
Život ohrožující manifestace systémových chorob pojiva a vaskulitid
Life threatening manifestations of connective tissue diseases and vasculitides
Rheumatic diseases may be presented at onset or in the course of diseases by acute serious illness, even life threatening and requiring admission to intensive care unit. The spectrum of symptoms and organs involvement is very wide, so as it corresponds to systemic character of diseases, which are the primary domain of rheumatology, but it also places high demands on knowledge of possible differential diagnosis of doctors working at intensive wards. This article gives an overview of most acute situation, which physicians can meet in patients with systemic diseases, including lupus erythematosus, antiphospolipid syndrome, systemic scleroderma, dermatomyositis/polymyositis and individual types of vasculitides group.
Key words:
Life-threatening events, SLE, antiphospholipid syndrome, scleroderma, idiopatic myositis, vasculitis
Autoři:
J. Vymětal; P. Horák; M. Skácelová
Působiště autorů:
III. interní klinika, nefrologická, revmatologická, endokrinologická, FN a LF UP Olomouc
Vyšlo v časopise:
Čes. Revmatol., 23, 2015, No. 3, p. 100-113.
Kategorie:
Přehledový článek
Souhrn
Řada revmatických onemocnění, ať již na svém počátku nebo v průběhu onemocnění, se může manifestovat závažným akutním, někdy i život ohrožujícím stavem a vyžadujícím přijetí na jednotku intenzivní péče. Spektrum příznaků bývá velmi různorodé a odpovídá povaze systémových onemocnění, která jsou primární doménou revmatologie, avšak současně kladou vysoké nároky na znalosti diferenciální diagnostiky lékařů pracujících na oborových jednotkách intenzivní péče. Tento článek předkládá přehled většiny akutních situací, se kterými se lékaři mohou u pacientů se systémovými onemocněními setkat, včetně systémového lupusu, antifosfolipidového syndromu, sklerodermie, dermatomyozitidy/polymyozitidy a skupiny vaskulitid a má být nápomocen k orientaci v této široké problematice.
Klíčová slova:
Život ohrožující stavy, systémový lupus, antifosfolipidový syndrom, sklerodermie, idiopatické myozitidy, vaskulitidy
Úvod
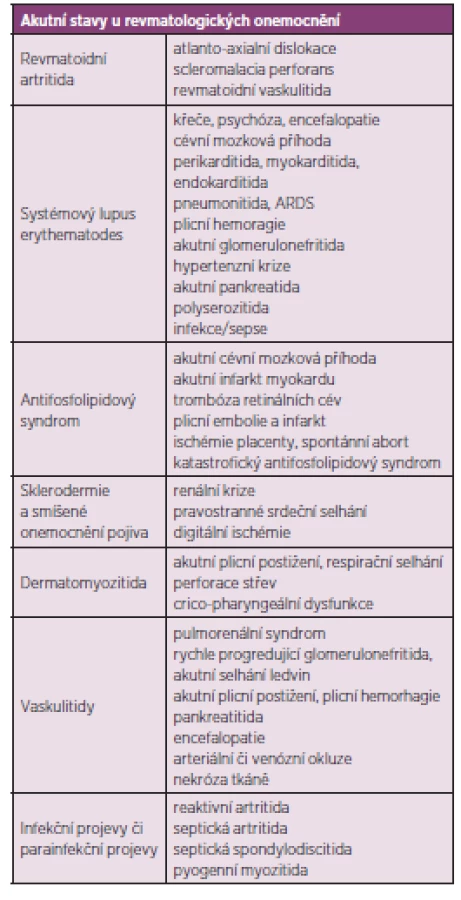
Revmatické choroby jsou často chronické, pomalu se rozvíjející a bez adekvátní léčby progredující nemoci. Nezřídka se však setkáváme s akutními projevy revmatických chorob, které mohou bezprostředně ohrozit zdraví či život postiženého a vyžadují okamžitou intervenci a intenzívní péči. Pacient s revmatologickým onemocněním může být dle dominujícího klinického obrazu primárně referován k přijetí na oddělení rozdílných odborností (interní, neurologické, kardiologické) a komplexnost problematiky systémových onemocnění vyžaduje v případě život ohrožující manifestace či komplikace multioborovou spolupráci včetně konzultace revmatologa a hospitalizaci na příslušné oborové JIP, resp. na oddělení anesteziologie a resuscitace, schopném zajistit komplexní intenzívní a resuscitační péči (tab. 1) shrnuje nejčastější akutní stavy u revmatických chorob.
1. Systémový lupus erythematodes (SLE)
Systémový lupus erythematodes (SLE) je autoimunitní zánětlivé onemocnění postihující zejména ženy v reprodukčním věku. Je charakterizované hyperaktivitou B buněk a nadprodukcí orgánově nespecifických autoprotilátek, z nichž mnohé se podílejí na tvorbě imunokomplexů. Jejich tkáňová či cévní depozita pak vedou k zánětlivému orgánovému postižení. Klinický obraz SLE je velmi pestrý, jedná se o vysoce heterogenní chorobu, kterou lze dělit do řady klinicky a laboratorně definovaných podtypů.
K častým projevům akutního vzplanutí patří systémové projevy, jako je horečka, únava, váhový úbytek. Průběh choroby je charakterizován střídáním remisí a exacerbací. Laboratorně je pro SLE typická tvorba orgánově nespecifických protilátek, které jsou namířeny proti nukleárním, cytoplazmatickým i povrchovým antigenům vlastních buněk. K nejčastějším projevům patří postižení kůže, kloubů, kardiovaskulárního systému, plic, glomerulů ledvin, centrálního nervového systému či krvetvorby. SLE může vyústit do selhání postiženého orgánu a závažná forma choroby je spojena s významnou mortalitou.
Život ohrožující manifestace SLE
Postižení srdce a kardiovaskulárního systému
Postižení srdce je v rámci SLE poměrně časté. Schematicky lze tuto problematiku rozdělit do čtyř okruhů:
- Chlopenní postižení
Nejčastěji se vyskytuje mitrální regurgitace, která je však zpravidla hemodynamicky málo významná. Závažnější formy chlopenního postižení jsou způsobeny chlopenními vegetacemi, které mohou mít velikost od malých uzlíků až po velké verukózní vegetace nazývané Libman Sacksova endokarditida. Vegetace mohou být spojeny s rozvojem chlopenní vady, nejčastěji opět s mitrální regurgitací, méně často pak s insuficiencí aortální chlopně (1). - Perikardiální postižení
Perikarditida, často spolu s pleuritidou a difúzním zánětem pobřišnice se řadí mezi tzv. serozitidy. Perikarditida je často asymptomatická (2). Může se však projevit perikardiální bodavou bolestí lokalizovanou za dolní částí sterna, spojenou s přítomností třecího šelestu. Velkoobjemové perikarditidy spojené s rizikem srdeční tamponády jsou u SLE vzácné. - Myokardiální dysfunkce
Projevuje se známkami srdečního selhání, tachykardií či arytmiemi, bývá spojena s kardiomegalií. Echokardiografické vyšetření nachází obvykle poruchu diastolické funkce, někdy i dilataci a difúzní hypokinezi s depresí systolické funkce levé komory. Akutní myokarditida je často doprovázena perikarditidou (3). - Ischemická choroba srdeční
Kardiovaskulární mortalita je v rámci SLE na jednom z předních míst v příčinách úmrtí pacientů, relativní riziko infarktu myokardu je 2,27, cévní mozkové příhody 2,05, a aterosklerózy dokonce 7,1 (4). Rozvoj aterosklerózy je spojený jednak s vyšší přítomností tradičních rizikových faktorů aterogeneze (obezita, kouření, metabolický syndrom), na jejím rozvoji se však podílejí i faktory specifické pro lupus (antifosfolipidové protilátky, aktivita choroby, endoteliální dysfunkce, vaskulitida, vliv léčiv).
Akutní plicní manifestace
- Lupusová pneumonitida je vzácnější manifestací choroby. Projevuje se horečkou, dušností, produktivním kašlem s menším množstvím sputa, tachypnoí, hemoptýzou, pleurální bolestí a hypoxémií. Fyzikální nález odhalí oboustranný bazální krepitus a v těžkých případech i centrální cyanózu. Na radiogramech plic bývají přítomny oboustranné skvrnité alveolární infiltráty. Vzhledem k typu manifestace je vždy třeba vyloučit infekci (5).
- Plicní hemoragie je vzácná, ale závažná a často fatální příhoda. Klinický průběh je podobný průběhu akutní lupusové pneumonitidy, je doprovázena rychlou progresí a alterací stavu nemocného s úzkostí. Je přítomná tachypnoe, hypoxémie, tachykardie, může se rozvinout syndrom dechové tísně dospělých ARDS (adult respiratory distress syndrome). Hemoragie může být buď intraalveolární či zevní a je často doprovázena i poměrně rychlým poklesem hemoglobinu. Histopatologicky je přítomno intraalveolární prokrvácení s intaktními erytrocyty a makrofágy nabitými hemosiderinem. Nemusí být přítomny zjevné známky vaskulitidy, někdy je popisovaná přítomnost mikroangiitidy, zánětu s nekrózou alveolárních kapilár, arteriol a malých muskulárních arteriol (6), obr. 1.
- Akutní reverzibilní hypoxémie: Patogeneze tohoto syndromu je nejasná. Snížená oxygenační kapacita plic může být vyvolána agregací neutrofilních leukocytů, jež obsahují rozpadové produkty komplementu, v plicních cévách (7, 8).

Postižení centrální nervového systému (CNS) – neuropsychiatrické manifestace
Údaje o prevalenci neuropsychiatrických projevů SLE se často rozcházejí dle úhlu pohledu autorů. Na počátku nemoci se vyskytují cca v 10 % případů, kdykoli v průběhu až v 80 % (9). Takto rozdílně stanovená prevalence neuropsychiatrických manifestací odráží existenci různých kritérií postižení CNS u SLE. American College of Rheumatology (ACR) formulovala definice a standardy pro diagnostiku 19 neuropsychiatrických syndromů v rámci SLE. Jejich plné definice a rozsáhlý popis jsou čtenáři k dispozici například na webové stránce: www.rheumatology.org/publications/ar/1999/aprilappendix.asp?aud=mem
- Cévní mozková příhoda: Riziko mozkového infarktu či náhlého úmrtí v jeho důsledku je u pacientů se systémovým lupusem signifikantně zvýšeno. Může se projevit jak klasickým iktem, tak tranzitorní ischemickou atakou. Riziko jejich recidivy je obvykle vysoké. Existuje silná asociace mezi těmito příhodami a antifosfolipidovými protilátkami (10). V souvislosti s antifosfolipidovým syndromem se vyskytují nejčastěji drobné, opakované a tranzitorní ischemické ataky, které mohou vést k podstatnému kognitivnímu postižení. Často je tato manifestace spojena s přítomností rozsáhlého liveda reticularis (Sneddonův syndrom) (11).
- Záchvaty křečí: Generalizované či parciální paroxysmy křečí mohou být i prvním příznakem choroby. Jejich příčina a etiologie je různá, může se jednat o důsledek zánětlivého postižení CNS či přítomnosti staré jizvy. K jejich riziku přispívají antifosfolipidové protilátky, metabolické poruchy, hypertenze, infekce, tumory, úrazy, ischemické mozkové příhody, vaskulopatie či toxicita léků (vysoké dávky antimalarik). Rozvoj křečí bez jasné lokální etiologie (s negativním angiogramem, CT, MRI) je pravděpodobně důsledkem vaskulopatie (12).
- Stavy akutní zmatenosti, které se mohou někdy v rámci SLE vyskytnout, jsou multifaktoriální. Je třeba odlišit podíl aktivity choroby od metabolických příčin či vedlejších účinků léčby. V klasické formě se jedná o komplexní poruchu s redukovanou schopností udržet pozornost a koncentraci. Bývají doprovázeny také poruchami chování, nálady, objevuje se psychomotorický neklid a různý stupeň poruchy vědomí. Tyto delirantní stavy, rozvíjející se během několika hodin či dnů, mohou vyústit do kómatu (13).
Postižení ledvin
Postižení ledvin přispívá významným způsobem k mortalitě a morbiditě nemocných se systémovým lupusem, na čemž se podílí jak samotné renální postižení, tak toxicita používaných léků. Diagnostika postižení ledvin se opírá o detailní klinické a laboratorní vyšetření doplněné v indikovaných případech o histologické vyšetření vzorku získaného při biopsii ledvin (14).
- Glomerulonefritida: U SLE je nejčastějším patogenetickým faktorem postižení ledviny glomerulární porucha. Histologická klasifikace WHO a její revize z roku 2004 rozlišuje 6 základních tříd glomerulonefritid a je v současnosti doplněna o indexy aktivity a chronicity (15). Značná část pacientů může vykazovat různý stupeň renální nedostatečnosti, která odvíjí od postižení glomerulů.
- Vaskulární postižení: Akutní renální selhání se často rozvíjí v souvislosti s přítomností kapilárních mikrotrombů při trombotické trombocytopenické purpuře/hemolyticko-uremickém syndromu (TTP/HUS) či u trombóz renálních arterií i žil v rámci významných komplikacích SLE a antifosfolipidového syndromu (16).
- Akutní intersticiální nefritida spojená s depozity imunokomplexů podél tubulární bazální membrány nekorelující s přítomností a aktivitou glomerulonefritidy může být další manifestací choroby. Jak proximální, tak distální tubulární syndromy jsou přítomny u řady nemocných se SLE, častokrát jsou asociovány s projevy sekundárního Sjögrenova syndromu a s pozitivitou anti-Ro a/nebo anti-La protilátek (17).
Akutní projevy SLE v graviditě
Díky soustavné léčbě a monitoraci nemocných žen s SLE se riziko vzplanutí během gravidity či po jejím ukončení do určité míry snížilo. Rizikovým faktorem zůstává jednak vysoká aktivita choroby před početím, jednak přítomnost ledvinného postižení. První manifestace lupusu během gravidity či bezprostředně po ní může být velmi těžká až katastrofická. Podezření by mělo vzniknout při výskytu odpovídající klinické symptomatologie v graviditě (vyrážka, artritida, alopecie, proteinurie s aktivním sedimentem, psychóza, chorea, pleuroperikarditida, vaskulitidy, atd.) U žen se SLE je vyšší riziko preeklampsie i hepatopatie. Preeklampsie je charakterizovaná náhlým vznikem hypertenze a proteinurie po 24. týdnu gravidity a je častější u prvních gravidit. Závažná klinická manifestace je důsledkem systémové endoteliální dysfunkce a mikroangiopatie, postiženými orgány mohou být CNS (křeče, eklampsie), játra (elevace jaterních indikátorových enzymů, porucha koagulace, HELLP syndrom – Hemolysis, Elevalted Liver enzymes, Low Plateles), ledvina (glomerulární postižení a proteinurie), mikroangiopatická hemolýza a trombocytopenie (18, 19).
Diagnostika choroby: Diagnostika choroby se opírá o správnou interpretaci řady anamnestických, klinických, laboratorních a paraklinických nálezů. Je třeba zejména vyzdvihnout roli stanovení autoprotilátek ANA, anti-dsDNA, ENA, antifosfolipidových protilátek či průkaz snížených hladin komplementu. Pozitivita těchto testů v souvislosti s klinickým obrazem pak vede ke stanovení diagnózy.
Diferenciální diagnostika živost ohrožujících stavů v rámci SLE zahrnuje vyloučení přítomnosti jiných systémových chorob pojiva či vaskulitid a zejména infekčních komplikací. Infekce probíhající v rámci systémového lupusu jsou často charakterizovány záludným průběhem, který je modifikován nasazenou imunosupresivní léčbou (20).
Léčba SLE je zaměřena na zvládnutí aktivní nemoci, na udržení remise onemocnění, na zabránění vzniku závažného orgánového postižení, na prevenci vzniku druhotných komplikací či na jejich léčbu. Strategie léčby SLE je velmi individuální. U pacienta v těžkém celkovém stavu s multiorgánovým selháváním jsou cíle i strategie léčby podřízeny snaze o přežití. U nemocných ve stabilním stavu je třeba ohodnotit aktivitu choroby, přítomnost orgánového postižení, kvalitu života, riziko toxicity zvažované či již nasazené léčby. Tabulka 2 sumarizuje zjednodušená doporučení pro léčbu jednotlivých forem SLE dle závažnosti stavu (21, 22, 23).

Prognóza: Je nesporné, že prognóza nemocných se systémovým lupusem se mění ruku v ruce s pokroky v jeho léčbě. V období před zavedením kortikoidů se pětileté přežívání od stanovení diagnózy pohybovalo kolem 50 %. Po zavedení glukokortikoidní léčby se pětileté přežívání zvýšilo na 75 %. Použití imunosupresní terapie zvyšuje dále pětileté přežívání na 85–90 %. Současně se však zvyšuje potřeba prevence a léčby závažných pozdních komplikací choroby, jako je osteoporóza, akcelerovaná ateroskleróza, zvýšené riziko nádorových onemocnění či léčba infekcí. Přes nesporné úspěchy imunosupresivní terapie tato u řady nemocných stále selhává a je navíc spojena s řadou nežádoucích účinků (24).
2. Antifosfolipidový syndrom
Antifosfolipidový syndrom (APS) je soubor příznaků spojený s přítomností protilátek proti fosfolipidům (lupus antikoagulans, antikardiolipinové protilátky, protilátky proti beta2glykoproteinu-1). Bývá přítomen u systémových chorob pojiva, zejména u SLE, kdy hovoříme o sekundárním antifosfolipidovém syndromu, nebo se vyskytuje samostatně jako tzv. primární antifosfolipidový syndrom (25).
Klinické příznaky: APS se projevuje poruchami krevního srážení spojenými s laboratorními odchylkami při vyšetření koagulace (prodloužení aPTT, falešně pozitivní Wassermannova reakce), klinicky pak častými trombózami arteriálního či venózního řečiště spojenými s poškozením postižených orgánů (pneumologické, neurologické, renální manifestace) a specifickou patologií spojenou s graviditou (úmrtí plodu, časté spontánní aborty). Často se vyskytuje rovněž trombocytopenie jako i další asociované příznaky.
Život ohrožující projevy antifosfolipidového syndromu jsou trombózy, plicní embolie, arteriální uzávěry, mozkové příhody, infarkty myokardu, mikroangiopatické postižení charakteru TTP/HUS i problematika gravidity spojená s rizikem potratu, úmrtí plodu či eklampsie a HELLP syndromu (26). Zvláštní pozornost si v této souvislosti zasluhuje tzv. katastrofický antifosfolipidový syndrom. Je spíše vzácnou manifestací této choroby s rozsáhlou trombotickou chorobou spojenou s multiorgánovým selháním. Jedná se často o fatální příhodu spojenou s rozvojem diseminované intravaskulární koagulace se zvýšením hladin fibrin-degradačních produktů a D-dimerů a s konsumpcí fibrinogenu (27). Klinická kritéria katastrofického APS zahrnují:
- Anamnézu antifosfolidového syndromu či přítomnosti antifosfolipidových protilátek.
- Tři či více epizod orgánových trombóz v posledním týdnu.
- Bioptické potvrzení mikrotrombu.
- Vyloučení jiných příčin orgánových trombóz či mikrotrombóz.
Léčba APS: Terapie primárního či sekundárního APS se zásadně neliší. U sekundární formy je často nezbytná i imunosuprese k potlačení aktivity základního patologického procesu. Nejčastěji se v léčbě užívá heparin (nízkomolekulární LMWH či nefrakcionovaný), warfarin a kyselina acetylsalicylová. Pacienti se systémovým lupusem užívají velmi často hydroxychlorochin, který má prospěšný efekt i v rámci antifosfolipidového syndromu. Léčba TTP/HUS v rámci antifosfolipidového syndromu se neliší od doporučení k léčbě idiopatických forem těchto závažných stavů a zahrnuje mimo symptomatické léčby také provedení opakovaných plazmaferéz. Léčba katastrofického APS spočívá v eliminaci možných vyvolávajících faktorů (infekce) a dále v kombinaci antikoagulační terapie (heparin, LMWH) s vysokými dávkami methylprednisolonu a opakovanou plazmaferézou v případě známek mikroangiopatie. Tyto postupy mohou být ještě doplněny o podávání intravenózních imunoglobulinů v dávce 400 mg/kg denně. Počet potřebných plazmaferéz se řídí klinickou odpovědí, dle některých prací dochází k 95% poklesu hladin antikardiolipinových protilátek po 5 plazmaferézách (28, 29). Existují také údaje o pozitivním efektu rituximabu (30). Velmi specifická je problematika léčby APS v graviditě (31, 32).
Prognóza: Katastrofická forma APS, zejména pokud je rozpoznána a léčena pozdě, je spojena s vysokou mortalitou.
3. Sklerodermie (SSc)
Systémová sklerodermie je chronické onemocnění pojivové tkáně postihující nejen kůži, ale i vnitřní orgány, jako např. ledviny, srdce, plíce a gastrointestinální trakt. Má dvě fenotypově vyhraněnější formy, difuzní systémovou sklerodermii a limitovanou systémovou sklerodermii. Onemocnění je charakterizováno fibrotizující sklerotizací periferních a viscerálních cév, nadprodukcí kolagenu v pojivové tkáni, změnami mikrovaskularizace a poruchou buněčné i humorální imunity. Raynaudův fenomén společně s trofickými změnami a tuhnutí kůže jsou dominantní příznaky choroby, postižení srdce, plic, gastrointestinálního traktu a ledvin závisí na formě a stadiu onemocnění (33).
Život ohrožující manifestace sklerodermie
Renální krize
Porucha funkce ledvin je u sklerodermie poměrně častým nálezem, většinou ve formě renální insuficience mírného stupně a s dobrou prognózou (34). Naproti tomu renální krize je akutní život ohrožující stav s incidencí 8–10 % u pacientů s limitovanou formou SSc, resp. 10–20 % u pacientů s difúzní formou choroby (35). Obvykle se rozvine během prvních pěti let od začátku onemocnění. Renální krize je důsledkem trombotické mikroangiopatie ledvin obdobné u TTP/HUS nebo APS.
Histologickým korelátem je proliferace a zesílení intimy arkuátních a interlobulárních arteriol a glomerulů vedoucí k zúžení až obliteraci cév se vznikem typického obrazu (onion-like) koncentrické hypertrofie arteriol (36). Prognóza renální krize je vážná, před zavedením ACE inhibitorů téměř všichni pacienti s těžkou renální insuficiencí umírali v průběhu 1 roku. Mezi základní klinické znaky renální krize patří náhlé selhání ledvin bez jakýchkoliv varovných příznaků, náhlý vznik středně těžké až těžké hypertenze, která může být provázena známkami emergentní hypertenzní krize (encefalopatie, levostranné srdeční selhání, cerebrovaskulární příhoda). Rizikovými faktory pro rozvoj renální krize jsou rychle progredující difúzní sklerodermie, přítomnost protilátek proti RNA-polymeráze, vysoké dávky glukokortikoidů či cyklosporinu. Nově vzniklá proteinurie a/nebo hematurie bývají většinou mírné, oligoanurie, popř. až anurie a rozvoj plicního edému mohou doplňovat celkový obraz společně s charakteristickým nálezem při renální biopsii (37).
Základními rysy renální krize jsou:
- Akutní selhání ledvin bez předchozích známek významnějšího onemocnění ledvin v anamnéze.
- Náhlý vznik středně těžké nebo těžké hypertenze, často spojený s projevy emergentní hypertenzní krize:
hypertonická encefalopatie (bolest hlavy, zmatenost, poruchy vizu),
hypertonická retinopatie (hemoragie, exsudáty),
akutní cévní mozková příhoda,
akutní levostranné srdeční selhání. - Diskrétní změny v močovém sedimentu (mírná proteinurie a/nebo hematurie).
Léčba: Hlavním léčebným zásahem je účinná korekce krevního tlaku (v průběhu 72 hodin), která vede k úpravě renálních funkcí až u 70% pacientů, což zlepšuje roční přežití na 80 %. Inhibitory angiotenzin konvertujícího enzymu (ACEI), které jsou jinak kontraindikovány u většiny případů jiných forem akutního selhání ledvin, se zdají naopak vhodné pro léčbu renální krize u systémové sklerózy. Ačkoliv nejširšími daty disponuje kaptopril, použitý ve většině studií, enalapril, ramipril a quinapril poskytují zřejmě podobnou ochranu. Terapie renální krize ACE inhibitory vede k výrazně lepšímu přežívání nemocných a zlepšení funkce ledvin i u již dialyzovaných nemocných, umožňující v některých případech i přerušení dialyzační léčby (38). Prognóza však stále zůstává nepříznivá a mortalita vysoká, zejména v důsledku pozdní diagnózy.
Intersticiální plicní onemocnění
Při rozvoji intersticiálního plicního onemocnění (IPO) u sklerodermie dochází k časnému poklesu plicních funkcí, zejména FVC. Byl zjištěn pokles FVC až o 32 % ročně během prvních dvou let (39). Nejčasněji intersticiální změny probíhají v subpleurálních oblastech dorzálních bazálních částí obou plicních křídel, u nemocných, u kterých se rozvíjí střední a těžká restrikční ventilační porucha, postupují změny i do horních plicních polí. Prognóza IPO je nepříznivá, zvýšená mortalita na IPO u sklerodermie je pozorována zejména ve druhých 5 letech od začátku choroby. Pacienti se závažným plicním postižením, definovaným jako FVC < 55 % a DLCO < 40 % náležitých hodnot, mají zřetelně horší prognózu s mortalitou až 42 % během 10 let od začátku nemoci (40).
Diagnóza: Diagnóza je založena na zhodnocení rentgenového snímku plic a spirometrie, včetně vyšetření difúzní kapacity a následném doplnění HRCT plic a bronchoalveolární laváže s cytologickou analýzou aspirátu, popř. také biopsie plicní tkáně.
Léčba: V léčbě aktivní alveolitidy se uplatňují nižší dávky glukokortikoidů a cyklofosfamid (41). V případě pomalé progrese plicního postižení má onemocnění lepší prognózu než idiopatické intersticiální plicní procesy.
Plicní arteriální hypertenze (PAH)
Plicní arteriální hypertenze je způsobena postižením plicní cirkulace, které se vyskytuje buď idiopaticky, nebo asociovaně s řadou chorob. Z asociované PAH představují významnou skupinu systémová onemocnění, a to nejčastěji právě systémová sklerodermie, u které se může vyskytovat samostatně, nebo provázet intersticiální onemocnění plic. Hemodynamicky je plicní hypertenze definována jako zvýšení středního tlaku v plicnici nad 25 mmHg v klidu, v případě systémové sklerodermie jde o plicní arteriální hypertenzi prekapilárního typu. Progredující námahová dušnost a únavnost jsou nejčastějšími příznaky, mohou se vyskytovat i anginózní bolesti jako důsledek ischémie pravé komory. Mezi vzácnější projevy patří chrapot způsobený útlakem levého n. recurrens dilatovaným kmenem plicnice, kašel a hemoptýza. Pokročilá PAH se projeví obrazem pravostranného srdečního selhání a vede k terminálnímu selhání pravé komory.
Diagnóza: Echokardiografie je neinvazivní vyšetřovací metodou volby a v současné době je u systémových onemocnění prováděna jako pravidelný screening. Slouží jak k posouzení velikosti a funkce pravé komory, tak i k odhadu tlaku v plicnici. Pro odhad stupně plicní hypertenze je rozhodující měření gradientu trikuspidální regurgitace a odhad tlaku v pravé síni z náplně dolní duté žíly. Selektovaní nemocní jsou indikováni k pravostranné katetrizaci srdce, která je pro potvrzení PAH nezbytná. K selekci a indikaci pacientů pro tuto invazivní metodu může být nápomocen algoritmus vycházející z DETECT study (42).
Léčba: Snahou je zahájit léčbu již v časném stadiu. Při splnění kritérií je indikována specifická léčba plicní arteriální hypertenze – antagonisté receptoru pro endotelin 1 (bosentan, macicentan), inhibitory fosfodiesterázy 5 (sildenafil) nebo intravenózní prostanoidy (43, 44). Specifická léčba je soustředěna do Center pro plicní hypertenzi. V terapii plicní hypertenze se využívá i antikoagulační léčba (warfarin) pro riziko plicní embolie.
4. Idiopatické myozitidy (Dermatomyozitida/polymyozitida)
Definice: Dermatomyozitida a polymyozitida (DM/PM) jsou chronické autoimunitní choroby charakterizované postižením příčně pruhovaného svalstva (polymyozitida) nebo svalstva a kůže (dermatomyozitida) s laboratorní elevací svalových enzymů. Onemocnění může postihovat také jícen, plíce nebo myokard. Predominantními příznaky jsou slabost proximálních svalových skupin a typické kožní příznaky (heliotropní exantém, Gottronovy papuly).
Plicní manifestace choroby je hlavní život ohrožující příhodou u DM/PM (45), která se často vyskytne u nemocných s pozitivitou antisyntetázových protilátek (např. anti-Jo). Dušnost a respirační insuficience u DM/PM může být jednak sekundární v důsledku omezení ventilace při slabosti dechových svalů, zejména bránice, jednak primární při difúzní fibrotizující alveolitidě s přechodem do plicní fibrózy. Pacienti mohou být ohroženi zvýšeným rizikem aspirace a související aspirační pneumonií, která je uváděna jako jedna z hlavních příčin úmrtí u pacientů s omezenou motilitou a poruchami polykání (46).
Diagnóza: Funkční vyšetření plic, radiogram hrudníku, HRCT plic a bronchoalveolární laváž jsou standardními metodami v diagnostice plicního postižení; hladiny svalových enzymů, EMG, munologické testy a popř. svalová biopsie pak diferencující DM/PM.
Léčba: Léčba DM/PM spočívá v podávání vysokých dávek kortikoidů, často v kombinaci s imunosupresivy (azathioprin, methotrexát), v indikovaných případech lze použít vysokodávkované polyklonální imunoglobuliny.
Prognóza: Plicní postižení je u dermato/polymyozitidy jedním z hlavních negativních prognostických faktorů, pacienti s DM a plicním postižením mají výrazně zřetelně horší prognózu než bez plicní manifestace (47).
5. Vaskulitidy
Vaskulitidy jsou poměrně heterogenní skupinou onemocnění, jejichž společným rysem je zánět a nekróza cévní stěny. Často je jedná o primární systémové onemocnění se závažným multiorgánovým postižením, postiženy mohou být cévy všech kalibrů. V některých případech bývá etiologie vaskulitidy sekundární, zánět cév může být součástí některých revmatických chorob, například revmatoidní artritidy či SLE, nebo se může vyskytnout v rámci jiných závažných chorob (sarkoidóza, malignity, závažné bakteriální infekce). Závažné či život ohrožující komplikace se u pacientů se systémovými vaskulitidami vyskytují poměrně často, mezi nejzávažnější patří postižení plic a ledvin. Tyto a další akutní stavy doprovázející jednotlivé typy vaskulitid jsou přehledně shrnuty v tabulce 3.

Diagnóza: V diagnostice vaskulitid je nutno klást důraz především na správnou interpretaci výsledků klinického a laboratorního vyšetření a zobrazovacích metod. Vzhledem k poměrně velké variabilitě příznaků bývá nutná mezioborová spolupráce mezi revmatologem, pneumologem, nefrologem a klinickým patologem. Důležitou součástí diagnostiky je i imunologické vyšetření, a to zejména vyšetření protilátek proti cytoplazmě neutrofilů (ANCA), jejichž pozitivita je typická pro granulomatózu s polyangiitidou (pozitivita protilátek proti proteináze 3 – c ANCA) a eozinofilní granulomatózu s polyangiitidou (pozitivita protilátek proti myeloperoxidáze – p ANCA), které bývají souhrnně nazývány „ANCA asociované vaskulitidy“ (48).
Plicní postižení
Závažné plicní postižení se vyskytuje u pacientů s ANCA asociovanými vaskulitidami a dále pak v rámci pulmorenálního (Goodpasteurova) syndromu (49).
Granulomatóza s polyangiitidou (dříve nazývaná Wegenerova granulomatóza) je ANCA asociovaná vaskulitida primárně postihující cévy malého průměru s častým postižením respiračního systému. V horních dýchacích cestách často nacházíme chronickou rinitidu a sinusitidu s granulomatózním zánětem; závažnou komplikací může být zborcení nosní přepážky a vznik sedlovité deformity nosu jako důsledek chronického zánětu paranazálních dutin. Poměrně častá bývá i perforace nosního septa. Postižení plic se může manifestovat febriliemi, dušností, kašlem, někdy i s hemoptýzou, pleurodynií, vzácně i tracheální obstrukcí. Závažná stenóza dýchacích cest je život ohrožujícím stavem, bývá nejčastěji důsledkem reparačních procesů, jejím charakteristickým klinickým projevem je stridor. Pro onemocnění jsou typické plicní infiltráty, které bývají rozsáhlé a mohou být doprovázeny rozpadem tkání, plicní hemoragií a respiračním selháním, které může vést až k nutnosti umělé plicní ventilace. Exacerbace onemocnění velmi často souvisí s infekcí dolních cest dýchacích (50). Důsledkem chronického postižení plic může být plicní fibróza.
Eozinofilní granulomatóza s polyangiitidou (syndrom Churg-Straussové) je nekrotizující ANCA pozitivní vaskulitida, která je asociována s bronchiálním astmatem, eozinofilií a granulomatózním zánětem respiračního traktu. Na radiogramech bývají typicky přítomny prchavé plicní infiltráty. Histologické vyšetření prokáže převážně extravaskulární granulomy s bohatou eozinofilní infiltrací a známkami nekrotizující vaskulitidy (51, 52).
Mikroskopická polyangiitida je nekrotizující ANCA asociovaná vaskulitida postihující drobné cévy kůže, ledvin a plic (47), která může vést až u 29 % pacientů k plicní hemoragii (obr. 2).

Pulmorenální (Goodpasteurův) syndrom je vaskulitida charakterizovaná renálním poškozením a alveolárním krvácením. Charakteristická je pro ni pozitivita protilátek proti bazální membráně glomerulů (anti-GBM) (54).
Renální postižení
Jedná se o poměrně častou, závažnou manifestací vaskulitid, zejména polyarteritis nodosa, ANCA asociovaných vaskulitid, Henoch-Schönleinovy purpury a kryoglobulinemické vaskulitidy.
Polyarteritis nodosa je vůbec první popsanou vaskulitidou. Jedná se o relativně vzácnou vaskulitidu postihující cévy malého a středního kalibru, která bývá poměrně často asociovaná s virovou hepatitidou B (55), pozitivita ANCA protilátek nebývá častá, vyskytuje se v méně než 5 % případů. Poškození ledvin, v některých případech doprovázené maligní hypertenzí, vzniká jako důsledek ischemické nefropatie a může vést až k selhání ledvin, jehož podkladem jsou renální infarkty a jizvení parenchymu ledvin. Pro tuto vaskulitidu je typický angiografický průkaz četných zúžení a mikroaneurysmat viscerálních a renálních arterií. Ruptury mikroaneurysmat jsou vzácné, mohou však komplikovat biopsii ledvin.
Renální ANCA asociované vaskulitidy (granulomatóza s polyangiitidou, eozinofilní granulomatóza s polyangiitidou a mikroskopická polyangiitida) se mohou manifestovat jako rychle progredující glomerulonefritida (RPGN) s histologickým průkazem pauciimunní glomerulonefrititidy s tvorbou srpků. Na rozdíl od glomerulonefritidy u systémového lupus erythematodes nejsou u pauciimunní glomerulonefritidy přítomna depozita imunoglobulinů nebo komplementových částic (obr. 3).

Henoch-Schönleinova purpura je poměrně častá vaskulitida malých cév s multiorgánovým postižením (kůže, gastrointestinální trakt, ledviny, klouby). Jedná se o ANCA negativní leukocytoklastickou vaskulitidu. Poškození ledvin se může projevit makroskopickou nebo mikroskopickou hematurií s mírnou proteinurií, v některých případech i s nefrotickým nebo nefritickým syndromem s různým stupněm renální insuficience. Postižení ledvin se může v některých případech manifestovat dříve než typická kožní purpura. V biopsii ledviny bývá typicky přítomna IgA nefropatie s poměrně variabilním morfologickým obrazem od fokální mesangioproliferativní glomerulonefritidy až po nekrotizující formy s tvorbou srpků. Charakteristickým rysem jsou mesangiální depozita IgA (56).
Kryoglobulinemická vaskulitida: Kryoglobuliny jsou imunoglobuliny, které reverzibilně precipitují v chladném prostředí (typicky při 4°C). Přítomnost kryoglobulinu typu I, který je tvořen monoklonálním imunoglobulinem bez protilátkové aktivity, je typická pro monoklonální gamapatie. Kryoglobuliny typu II a III jsou smíšené, jsou tvořeny minimálně dvěma různými imunoglobuliny. Kryoglobuliny II. typu jsou převážně smíšené mono-, či polyklonální protilátky, jejichž M komponenta má většinou povahu revmatoidního faktoru. Kryoglobuliny III. typu jsou smíšené, obsahují celou řadu polyklonálních imunoglobulinů, poměrně často bývají přítomny u systémových onemocnění pojiva, leukémií, onemocnění hepatobiliárního traktu a infekcí. Naopak u kryoglobulinémie II. typu je častá asociace s virovou hepatitidou C (57). Typickým kožním projevem smíšené kryoglobulinémie je kožní purpura s leukocytoklastickou vaskulitidou, kožní léze mohou vést až k rozsáhlým ulceracím a těžkým mutilacím. Dalšími projevy choroby mohou být únava, horečky, Raynaudův syndrom a artralgie, dále pak i periferní neuropatie s dysesteziemi a anestezií. Při infekci virem hepatitidy C bývá přítomna hepatosplenomegalie. Poškození ledvin se může projevit proteinurií a mikroskopickou hematurií a různým stupněm renální insuficience. Klinickým příznakem pak může být nefrotický nebo nefritický syndrom a hypertenze (58), závažná renální insuficience bývá častější u mužů a starších nemocných. Při laboratorním vyšetření prokážeme přítomnost smíšeného kryoglobulinu, významnou konsumpci složek komplementu a pozitivitu revmatoidních faktorů. Renální biopsie, nejčastěji prováděná u pacientů se smíšenou kryoglobulinemií II. typu, prokáže membranoproliferativní glomerulonefritidu s výraznou neutrofilní a monocytární infiltrací a dvojitou konturou bazální membrány. V akutní fázi nefritického syndromu bývají přítomna intraluminální amorfní eozinofilní depozita. Důležitou součástí péče o pacienty je vyvarování se hypotermie, zejména při použití extrakorporálních metod, jako je například hemodialýza či kontinuální eliminační metody.
Léčba: Léčebná strategie záleží na typu onemocnění a rozsahu orgánového postižení. V terapii se využívají glukokortikoidy, cyklofosfamid, azathioprin, methotrexát, cyklosporin A a imunoglobuliny, v poslední době se začínají uplatňovat nové léčebné přístupy, jako je podávání mykofenolát mofetilu a rituximabu. U pacientů s těžkými, život ohrožujícími formami ANCA asociovaných vaskulitid, zejména v případě RPGN či plicních hemoragií, může kombinace agresivní imunosupresivní terapie vysokodávkovanými glukokortikoidy a cyklofosfamidem s plazmaferézou významně zlepšit prognózu nemocného.
Závěr
Akutní stavy u revmatických chorob, zejména u systémových chorob pojiva a u vaskulitid, představují velkou diferenciálně diagnostickou výzvu. Častokrát je nutné vzhledem k akutní manifestace orgánového postižení pomýšlet na celou řadu stavů. Jedná se o onemocnění, která snad jako žádná jiná překračují hranice řady medicínských oborů, a proto je diagnostický proces výrazně multidisciplinární záležitostí, který by však měl být koordinovaný lékařem se zkušeností v diagnostice a léčbě těchto stavů.
Grant: Podpořeno grantem IGA 15-28659A
Adresa pro korespondenci:
MUDr. Jiří Vymětal
III. interní klinika nefrologická, revmatologická a endokrinologická, FN a LF UP
I.P. Pavlova 6
779 00 Olomouc
e-mail: jiri.vymetal@fnol.cz
Zdroje
1. Galve E, Candell-Riera J, Pigrau C, Permanyer-Miralda G, Garcia-Del-Castillo H, Soler-Soler J. Prevalence, morphologic types, and evolution of cardiac valvular disease in systemic lupus erythematosus. N Engl J Med 1988; 319(13): 817–23.
2. Doria A, Iaccarino L, Sarzi-Puttini P, Atzeni F, Turriel M, Petri M. Cardiac involvement in systemic lupus erythematosus. Lupus 2005; 14(9): 683–6.
3. Moder KG, Miller TD, Tazelaar HD. Cardiac involvement in systemic lupus erythematosus. Mayo Clin Proc 1999; 74(3): 275–84.
4. Smržová A, Horák P, Skácelova M, Žurek M, Fryšáková L, Vymětal J, et al. Cardiovascular events in patients with systemic lupus erytematodes. Cor vasa [serial online] 2014 Mar 24; [Cited 2014 October 20]; 56: e145-e152 Available from: http://www.sciencedirect.com/science/article/pii/S0010865014000277
5. Horák P, Ciferská H, Krajsová B. Orgánové manifestace revmatických chorob. In: Pavelka K, Vencovský J, Horák P, Šenolt L, Mann H, Štěpán J, et al (eds). Revmatologie. Praha: Maxdorf-Jessenius; 2012. p. 106–37.
6. Martinez-Martinez MU, Abud-Mendoza C. Diffuse alveolar hemorrhage in patients with systemic lupus erythematosus. Clinical manifestations, treatment, and prognosis. Reumatol Clin 2014; 10(4): 248–53.
7. Martinez-Taboada VM, Blanco R, Armona J, Fernandez-Sueiro JL, Rodriguez-Valverde V. Acute reversible hypoxemia in systemic lupus erythematosus: a new syndrome or an index of disease activity? Lupus 1995; 4(4): 259–62.
8. Abramson SB, Dobro J, Eberle MA, Benton M, Reibman J, Epstein H, et al. Acute reversible hypoxemia in systemic lupus erythematosus. Ann Intern Med 1991; 114(11): 941–7.
9. Brey RL, Holliday SL, Saklad AR, Navarrete MG, Hermosillo-Romo D, Stallworth CL et al. Neuropsychiatric syndromes in lupus: Prevalence using standardized definitions. Neurology 2002; 58(8): 1214–20.
10. Koskenmies S, Vaarala O, Widen E, Kere J, Palosuo T, Julkunen H. The association of antibodies to cardiolipin, beta 2-glycoprotein I, prothrombin, and oxidized low-density lipoprotein with thrombosis in 292 patients with familial and sporadic systemic lupus erythematosus. Scand J Rheumatol 2004; 33(4): 246–52.
11. Sneddon IB. Cerebrovascular lesions and livedo reticularis. Br J Dermatol 1965; 77 : 180–5.
12. Cimaz R, Meroni PL, Shoenfeld Y. Epilepsy as part of systemic lupus erythematosus and systemic antiphospholipid syndrome (Hughes syndrome). Lupus 2006; 15(4): 191–7.
13. Bertsias GK, Ioannidis JP, Aringer M, Bollen E, Bombardieri S, Bruce IN, et al. EULAR recommendations for the management of systemic lupus erythematosus with neuropsychiatric manifestations: report of a task force of the EULAR standing committee for clinical affairs. Ann Rheum Dis 2010; 69(12): 2074–82.
14. Bertsias GK, Tektonidou M, Amoura Z, Aringer M, Bajema I, Berden JH, et al. Joint European League Against Rheumatism and European Renal Association–European Dialysis and Transplant Association (EULAR/ERA-EDTA) recommendations for the management of adult and paediatric lupus nephritis. Ann Rheum Dis 2012; 71(11): 1771–82.
15. Weening JJ, D'Agati VD, Schwartz MM, Seshan SV, Alpers CE, Appel GB, et al. The classification of glomerulonephritis in systemic lupus erythematosus revisited. J Am Soc Nephrol 2004; 15(2):241–50.
16. Hughson MD, Nadasdy T, McCarty GA, Sholer C, Min KW, Silva F. Renal thrombotic microangiopathy in patients with systemic lupus erythematosus and the antiphospholipid syndrome. Am J Kidney Dis 1992; 20(2): 150–8.
17. Dhingra S, Qureshi R, Abdellatif A, Gaber LW, Truong LD. Tubulointerstitial nephritis in systemic lupus erythematosus: innocent bystander or ominous presage. Histol Histopathol 2014; 29(5): 553–65.
18. Petri M, Howard D, Repke J. Frequency of lupus flare in pregnancy. The Hopkins Lupus Pregnancy Center experience. Arthritis Rheum 1991; 34(12): 1538–45.
19. Stojan G, Baer AN. Flares of systemic lupus erythematosus during pregnancy and the puerperium: prevention, diagnosis and management. Expert Rev Clin Immunol 2012; 8(5): 439–53.
20. Horák P, Tegzová D, Závada J, Olejárová M, Skácelová M, Smržová A, et al. Doporučení České revmatologické společnosti pro diagnostiku a sledování nemocných se systémovým lupus erythematodes. Čes Revmatol 2013; 21(2): 59–70.
21. Horák P, Tegzová D, Závada J, Olejárová M, Skácelová M, Smržová A, et al. Doporučení České revmatologické společnosti pro léčbu nemocných se SLE. Čes Revmatol 2013; 21(2): 110–122.
22. Xiong W, Lahita RG. Pragmatic approaches to therapy for systemic lupus erythematosus. Nat Rev Rheumatol 2014; 10(2): 97–107.
23. Dall'era M, Chakravarty EF. Treatment of mild, moderate, and severe lupus erythematosus: focus on new therapies. Curr Rheumatol Rep 2011; 13(4): 308–16.
24. Ruiz-Arruza I, Ugarte A, Cabezas-Rodriguez I, Medina JA, Moran MA, Ruiz-Irastorza G. Glucocorticoids and irreversible damage in patients with systemic lupus erythematosus. Rheumatology (Oxford) 2014; 53(8):1470–6.
25. Miyakis S, Lockshin MD, Atsumi T, Branch DW, Brey RL, Cervera R, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost 2006; 4(2): 295–306.
26. Ruffatti A, Salvan E, Del Ross T, Gerosa M, Andreoli L, Maina A, et al. Treatment strategies and pregnancy outcomes in antiphospholipid syndrome patients with thrombosis and triple antiphospholipid positivity. A European multicentre retrospective study Thromb Haemost. 2014; 112(4):727–35.
27. Nayer A, Ortega LM. Catastrophic antiphospholipid syndrome: a clinical review. J Nephropathol 2014; 3(1): 9–17.
28. Keeling D, Mackie I, Moore GW, Greer IA, Greaves M British Committee for Standards in Haematology. Guidelines on the investigation and management of antiphospholipid syndrome. Br J Haematol 2012; 157(1): 47–58.
29. Ruiz-Irastorza G, Cuadrado MJ, Ruiz-Arruza I, Brey R, Crowther M, Derksen R, et al. Evidence-based recommendations for the prevention and long-term management of thrombosis in antiphospholipid antibody-positive patients: Report of a Task Force at the 13th International Congress on Antiphospholipid Antibodies. Lupus 2011; 20(2): 206–218.
30. Berman H, Rodríguez-Pintó I, Cervera R, Morel N, Costedoat-Chalumeau N, Erkan D, et al. Rituximab use in the catastrophic antiphospholipid syndrome: Descriptive analysis of the CAPS registry patients receiving rituximab. Autoimmun Rev 2013; 12(11): 1085–90.
31. The American College of Obstetricians and Gynecologists. ACOG Practice Bulletin No. 118: antiphospholipid syndrome. Obstet Gynecol 2011; 117(1): 192–9.
32. Di Prima FAF, Valenti O, Hyseni E, Giorgio E, Faraci M, Renda E, et al. Antiphospholipid Syndrome during pregnancy: the state of the art. J Prenat Med. 2011; 5(2): 41–53.
33. Bečvář R, Soukup T, Štork J, Suchý D, Němec P, Jansa P, et al. Doporučení České revmatologické společnosti pro diagnostiku systémové sklerodermie. Čes Revmatol 2014; 22(2): 51–68.
34. Steen VD, Syzd A, Johnson JP, Greenberg A, Medsger TA Jr. Kidney disease other than renal crisis in patients with diffuse scleroderma. J Rheumatol 2005; 32(4): 649–55.
35. Denton CP, Lapadula G, Mouthon L, Müller-Ladner U. Renal complications and scleroderma renal crisis. Rheumatology (Oxford) 2009; 48(Suppl 3): S32–5.
36. Batal I, Domsic RT, Medsger TA, Bastacky S. Scleroderma renal crisis: a pathology perspective. Int J Rheumatol [serial on the internet]. 2010 Jul 28; [Cited 2014 October 20]. Available from: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2958499/
37. Mouthon L, Bussone G, Berezné A, Noël LH, Guillevin L. Scleroderma renal crisis. J Rheumatol 2014; 41(6): 1040–8.
38. Steen VD. Kidney involvement in systemic sclerosis. Presse Med 2014; 43(10 Pt 2): e305-14. Available from: http://www.em-consulte.com/article/928057/article/kidney-involvement-in-systemic-sclerosis
39. Steen VD, Conte C, Owens GR, Medsger TA Jr. Severe restrictive lung disease in systemic sclerosis. Arthritis Rheum 1994; 37(9):1283–9.
40. Luo Y, Xiao R. Interstitial Lung Disease in Scleroderma: Clinical Features and Pathogenesis. Rheumatology 2011 doi:10.4172/2161-1149. S1-002. Available from: http://omicsonline.org/interstitial-lung-disease-in-scleroderma-clinical-features-and-pathogenesis-2161-1149.S1-002.pdf
41. Wells AU. Interstitial lung disease in systemic sclerosis. Presse Med 2014; (10 Pt 2):e329–43.
42. Coghlan JG, Denton CP, Grünig E, Bonderman D, Distler O, Khanna D, et al. Evidence-based detection of pulmonary arterial hypertension in systemic sclerosis: the DETECT study. Ann Rheum Dis 2014; 73(7):1340–9.
43. Jansa P, Becvar R, Ambroz D, Palecek T, Tomcik M, Skacelova S, et al. Pulmonary arterial hypertension associated with systemic sclerosis in the Czech Republic. Clin Rheumatol 2012, 31(3): 557–61.
44. Aschermann M, Jansa P. Medikamentózní léčba plicní arteriální hypertenze v roce 2014. Vnitr Lek 2014; 60(4): 282–8.
45. Hallowell RW, Ascherman DP, Danoff SK. Pulmonary manifestations of polymyositis/dermatomyositis. Semin Respir Crit Care Med 2014; 35(2): 239–48.
46. Maldonado F, Patel RR, Iyer VN, Yi ES, Ryu JH. Are respiratory complications common causes of death in inflammatory myopathies? An autopsy study. Respirology 2012; 17(3): 455–60.
47. Fujisawa T, Hozumi H, Kono M, Enomoto N, Hashimoto D, Nakamura Y, et al. Prognostic factors for myositis-associated interstitial lung disease. PLoS One [on the internet] 2014 Jun 6; [cited 2014 Oct 20]; Available from: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4048238/?report=classic
48. Hruskova Z, Casian AL, Konopasek P, Svobodova B, Frausova D, Lanska V, et al. Long-term outcome of severe alveolar haemorrhage in ANCA-associated vasculitis: a retrospective cohort study. Scand J Rheumatol. 2013; 42(3): 211–4.
49. Niles JL, Böttinger EP, Saurina GR, Kelly KJ, Pan G, Collins AB, et al. The syndrome of lung hemorrhage and nephritis is usually an ANCA-associated condition. Arch Intern Med 1996; 156(4): 440–5.
50. Capizzi SA, Specks U. Does infection play a role in the pathogenesis of pulmonary vasculitis? Semin Respir Infect 2003; 18(1): 17–22.
51. Guillevin L, Cohen P, Gayraud M, Lhote F, Jarrousse B, Casassus P. Churg-Strauss syndrome. Clinical study and long-term follow-up of 96 patients. Medicine (Baltimore) 1999; 78(1): 26–37.
52. Vaglio A, Buzio C, Zwerina J. Eosinophilic granulomatosis with polyangiitis (Churg-Strauss): state of the art. Allergy 2013; 68(3): 261–73.
53. Guillevin L, Durand-Gasselin B, Cevallos R, Gayraud M, Lhote F, Callard P, et al. Microscopic polyangiitis: clinical and laboratory findings in eighty-five patients. Arthritis Rheum 1999; 42(3): 421–30.
54. Hellmark T, Segelmark M. Diagnosis and classification of Goodpasture's disease (anti-GBM). J Autoimmun 2014; 48–49 : 108–12.
55. Guillevin L, Mahr A, Callard P, Godmer P, Pagnoux C, Leray E. French Vasculitis Study Group. Hepatitis B virus-associated polyarteritis nodosa: clinical characteristics, outcome, and impact of treatment in 115 patients. Medicine (Baltimore) 2005; 84(5): 313–22.
56. Pillebout E, Thervet E, Hill G, Alberti C, Vanhille P, Nochy D. Henoch-Schönlein Purpura in adults: outcome and prognostic factors. J Am Soc Nephrol 2002; 13(5): 1271–8.
57. Misiani R, Bellavita P, Fenili D, Borelli G, Marchesi D, Massazza M, et al. Hepatitis C virus infection in patients with essential mixed cryoglobulinemia. Ann Intern Med 1992; 117(7): 573–7.
58. Zaidan M, Mariotte E, Galicier L, Arnulf B, Meignin V, Vérine J, et al. Vasculitic emergencies in the intensive care unit: a special focus on cryoglobulinemic vasculitis. Ann Intensive Care. 2012; 2(1):31–9.
Štítky
Dermatologie Dětská revmatologie RevmatologieČlánek vyšel v časopise
Česká revmatologie

2015 Číslo 3
- Ingenol-mebutát rozšiřuje možnosti léčby aktinické keratózy
- Riziko roztroušené sklerózy u pacientů s psoriázou
- Proces hojení ran krok za krokem a co ho může zkomplikovat
- Léčba chronické blefaritidy vyžaduje dlouhodobou péči
- Léčba zánětů spojivek a mazových žlázek víčka v primární péči
Nejčtenější v tomto čísle
- Ultrazvuková detekce entezitid u pacientů se spondyloartritidou
- Oční komplikace u pacientů s revmatoidní artritidou
- Život ohrožující manifestace systémových chorob pojiva a vaskulitid
- Abstrakta z XII. semináře mladých revmatologů
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy