oszutowska D et al. Constitutional CHEK2 mutations are associated with a decreased risk of lung and laryngeal cancers. Carcinogenesis 2008; 29(4): 762–765.
28. Antoni L, Sodha N, Collins I et al. CHK2 kinase: cancer susceptibility and cancer therapy – two sides of the same coin? Nat Rev Cancer 2007; 7(12): 925–936.
29. Weischer M, Bojesen SE, Ellervik C et al. CHEK2*1100delC genotyping for clinical assessment of breast cancer risk: meta-analyses of 26,000 patient cases and 27,000 controls. J Clin Oncol 2008; 26(4): 542–548.
30. Narod SA. Testing for CHEK2 in the cancer genetics clinic: ready for prime time? Clin Genet 2010; 78(1): 1–7.
31. Macháčkova E, Vašíčková P, Sťahlová Hrabincová E et al. Zárodečné mutace v genu CHEK2 u českých pacientů s nádorovou predispozicí. Abstrakt 005. In: Edukační sborník. Brno: XXXVI Brněnské onkologické dny a XXVI Konference pro sestry a laboranty 2012.
32. Varon R, Vissinga C, Platzer M et al. Nibrin, a novel DNA double-strand break repair protein, is mutated in Nijmegen breakage syndrome. Cell 1998; 93(3): 467–476.
33. Williams RS, Dodson GE, Limbo O et al. NBS1 flexibly tethers Ctp1 and Mre11-RAD50 to coordinate DNA double-strand break processing and repair. Cell 2009; 139(1): 87–99.
34. Seemanova E, Sperling K, Neitzel H et al. Nijmegen breakage syndrome (NBS) with neurological abnormalities and without chromosomal instability. J Med Genet 2006; 43(3): 218–224.
35. Seemanova E, Jarolim P, Seeman P et al. Increased risk of malignancies in heterozygotes in families of patients with Nijmegen breakage syndrome. Čas Lék Česk 2006; 145(2): 138–143.
36. Pardini B, Naccarati A, Polakova V et al. NBN 657del5 heterozygous mutations and colorectal cancer risk in the Czech Republic. Mutat Res 2009; 666(1–2): 64–67.
37. Mateju M, Kleiblova P, Kleibl Z et al. Germline mutations 657del5 and 643C>T (R215W) in NBN are not likely to be associated with increased risk of breast cancer in Czech women. Breast Cancer Res Treat 2012; 133(2): 809–811.
38. Xia B, Sheng Q, Nakanishi K et al. Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2. Mol Cell 2006; 22(6): 719–729.
39. Xia B, Dorsman JC, Ameziane N et al. Fanconi anemia is associated with a defect in the BRCA2 partner PALB2. Nat Genet 2007; 39(2): 159–161.
40. Rahman N, Seal S, Thompson D et al. PALB2, which encodes a BRCA2-interacting protein, is a breast cancer susceptibility gene. Nat Genet 2007; 39(2): 165–167.
41. Hellebrand H, Sutter C, Honisch E et al. Germline mutations in the PALB2 gene are population specific and occur with low frequencies in familial breast cancer. Hum Mutat 2011; 32(6): E2176–E2188.
42. Casadei S, Norquist BM, Walsh T et al. Contribution of inherited mutations in the BRCA2-interacting protein PALB2 to familial breast cancer. Cancer Res 2011; 71(6): 2222–2229.
43. Erkko H, Dowty JG, Nikkilä J et al. Penetrance analysis of the PALB2 c.1592delT founder mutation. Clin Cancer Res 2008; 14(14): 4667–4671.
44. Tischkowitz M, Capanu M, Sabbaghian N et al. Rare germline mutations in PALB2 and breast cancer risk: a population-based study. Hum Mutat 2012; 33(4): 674–680.
45. Janatova M, Pohlreich P, Kotlas J. PALB2 mutations in familial breast cancer in the Czech republic. Eur J Cancer 2011; 47 (Suppl 1): S133.
46. Walsh T, King MC. Ten genes for inherited breast cancer. Cancer Cell 2007; 11(2): 103–105.
47. Walsh T, Casadei S, Lee MK et al. Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing. Proc Natl Acad Sci U S A 2011; 108(44): 18032–18037.
48. Brownstein Z, Friedman LM, Shahin H et al. Targeted genomic capture and massively parallel sequencing to identify genes for hereditary hearing loss in middle eastern families. Genome Biol 2011; 12(9): R89.
Štítky
Dětská onkologie Chirurgie všeobecná OnkologieČlánek vyšel v časopise
Klinická onkologie

2012 Číslo Supplementum
- Aktuální evropská doporučení pro léčbu renální koliky v důsledku urolitiázy
- Incidence mozkových metastáz a intrakraniální aktivita sotorasibu u pacientů s NSCLC s mutací G12C onkogenu KRAS
- Přibývající důkazy o přínosu léčebného konopí u pacientů s chronickou bolestí
- Předchozí zánětlivá onemocnění plic zvyšují riziko rozvoje nádoru plic
- "Železem saturovaný" hovězí laktoferin zlepšuje chemoterapeutický účinek tamoxifenu u léčby basal-like karcinomu prsu u myší
Nejčtenější v tomto čísle
- Syndrom Birt-Hogg-Dubé
- Klinický význam analýz genů středního rizika pro hodnocení rizika vzniku karcinomu prsu a dalších nádorů v České republice
- Hereditární difuzní karcinom žaludku
- Klinické dysmorfické syndrómy s tumorigenézou
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy