
The Activities of Current Antimalarial Drugs on the Life Cycle Stages of : A Comparative Study with Human and Rodent Parasites
Background:
Malaria remains a disease of devastating global impact, killing more than 800,000 people every year—the vast majority being children under the age of 5. While effective therapies are available, if malaria is to be eradicated a broader range of small molecule therapeutics that are able to target the liver and the transmissible sexual stages are required. These new medicines are needed both to meet the challenge of malaria eradication and to circumvent resistance.
Methods and Findings:
Little is known about the wider stage-specific activities of current antimalarials that were primarily designed to alleviate symptoms of malaria in the blood stage. To overcome this critical gap, we developed assays to measure activity of antimalarials against all life stages of malaria parasites, using a diverse set of human and nonhuman parasite species, including male gamete production (exflagellation) in Plasmodium falciparum, ookinete development in P. berghei, oocyst development in P. berghei and P. falciparum, and the liver stage of P. yoelii. We then compared 50 current and experimental antimalarials in these assays. We show that endoperoxides such as OZ439, a stable synthetic molecule currently in clinical phase IIa trials, are strong inhibitors of gametocyte maturation/gamete formation and impact sporogony; lumefantrine impairs development in the vector; and NPC-1161B, a new 8-aminoquinoline, inhibits sporogony.
Conclusions:
These data enable objective comparisons of the strengths and weaknesses of each chemical class at targeting each stage of the lifecycle. Noting that the activities of many compounds lie within achievable blood concentrations, these results offer an invaluable guide to decisions regarding which drugs to combine in the next-generation of antimalarial drugs. This study might reveal the potential of life-cycle–wide analyses of drugs for other pathogens with complex life cycles.
: Please see later in the article for the Editors' Summary
Published in the journal:
. PLoS Med 9(2): e32767. doi:10.1371/journal.pmed.1001169
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pmed.1001169
Summary
Background:
Malaria remains a disease of devastating global impact, killing more than 800,000 people every year—the vast majority being children under the age of 5. While effective therapies are available, if malaria is to be eradicated a broader range of small molecule therapeutics that are able to target the liver and the transmissible sexual stages are required. These new medicines are needed both to meet the challenge of malaria eradication and to circumvent resistance.
Methods and Findings:
Little is known about the wider stage-specific activities of current antimalarials that were primarily designed to alleviate symptoms of malaria in the blood stage. To overcome this critical gap, we developed assays to measure activity of antimalarials against all life stages of malaria parasites, using a diverse set of human and nonhuman parasite species, including male gamete production (exflagellation) in Plasmodium falciparum, ookinete development in P. berghei, oocyst development in P. berghei and P. falciparum, and the liver stage of P. yoelii. We then compared 50 current and experimental antimalarials in these assays. We show that endoperoxides such as OZ439, a stable synthetic molecule currently in clinical phase IIa trials, are strong inhibitors of gametocyte maturation/gamete formation and impact sporogony; lumefantrine impairs development in the vector; and NPC-1161B, a new 8-aminoquinoline, inhibits sporogony.
Conclusions:
These data enable objective comparisons of the strengths and weaknesses of each chemical class at targeting each stage of the lifecycle. Noting that the activities of many compounds lie within achievable blood concentrations, these results offer an invaluable guide to decisions regarding which drugs to combine in the next-generation of antimalarial drugs. This study might reveal the potential of life-cycle–wide analyses of drugs for other pathogens with complex life cycles.
: Please see later in the article for the Editors' Summary
Introduction
Malaria remains one of the most widespread infectious diseases of our time. The latest estimates reveal that ∼250 million people are infected with malaria across the globe, of whom ∼800,000 die every year [1], the vast majority being young children. In 2007, the malaria eradication agenda was adopted by many researchers in the antimalarial community and target product profiles for new antimalarial medicines were defined [2]. Most available antimalarials were designed to target the pathogenic blood stages in humans and to address the constant threat of drug resistance [3]. However, to meet the objective of malaria eradication, medicines that block parasite transmission [4] and eliminate the asymptomatic and sometimes cryptic hepatic forms also need to be developed. The bottleneck populations of liver and sexual stage parasites [5] represent potential pathogen vulnerabilities that could be targeted by small molecules; the first such bottleneck is at the liver stage. Within minutes of being released by the bite of an infected female Anopheles mosquito, Plasmodium sporozoites reach the mammalian liver, where they invade hepatocytes and either lie dormant or develop over several days, eventually forming the schizonts that are the prelude to a blood stage infection [6]. Molecules that efficiently target the parasite stages in the liver would offer protection from infection and could theoretically eliminate the cryptic hypnozoite (dormant parasite) infection reservoirs formed by P. vivax and P. ovale. Because only 100 or so sporozoites may be introduced by a bite, there are likely to be many orders of magnitude fewer parasites at this stage than in an active blood stage infection, reducing the possibility of resistance arising. A second bottleneck occurs during sexual development. At each round of schizogony ∼1% of merozoites differentiate into gametocytes [7], and it is these developmentally arrested cells that are transmitted to the mosquito. Mature gametocytes are sexually dimorphic, forming microgametocytes and macrogametocytes that escape the red blood cell (RBC) and produce male and female gametes in the blood meal of the mosquito by processes known as exflagellation [8] and activation, respectively. Following fertilization the zygote differentiates into a motile and invasive ookinete within which the briefly diploid genome undergoes meiosis. These processes occur within an environment almost totally derived from host blood, which can therefore provide a novel and ideal conduit for the delivery of drugs to inhibit parasite transmission to the mosquito. Having crossed the mosquito midgut wall [9], the very few surviving ookinetes differentiate into oocysts, which undergo endomitosis, eventually producing thousands of daughter sporozoites. The sporozoites migrate from the midgut of the mosquito to its salivary glands where the lifecycle begins again.
Given that it would be highly desirable for candidate drugs to have activity against hepatic and sexual forms of the malarial parasite, it is surprising that few clinical trials, to date, have examined whether gametocyte carriage can be reduced following drug treatment. The only drugs found to be effective at reducing gametocyte carriage include artemisinin [10], artemisinin combination therapies (ACTs) [11],[12], methylene blue [13], and primaquine [12],[14]. Additionally, few studies have investigated the impact of drugs on the transmission of parasites from human blood to the mosquito vector [15]–[18], nor have many been designed to evaluate antihepatic stage activity. In the context of malaria eradication these gaps in our understanding of the full potential of the drug armoury are problematic.
Here we report the development of a series of novel assays against liver, sexual blood, and mosquito stages of the malaria parasite, using both drug-susceptible and drug-resistant parasite strains. We applied these assays to the current portfolio of schizonticidal compounds, consisting of 50 anti-infectives currently in use or under development..
Methods
Ethics Statement
All work involving laboratory animals for the host-to-mosquito transmission studies was performed in accordance with the European Union (EU) regulations “EU Directive 86/609/EEC” and within the regulations of the United Kingdom Animals (Scientific Procedures) Act 1986, sanctioned by UK Home Office Licence PLL70/6347 awarded in January 2008. Protocol design and implementation was guided by the principle of the three Rs (reduction, refinement, and replacement) and are of mild-to-moderate severity. Protocols are regularly reviewed and revised following approval by the Imperial College Ethics Review Committee.
Parasite Maintenance
P. berghei parasites constitutively expressing GFP (PbGFPcon) [19]–[21] were routinely maintained as described previously [22]. Only blood showing exflagellating parasites was used in the transmission assays.
P. falciparum NF54 strain parasites were maintained in culture as described previously [23]. Gametocyte cultures were produced as described [24].
P. falciparum In Vitro Antimalarial Activity
In vitro antimalarial activity was measured using the [3H]-hypoxanthine incorporation assay [25] with various strains of P. falciparum obtained from MR4. Results were expressed as the concentration resulting in 50% inhibition (IC50).
P. falciparum Exflagellation Assay
Compounds were added to mature gametocyte of drug-sensitive P. falciparum cultures that showed the ability to exflagellate. After 24 h, exflagellation was triggered by a temperature decrease to ∼21°C and observed 20 min later under the microscope. Highly motile exflagellation centres were recorded for ∼50–150 adjacent fields of view and reported per 10,000 RBCs.
P. berghei Ookinete Development Assay – Slide Method
Compounds and P. berghei gametocyte-infected blood were dispensed in a 96-well plate containing ookinete medium [26]. After 24 h at 19°C, Giemsa-stained ookinetes were counted under the microscope (×40) as previously described [27],[28].
Standard Membrane Feed Assay – P. berghei
Membrane feeds were performed as described previously [22]. Briefly, PbGFPcon-infected mouse blood was mixed with compounds, immediately placed into membrane feeders (39°C) and offered for 30 min to 80–100 overnight-starved A. stephensi (SDA 500 strain). After 7–9 d at 19°C/80%RH, mosquito midguts were dissected out. Midguts were fixed with paraformaldehyde and oocyst number was determined microscopically by semi-automated analysis as previously described [29].
Standard Membrane Feed Assay – P. falciparum
Gametocyte cultures were produced according to the same protocol as the Pf exflagellation assay. Pooled gametocyte cultures [30] were evenly divided between compounds in fresh medium and incubated at 37°C for 24 h. The parasite pellets were then resuspended in fresh RBCs and human serum treated with the compounds to a 50% haematocrit and immediately fed to mosquitoes as described above and then maintained at 27°C/60%RH for 10–12 d before dissection and counting.
Liver Stage Assay
7.5×103 HepG2 cells in 50 µl of medium (1.5×105 cells/ml) were seeded in 384 well plates (Aurora 384 IQ-EB Black/Clear Plates) 20–26 h prior to the actual infection [31]. 2 h prior to infection, 50 nl of compound in DMSO (0.1% final DMSO concentration per well) were transferred with a PinTool (GNF Systems) into the assay plates (10 µM final concentration). P. yoelii sporozoites were freshly dissected from infected A. stephensi mosquito salivary glands and filtered twice with a 40-µm strainer. The HepG2 cells were then infected with 8×103 sporozoites per well. After infection and 1-h incubation at 37°C, the cultures were washed, new media and compound added, and further incubated with 5-fold increased concentration of penicillin/streptomycin for 48 h at 37°C before exoerythrocytic forms (EEFs) quantification of infected cells by immunofluorescence.
EEF immunofluorescence quantification
After washing with 1×PBS and fixation of the cells with 4% paraformaldehyde solution (EMS), membranes were permeabilised with 0.5% Triton-X-100 (Thermo Fisher Scientific) and EEFs were stained using a mouse polyclonal serum raised against the Plasmodium heat shock protein 70 (HSP70), an Alexa goat antimouse IgG, Fca-specific DyLight 649 secondary antibody (Invitrogen), and the Hoechst 33342 nucleic acid dye (Invitrogen). Stained EEFs were then quantified using the Opera Confocal High Content Screening system (PerkinElmer). Images were collected using a 20× magnification at a binning of 2 using a 365 Xeon arc lamp illumination to detect the nuclei and 635-nm laser line to excite DyLight649.
High-content imaging of infected HepG2-CD81 cells was performed as described in Meister et al. [31]. Wells were analysed using a custom Acapella (PerkinElmer) script parameterized for this assay. In brief, images from fields inside the well were first discarded as out of focus when the intensity in the nuclear area was too low. Then hepatic cells were counted by detecting the nuclei labelled with Hoechst 33342 and parasites were segmented using the HSP70 immuno-labelling. Morphology-based (e.g., area, roundness) and intensity-based features were calculated for each object detected including the hepatocyte nuclei and the parasites. Parasitemia was set as the ratio between parasite number (Alexa fluor positive) and the hepatocyte nuclei count, determined at the same time.
Results
Schizonticidal Activities of Compounds against Strains of P. falciparum with Known Drug Resistance Markers
A collection containing all antimalarials approved for use in humans and those in clinical development, anti-infectives, and other controls (Figure 1) was profiled simultaneously on asexual blood stage parasites in a standardized growth inhibition assay (GIA) [25] using seven strains of P. falciparum exhibiting diversity in the molecular causes of resistance and geographical origins (Table S1). The half-maximal inhibitory concentrations (IC50) were determined for each molecule (Table S2). Known mutations in pfcrt, pfmdr1, pfdhfr, and pfdhps correlated with a loss of potency of at least 10-fold for the relevant drugs. As expected the 4-aminoquinolines (4-AQs) chloroquine and hydroxychloroquine showed a 6–100-fold reduction in potency against all drug-resistant strains containing the mutated chloroquine transporter (PfCRT) (Figure 2B). Amodiaquine, tert-butyl-isoquine pyronaridine, piperaquine, and naphthoquine were potent against all parasite lines (IC50 = 2–10 nM), as were the endoperoxides including the natural (artemisinin), the semi-synthetic (artesunate), or the fully synthetic peroxides (ozonides), all with IC50 values of 1–15 nM (Figure 2A).

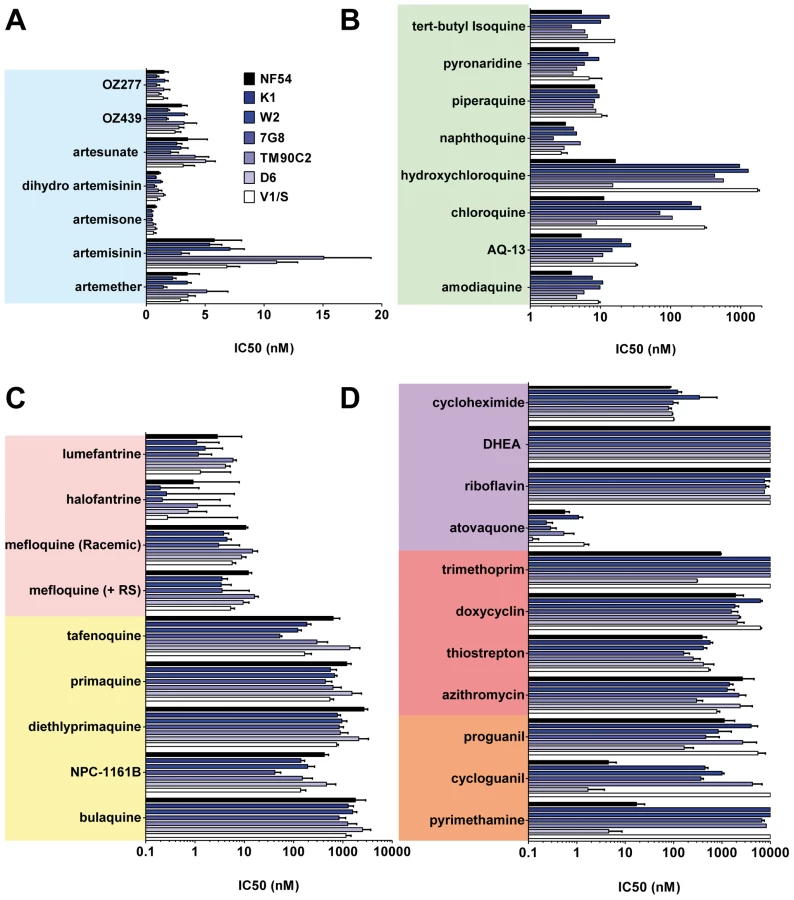
Compared to the 4-AQs, the 8-AQs (8-AQs) primaquine, diethylprimaquine, and bulaquine were less potent (IC50 = 0.5–2.5 µM) (Figure 2C) against both drug-resistant and -sensitive strains. Tafenoquine and NPC-1161B exhibited IC50 values in the 500-nM range against NF54 and in the 50-nM range against 7G8. IC50 values of both racemic mefloquine and the +RS isomer were below 10 nM. Halofantrine and lumefantrine displayed potencies below 4 nM against the sensitive strains NF54 and D6 and in the case of halofantrine, 0.3 nM against the multi–drug-resistant strains K1, W2, and 7G8. Resistance to pyrimethamine was verified in all drug-resistant strains (Figure 2D). Atovaquone a drug active in the subnanomolar range against P. falciparum blood stages showed at most a 10-fold difference in potency between the strains D6 and V1/S. In contrast, a ≥100-fold loss of potency was observed for cycloguanil between sensitive and resistant strains. The antibiotics azithromycin and trimethoprim, protein synthesis inhibitors, and other molecules such as dehydroepiandrosterone (DHEA), riboflavin, doxycyclin, and the prodrug proguanil showed IC50 values in the 1–10-µM range with no major differences between strains.
The Identification of Drugs That Additionally Block Transmission from Human Host to Mosquito Vector
Eradicating malaria will require medicines that prevent transmission of the parasite between humans and mosquitoes. Potentially the severe population bottleneck experienced as the parasite progresses from the mature gametocyte in the human host through gametogenesis and fertilization in the mosquito blood meal to the oocyst in the mosquito haemocoele offers the most vulnerable target for intervention. We developed assays for each of these events (Figure 3A). To integrate both the early sexual stages (gametocyte maturation and gametogenesis) and the late vector stage (sporogony) into the drug-testing cascade, we measured the exflagellation of male gametes in vitro (P. falciparum), ookinete formation in vitro (P.berghei), and the production of oocysts in A. stephensi (P. berghei and P. falciparum). Of these assays, we found analysis of P. berghei ookinete production in vitro was the most robust approach to identify molecules potentially targeting the early development of Plasmodium parasites in the mosquito. Forty-six molecules were tested at a concentration of 10 µM (Figure 3B). The most potent molecules were cycloheximide (blood stage IC50 of 25 nM) and atovaquone (IC50 = 65 nM). Thiostrepton (IC50 = 8 µM) and pyronaridine (IC50 = 6 µM) were less potent (Figure 3C). The latter two molecules and pyrimethamine also inhibited P. falciparum exflagellation by more than 80%, as did sulfamethoxazole and mefloquine (+RS). While displaying insignificant activity in the P. berghei ookinete formation assay (Figure 3C), all endoperoxides, with the sole exception of artemether, inhibited P. falciparum exflagellation by >65% (Figure 3D). Similarly all 4-AQs inhibited this event by >60%, with the exception of hydroxychloroquine and chloroquine, which enhanced exflagellation by at least 20% [16].

While recognising that the drugs being evaluated are not subject to metabolic degradation/activation by the mammalian hosts, among the 8-AQs, NPC-1161B and diethylprimaquine inhibited exflagellation by more than 70%. In marked contrast primaquine and the amino alcohol halofantrine unexpectedly stimulated exflagellation by 15% and 45%, respectively. All main chemical classes were then evaluated specifically against the vector stages by analysing the inhibition of P. berghei oocyst formation in vivo. Only NPC-1161B and lumefantrine exhibited >90% inhibition when tested at 10 µM (Figure S1). The transmission-blocking potential of molecules that were active against exflagellation and/or P. berghei sporogony was then assessed against the production of P. falciparum oocysts, which is the most difficult and lowest throughput, yet highest content analysis by encompassing all vector stages from gametocyte uptake to sporogony (see Figure 3A). In this assay, most endoperoxides inhibited oocyst production by >75%; NPC-1161B and mefloquine (+RS) totally blocked transmission at this stage (Figure 3E). Strikingly, the Coartem component, lumefantrine, and halofantrine impaired sporogony in both P. berghei and P. falciparum, while both mediated only moderate or no inhibition of exflagellation. This finding suggests that these molecules might act specifically on oocysts and not on gametogenesis, a behaviour that could be relevant to transmission reduction in the field given the long half-life of these molecules. As exflagellation is a component process of development within the gut of the mosquito in the P. falciparum oocyst assay (see Figure S2), these results, not unexpectedly, show at least partial concordance with the P. falciparum exflagellation assay.
The Identification of Drugs That Suppress Transmission from the Mosquito to the Human Host
When an infected mosquito bites a host, ∼100 sporozoites may be injected into the dermis from where they rapidly invade liver cells [32],[33]. This infective step represents the second bottleneck during transmission and therefore another potentially vulnerable point for intervention. In the absence of a practical liver stage assay measuring the formation of P. falciparum/P. vivax liver schizonts, an equivalent assay was developed in P. yoelii and used to assess the activity of the collection of molecules against this specific stage. Specifically, P. yoelii sporozoites were dissected from the salivary glands of infected mosquitoes and were allowed to invade human hepatocarcinoma cells expressing the CD81 protein. The development of the liver schizonts was monitored by immunofluorescence staining using an HSP70 antibody specific to the parasite (Figure 4A). As only 1% of the hepatocytes are infected in these circumstances, high content imaging was used to quantify growth inhibition of parasite schizonts (Figure 4B, 4D). Quantification of the total immunofluorescence per well is shown in Figure 4C. Dose response analysis using serially diluted compounds showed that cycloguanil, pyrimethamine, P218, and atovaquone all displayed IC50 values below 10 nM (Figure 4B, 4D). Methylene blue and artemisone demonstrated IC50 values of <100 nM. All endoperoxides tested in this assay, except artemisinin, exhibited IC50 values <3 µM, as did amodiaquine, AQ-13, pyronaridine, and naphthoquine. Of the 8-AQs, NPC-1161B was the only drug active in the submicromolar range against P. yoelii liver stages. Deferoxamine, thiostrepton, trimethoprim, and quinidine exhibited submicromolar potencies. Cycloheximide and thiostrepton, although showing IC50 values below 200 nM (Table 1), retarded the growth of HepG2-CD81 cells.


Discussion
Modes of Action of Major Classes of Antimalarials beyond the Asexual Blood Stage
The IC50 values of the known schizonticides against the asexual blood stages determined in this study correlate well with those reported in recent work examining 185 culture-adapted parasite strain lines treated with seven antimalarials [34].
Many schizonticidal drugs are hypothesised to interfere with haemoglobin metabolism. Our study shows that some of these drugs have activities against parasite stages that lack haemoglobin metabolism, e.g., the liver schizont, mature gametocyte, and sporogonic stages. This finding raises interesting questions about the mode(s) of action of these compounds beyond the asexual blood stage and our understanding of parasite metabolism. For example, natural, semi-synthetic and synthetic endoperoxides (artemisinin, DHA, artesunate, OZ277, and OZ439) are not only fast-acting molecules [35], but are among the most potent antimalarials currently used against the asexual blood stages [36]. They are thought to act by alkylating haem and other vital biomolecules [37],[38] (e.g., Pf TCTP) [39], and degrading phospholipids in parasite membranes [40]. The latter mechanism would be expected to have a major impact on all vegetative/replicating stages of Plasmodium's life cycle, e.g., asexual blood stage, liver schizont, oocyst, and microgametogenesis; and this is consistent with our results showing endoperoxide activity directly or indirectly against P. falciparum exflagellation, oocyst production, and P. yoelii liver schizont development. The lesser impact of this chemical class on ookinete and oocyst development in P. berghei might suggest species to species differences. It is also worth noting that because drugs are applied in human blood containing mature gametocytes prior to triggering exflagellation, inhibition of exflagellation by these molecules could be the direct consequence of a gametocytocidal property rather than solely a specific effect on gamete formation. Our data confirmed that 4-AQs are highly active against asexual blood-stage parasites in vitro, while the 8-AQs are not, indicating that subtle changes in the AQ core structure can result in major differences in the mode of action of some key antimalarials, although our data cannot address the possibility that the 8-AQ metabolites could be more active. Interestingly, molecules such as amodiaquine, pyronaridine, and tert-butyl-isoquine that target inter alia haem degradation also inhibited P. falciparum exflagellation. We are unaware of any prior data suggesting that haem degradation is essential to male gamete formation. This finding raises fascinating questions as to whether their modes of action in the mosquito blood meal are targeting the same, or different, molecular mechanisms. One working hypothesis could be that the major targets mediating the effect of these molecules in the sexual stages might also contribute to the effect seen in the asexual stages.
The 8-AQs are known to be active on the relapsing “hypnozoite” liver form of P. vivax following metabolic activation of the parent compound by liver enzymes [41]. In our assay where we do not anticipate any metabolism of any drugs, we note that the 8-AQ NPC-1161B was additionally shown to inhibit exflagellation in vitro and oocyst production in the mosquito vector. This result suggests either that NPC-1161B does not require metabolic activation or the drug exhibits poly-pharmacology by acting through another metabolite-independent mechanism. The antifolates P218 and pyrimethamine were found to be potent against rapidly replicating blood and liver stages and much less so against early vector stages, observations consistent with the essential role of the folate pathway in DNA synthesis [42].
When mature gametocytes are ingested, exflagellation is activated by a reduction in temperature and the presence of xanthurenic acid in the gut of the mosquito [43]. Inhibition of exflagellation by thiostrepton and cycloheximide is consistent with the observation that protein synthesis is a key component of these dramatic morphological changes [27], and all vegetative stages in the life cycle. Similarly our data confirm previous studies [44]–[46] showing the electron transport chain can be efficiently targeted by atovaquone in both the vector and the liver. For such stage-transcendent pathways, it could be hypothesised that the much lower parasite burden observed in the vector and the liver would increase the probability that treatments would eliminate infections when compared to targeting the abundant blood stage parasites. An important consequence would be that drugs specifically targeting the small “bottleneck” populations might delay significantly the selection of drug-resistant parasites.
“Management” of Drug Delivery to the Mosquito Blood Meal
While both our studies and the comprehensive treatise of Peters [47] show unequivocally that for many compounds effective delivery into the blood meal of the mosquito can be achieved, management of such delivery in the field is not trivial. In this context we note that the Cmax following administration of therapeutic doses for many antimalarials overlap the predicted potencies of the same antimalarials determined here against early vector stages (0.5–10 µM). For instance, in reported clinical trials, Cmax was shown to vary from 0.1 µM to 13.9 µM for artemether and lumefantrine administered at therapeutic doses, respectively. In the case of new molecules such as OZ439, Cmax values obtained in blood at relevant doses ranged between 1 and 2.5 µM (Table S3) [48]–[55].
Combination Therapies
To support the eradication agenda, new combination therapies will have to address three major issues: transmission of the pathogen, radical cure of P. vivax malaria, and the emergence of drug resistance. Ideally, new drug combinations should contain both schizonticidal and transmission-blocking components. Preferably fast-acting and long-lasting schizonticides would be combined with another agent that would target the parasite either at the sexual/vector stages, liver stage, or both. In the case of the liver stage, a co - or post-treatment prophylaxis would be provided as an end game scenario. From a pharmacokinetic perspective, when considering P. falciparum, some might speculate that this additional transmission-blocking component should be stable enough to exert its inhibitory capacity over several days against vector stages and/or liver schizont development to protect against a re-infection. However, considering that the schizonticidal component would kill both asexual blood stages and young gametocytes (<6-d old), a second coadministered drug efficiently eliminating mature (>6-d) gametocytes could potentially clear the host of all parasites; i.e., a long half-life would not be required [56]. A sustained stability would however be required for drugs targeting the parasite exclusively in the mosquito. Our study suggests that molecules such as atovaquone that inhibit the electron transport chain in the parasite mitochondria could be suitable candidates, but ideally should lack delayed onset of action and be difficult to raise resistance against [57]. To avoid triple therapies and reduce the risk of drug resistance, dual-activity molecules like amodiaquine, which inhibits haemoglobin digestion in the asexual blood stages and potentially inhibits gametocyte maturation/gamete exflagellation by a different mechanism, could be used in combination with a second antimalarial. Such polyvalent multistage activity has significant benefit to overall drug impact. Our study highlights that molecules such as amodiaquine, naphthoquine, tert-butyl-isoquine, and piperaquine do not lose potency when tested against chloroquine resistant strains. Therefore, to mitigate or defer the risk of drug resistance these molecules might be proposed as potential candidates for partnering new antimalarials such as OZ439. An important consideration would be to favour molecules that have never been used as monotherapy to avoid facing parasites that have previously acquired drug resistance. NPC-1161B inhibited both exflagellation and oocyst production; new molecules with similar properties but devoid of haemolytic liability of the 8-AQs in glucose-6-phosphate dehydrogenase (G6PD)–deficient patients could be interesting candidates as specific transmission-blocking agents.
While being strong inhibitors of blood-stage parasites, some molecules such as chloroquine reportedly enhance gametocytogenesis—a property that might have facilitated the spread of drug-resistant parasites [15],[58]. The panel of assays that we applied in this study has confirmed such “collateral” activity, in this case one that could prejudice the rational implementation in any elimination/eradication strategy.
Other Human Malarias
Although many have argued for the use of drug screens using nonhuman malarias [47], we recognize that to some, including assays with rodent malaria species might be considered suboptimal. Recent observations identifying interspecific variations include cysteine proteases in rodent plasmodia that show subtle active site differences to those in P. falciparum, leading to questions about the use of these models [59]. A critical role of amino acid 23 mediates activity and specificity of vinckepain-2, a papain-family cysteine protease of rodent malaria parasites [60]. P. yoelii 17X strain is intrinsically partially resistant to chloroquine and is therefore a poor model for studying acquisition of P. falciparum chloroquine resistance. Nevertheless we must recall that there are five species of Plasmodium that infect man and their biologies are patently different, therefore detecting drugs that may be active against multiple species in initial screens may offer long-term potential. Our assays provide, to our knowledge, the most comprehensive global overview of antimalarial drug action to date within the constraints imposed by the current state of culture methodologies for all life stages of all mammalian malaria parasites. Ideally, antimalarials developed against P. falciparum would have an even broader clinical usefulness if proven to be as effective against P. vivax [61]. The potencies of some antimalarials against the asexual blood stage of cultivated P. falciparum and P. vivax field isolates show a very good correlation (Table S4) [62]–[67]. This observation suggests that most of the pathways inhibited by antimalarials in P. falciparum are conserved and may offer valid targets in P. vivax. Moreover, the endoperoxide OZ439, which is currently evaluated in phase IIa clinical trials, has recently demonstrated equivalent efficacy in the treatment of P. falciparum and P. vivax patients (personal communication, MMV).
Drug Gaps and Future Steps
Our work has revealed previously unforeseen opportunities in the current discovery and development pipeline for new antimalarials. We demonstrated that drugs in the current portfolio, like pyronaridine and atovaquone, can also target liver and sexual stages in addition to asexual blood stages. Safe and stable drugs with similar multistage potential are now required. Developing drugs with long half-lives like mefloquine and chloroquine is essential to ensure that in patient blood the exposure of these drugs will remain above the minimum inhibitory concentration for several erythrocytic cycles and should ideally cover the period of gametocyte maturation. Additionally, new chemical scaffolds (e.g., nonendoperoxide) with fast killing potential are needed for a “one dose cure.”
The search for new drugs would be enhanced by the continued development of P. falciparum and P. vivax culture systems for every parasite life stage. It is critical to the malaria eradication agenda that these assays are able to identify drugs, such as the 8-AQs, with the capacity to safely eliminate P. vivax hypnozoites from the liver: this objective will also require the implementation and validation of in vitro and in vivo G6PD deficiency-dependent hemolysis assays. In parallel with efforts to discover innovative drugs for radical cure (elimination of P. vivax hypnozoites from the liver), new molecules blocking the onward development of mature (stage V) gametocytes are the other major priority in antimalarial discovery. There is therefore an urgent need to develop and validate high throughput screening assays allowing new libraries to be tested against P. falciparum and P. vivax gametocytes/transmission. These assays could then prioritise compounds for examination in preclinical studies in small mammals and then in standard membrane feeding assays (SMFA) using patient blood to find drugs blocking transmission in the field/clinical situation. Fields studies will remain essential to carefully examine any correlation between activities of new molecules against stabilized laboratory parasite strains and against field isolates.
Conclusions
For the first time, the main chemical classes of current and future antimalarials have been profiled simultaneously in standardized conditions against three Plasmodium species with respect to every major cellular strategy of the malarial life cycle, e.g., vegetative replication, dispersal, and sex (Figure 5). The present study provides the antimalarial research community with a reference set of methods and data, which may serve as a benchmark for newly discovered molecules when profiled against the entire life cycle of Plasmodium. This information might guide decisions regarding which molecules could be optimally combined to provide the next generation of drugs that will succeed to artemisinin combination therapies (ACTs) [68] and support the eradication of malaria. This comprehensive approach to drug discovery has potential utility for targeting other pathogens with complex life cycles.

Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. World Health Organization 2010 World malaria report. Available: http://www.who.int/malaria/world_malaria_report_2010/en/. Accessed 6 December 2011
2. Medicines for Malaria Venture (MMV) 2010 MMV target product profiles. Available: http://www.mmv.org/research-development/essential-information-scientists/target-product-profiles. Accessed 5 December 2011
3. FidockDA 2010 Drug discovery: priming the antimalarial pipeline. Nature 20 297 298
4. WellsTNCAlonsoPLGutteridgeWE 2009 New medicines to improve control and contribute to the eradication of malaria. Nat Rev Drug Discov 8 879 891
5. SindenRE 2010 A biologist's perspective on malaria vaccine development. Hum Vaccin 6 3 11
6. GreenwoodBMFidockDAKyleDEKappeSHIAlonsoPL 2008 Malaria: progress, perils, and prospects for eradication. J Clin Invest 118 1266 1276
7. BakerDA 2010 Malaria gametocytogenesis. Mol Biochem Parasitol 172 57 65
8. SindenRETalmanAMarquesSRWassMNSternbergMJ 2010 The flagellum in malarial parasites. Curr Opin Microbiol 13 491 500
9. VinetzJM 2005 Plasmodium ookinete invasion of the mosquito midgut. Curr Top Microbiol Immunol 295 357 382
10. PiyaphaneeWKrudsoodSTangpukdeeNThanachartwetWSilachamroonU 2006 Emergence and clearance of gametocytes in uncomplicated Plasmodium falciparum malaria. Am J Trop Med Hyg 74 432 435
11. StepniewskaKPriceRNSutherlandCJDrakeleyCJvon SeidleinL 2008 Plasmodium falciparum gametocyte dynamics in areas of different malaria endemicity. Malar J 7 249
12. BousemaTOkellLShekalagheSGriffinJTOmarS 2010 Revisiting the circulation time of Plasmodium falciparum gametocytes: molecular detection methods to estimate the duration of gametocyte carriage and the effect of gametocytocidal drugs. Malar J 9 136
13. CoulibalyBZoungranaAMockenhauptFPSchirmerRHKloseC 2009 Strong gametocytocidal effect of methylene blue-based combination therapy against falciparum malaria: a randomised controlled trial. PLoS One 4 e5318 doi:10.1371/journal.pone.0005318
14. ShekalagheSDrakeleyCGoslingRNdaroAvan MeegerenM 2007 Primaquine clears submicroscopic Plasmodium falciparum gametocytes that persist after treatment with sulphadoxine-pyrimethamine and artesunate. PLoS One 2 e1023 doi:10.1371/journal.pone.0001023
15. HallettRLSutherlandCJAlexanderNOrdRJawaraM 2004 Combination therapy counteracts the enhanced transmission of drug-resistant malaria parasites to mosquitoes. Antimicrob Agents Chemother 48 3940 3943
16. ButcherGA 1997 Antimalarial drugs and the mosquito transmission of Plasmodium. Int J Parasitol 27 974 987
17. KonAvan de Vegte-BolmerMSiebelink-StoterRvan GemertGJDaraA 2010 Sulfadoxine-pyrimethamine impairs Plasmodium falciparum gametocyte infectivity and Anopheles mosquito survival. Int J Parasitol 40 1221 1228
18. ColemanRE 1990 Sporontocidal activity of the antimalarial WR-238605 against Plasmodium berghei ANKA in Anopheles stephensi. Am J Trop Med Hyg 42 196 205
19. JanseCJFranke-FayardBMairGRRamesarJThielC 2006 High efficiency transfection of Plasmodium berghei facilitates novel selection procedures. Mol Biochem Parasitol 145 60 70
20. DawesEJZhuangSSindenREBasáñezM 2009 The temporal dynamics of Plasmodium density through the sporogonic cycle within Anopheles mosquitoes. Trans R Soc Trop Med Hyg 103 1197 1198
21. Franke-FayardBTruemanHRamesarJMendozaJvan der KeurM 2004 A Plasmodium berghei reference line that constitutively expresses GFP at a high level throughout the complete life cycle. Mol Biochem Parasitol 137 23 33
22. SindenRE 1997 Infection of mosquitoes with rodent malaria. Molecular biology of insect disease vectors: a methods manual. CramptonJMBeardCBLouisC London; New York Chapman and Hall 67 91
23. TragerWJensenJB 1976 Human malaria parasites in continuous culture. Science 193 673 675
24. KaushalDCCarterRMillerLHKrishnaG 1980 Gametocytogenesis by malaria parasites in continuous culture. Nature 286 490 492
25. VennerstromJLArbe-BarnesSBrunRCharmanSAChiuFC 2004 Identification of an antimalarial synthetic trioxolane drug development candidate. Nature 430 900 904
26. SindenREHartleyRHWingerL 1985 The development of Plasmodium ookinetes in vitro: an ultrastructural study including a description of meiotic division. Parasitology 91 227 244
27. ToyéPJSindenRECanningEU 1977 The action of metabolic inhibitors on microgametogenesis in Plasmodium yoelii nigeriensis. Z Parasitenkd 53 133 141
28. AlaviYAraiMMendozaJTufet-BayonaMSinhaR 2003 The dynamics of interactions between Plasmodium and the mosquito: a study of the infectivity of Plasmodium berghei and Plasmodium gallinaceum, and their transmission by Anopheles stephensi, Anopheles gambiae and Aedes aegypti. Int J Parasitol 33 933 943
29. DelvesMJSindenRE 2010 A semi-automated method for counting fluorescent malaria oocysts increases the throughput of transmission blocking studies. Malar J 9 35
30. CarterRRanford-CartwrightLAlanoP 1993 The culture and preparation of gametocytes of Plasmodium falciparum for immunochemical, molecular, and mosquito infectivity studies. Methods Mol Biol 21 67 88
31. MeisterSPlouffeDMKuhenKLBonamyGMCWuT 2011 Imaging of Plasmodium liver stages to drive next-generation antimalarial drug discovery. Science 334 1372 1377
32. ShinSCVanderbergJPTerzakisJA 1982 Direct infection of hepatocytes by sporozoites of Plasmodium berghei. J Protozool 29 448 454
33. MotaMMRodriguezA 2004 Migration through host cells: the first steps of Plasmodium sporozoites in the mammalian host. Cell Microbiol 6 1113 1118
34. MuJMyersRAJiangHLiuSRicklefsS 2010 Plasmodium falciparum genome-wide scans for positive selection, recombination hot spots and resistance to antimalarial drugs. Nat Genet 42 268 271
35. WhiteNC 1997 Assessment of the pharmacodynamics properties of antimalarial drugs in vivo. Antimicrob Agents Chemother 41 1413 1422
36. DondorpAMFanelloCIHendriksenICEGomesESeniA 2010 Artesunate versus quinine in the treatment of severe falciparum malaria in African children (AQUAMAT): an open-label, randomised trial. Lancet 376 1647 1657
37. O'NeillPMBartonVEWardSA 2010 The molecular mechanism of action of artemisinin—the debate continues. Molecules 15 1705 1721
38. KlonisNCrespo-OrtizMPBottovaIAbu-BakarNKennyS 2011 Artemisinin activity against Plasmodium falciparum requires hemoglobin uptake and digestion. Proc Natl Acad Sci U S A 108 11405 11410
39. BhisutthibhanJPanXHosslerPAWalkerDJYowellCA 1998 The plasmodium falciparum translationally controlled tumor protein homolog and its reaction with the antimalarial drug artemisinin. J Biol Chem 273 16192 16198
40. KumuraNFurukawaHOnyangoANIzumiMNakajimaS 2009 Different behavior of artemisinin and tetraoxane in the oxidative degradation of phospholipid. Chem Phys Lipids 160 114 120
41. WellsTNCBurrowsJNBairdJK 2009 Targeting the hypnozoite reservoir of Plasmodium vivax: the hidden obstacle to malaria elimination. Trends in Parasitol 26 145 151
42. GerbergEJ 1971 Evaluation of antimalarial compounds in mosquito test systems. Trans R Soc Trop Med Hyg 65 358 363
43. BillkerOLindoVPanicoMAEtienneEAPaxtonT 1998 Identification of xanthurenic acid as the putative inducer of malaria development in the mosquito. Nature 392 289 292
44. FowlerREBillingsleyPFPudneyMSindenRE 1994 Inhibitory action of the anti-malarial compound atovaquone (566C80) against Plasmodium berghei ANKA in the mosquito, Anopheles stephensi. Parasitology 108 383 388
45. FowlerRESindenREPudneyM 1995 Inhibitory activity of the anti-malarial atovaquone (566C80) against ookinetes, oocysts, and sporozoites of Plasmodium berghei. J Parasitol 81 452 458
46. BoysenKEMatuschewskiK 2011 Arrested oocyst maturation in Plasmodium parasites lacking type II NADH:ubiquininone dehydrogenase. J Biol Chem 286 32661 32671
47. PetersW 1987 Chemotherapy and drug resistance in malaria. Volume 1, 2nd edition London Academic Press
48. MoehrleJJArbe-BarnesSSiethoffCCraftJCDuparcS 2010 Novel synthetic ozonide OZ439: tolerability and pharmacokinetics in healthy volunteers. American Society of Tropical Medicine and Hygiene; November 3–7, 2010; Atlanta (Georgia, US); poster 1244. Available: http://www.astmh.org/AM/Template.cfm?Section=Abstracts_and_Education1&Template=/CM/ContentDisplay.cfm&ContentID=2847. Accessed 5 December 2011
49. McGreadyRStepniewskaKLindegardhNAshleyEALaY 2006 The pharmacokinetics of artemether and lumefantrine in pregnant woman with uncomplicated falciparum malaria. Eur J Clin Pharmacol 62 1021 1031
50. RamharterMKurthFSchreierACNemethJvon GlasenappI 2008 Fixed-dose pyronaridine-artesunate combination for treatment of uncomplicated falciparum malaria in pediatric patients in Gabon. J Infect Dis 198 911 919
51. ChinhNTQuangNNThanhNXDaiBTraversT 2008 Pharmacokinetics of the antimalarial drug piperaquine in healthy Vietnamese subjects. Am J Trop Med Hyg 79 620 623
52. NyuntMMAdamIKayentaoKvan DijkJThumaP 2010 Pharmacokinetics of sulfadoxine and pyrimethamine in intermittent preventive treatment of malaria in pregnancy. Clin Pharmacol Ther 87 226 234
53. SabchareonAAttanathPPhanuaksookPChanthavanichPPoonpanichY 1998 Efficacy and pharmacokinetics of atovaquone and proguanil in children with multidrug-resistant Plasmodium falciparum malaria. Trans R Soc Trop Med Hyg 92 201 206
54. LeeSJMcGreadyRFernandezCStepniewskaKPawMK 2008 Chloroquine pharmacokinetics in pregnant and nonpregnant women with vivax malaria. Eur J Clin Pharmacol 64 987 992
55. RijkenMJMcGreadyRJullienVTarningJLindegardhN 2011 Pharmacokinetics of amodiaquine and desethylamodiaquine in pregnant and postpartum women with Plasmodium vivax malaria. Antimicrob Agents Chemother 55 4338 4342
56. WilairatanaPTangpukdeeNKrudsoodS 2010 Long term primaquine administration to reduce Plasmodium falciparum gametocyte transmission in hypoendemic areas. Southeast Asian J Trop Med Public Health 41 1306 1311
57. BartonVFisherNBiaginiGAWardSAO'NeillPM 2010 Inhibiting Plasmodium cytochrome bc1: a complex issue. Curr Opinion in Chem Biol 14 440 446
58. HoghBGamage-MendisAButcherGAThompsonRBegtrupK 1998 The differing impact of chloroquine and pyrimethamine/sulfadoxine upon the infectivity of malaria species to the mosquito vector. Am J Trop Med Hyg 58 176 182
59. SinghAWalkerKJSijwaliPSLauALRosenthalPJ 2007 A chimeric cysteine protease of Plasmodium berghei engineered to resemble the Plasmodium falciparum protease falcipain-2. Protein Eng Des Sel 20 171 177
60. SinghAShenaiBKChoeYGutJSijwaliPS 2002 Critical role of amino acid 23 in mediating activity and specificity of vinckepain-2, a papain-family cysteine protease of rodent malaria parasites. Biochem J 368 273 281
61. DouglasNMAnsteyNMAngusBJNostenFPriceRN 2010 Artemisinin combination therapy for vivax malaria. Lancet Infect Dis 10 405 416
62. KockenCvan der WelAArbe-BarnesSBrunRMatileH 2006 Plasmodium vivax: in vitro susceptibility of blood stages to synthetic trioxolane compounds and the diamidine DB75. Exp Parasitol 113 197 200
63. LuxDPrajakwongSKollaritschHWernsdorferGWernsdorferWH 2003 In-vitro sensitivity testing of Plasmodium vivax: response to lumefantrine and chloroquine in northwestern Thailand. Wien Klin Wochenschr 115 50 54
64. PriceRMarfurtJChalfeinFKenangalemEPieraKA 2010 In vitro activity of pyronaridine against multidrug-resistant Plasmodium falciparum and Plasmodium vivax. Antimicrob Agents Chemother 54 5146 5150
65. TreiberMWernsdorferGWiedermannUCongpuongKSirichaisinthopJ 2011 Sensitivity of Plasmodium vivax to chloroquine, mefloquine, artemisinin and atovaquone in north-western Thailand. Wien Klin Wochenschr 123 Suppl 1 20 25
66. RussellBMUdomsangpetchRRieckmannKHKoteckaBMColemanRE 2003 Simple in vitro assay for determining the sensitivity of Plasmodium vivax isolates from fresh human blood to antimalarials in areas where P. vivax is endemic. Antimicrob Agents Chemother 47 170 173
67. LuFGaoQChotivanichKXiaHCaoJ 2011 In vitro anti-malarial drug susceptibility of temperate Plasmodium vivax from central China. Am J Trop Med Hyg 85 197 201
68. NostenFWhiteNJ 2007 Artemisinin-based combination treatment of falciparum malaria. Am J Trop Med Hyg 77 181 192
Štítky
Interní lékařstvíČlánek vyšel v časopise
PLOS Medicine
2012 Číslo 2
- Proces hojení ran krok za krokem a co ho může zkomplikovat
- Limity glykovaného hemoglobinu a význam dalších glykovaných proteinů
- Korelace dávky levothyroxinu s titrem autoimunitních protilátek u primární hypotyreózy
- Plicní hypertenze – syndrom mnoha tváří – vyžaduje přesnou diagnostiku a specializovanou léčbu
- Rána vizitkou (nejen) chirurga
Nejčtenější v tomto čísle
- The Activities of Current Antimalarial Drugs on the Life Cycle Stages of : A Comparative Study with Human and Rodent Parasites
- Association between Clean Delivery Kit Use, Clean Delivery Practices, and Neonatal Survival: Pooled Analysis of Data from Three Sites in South Asia
- Prevalence, Distribution, and Impact of Mild Cognitive Impairment in Latin America, China, and India: A 10/66 Population-Based Study
- Characterisation of Hospital Ward–Based Transmission Using Extensive Epidemiological Data and Molecular Typing
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy