
A Missense Mutation in the Gene in Dachshunds with Osteogenesis Imperfecta
Osteogenesis imperfecta (OI) is a hereditary disease occurring in humans and dogs. It is characterized by extremely fragile bones and teeth. Most human and some canine OI cases are caused by mutations in the COL1A1 and COL1A2 genes encoding the subunits of collagen I. Recently, mutations in the CRTAP and LEPRE1 genes were found to cause some rare forms of human OI. Many OI cases exist where the causative mutation has not yet been found. We investigated Dachshunds with an autosomal recessive form of OI. Genotyping only five affected dogs on the 50 k canine SNP chip allowed us to localize the causative mutation to a 5.82 Mb interval on chromosome 21 by homozygosity mapping. Haplotype analysis of five additional carriers narrowed the interval further down to 4.74 Mb. The SERPINH1 gene is located within this interval and encodes an essential chaperone involved in the correct folding of the collagen triple helix. Therefore, we considered SERPINH1 a positional and functional candidate gene and performed mutation analysis in affected and control Dachshunds. A missense mutation (c.977C>T, p.L326P) located in an evolutionary conserved domain was perfectly associated with the OI phenotype. We thus have identified a candidate causative mutation for OI in Dachshunds and identified a fifth OI gene.
Published in the journal:
. PLoS Genet 5(7): e32767. doi:10.1371/journal.pgen.1000579
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1000579
Summary
Osteogenesis imperfecta (OI) is a hereditary disease occurring in humans and dogs. It is characterized by extremely fragile bones and teeth. Most human and some canine OI cases are caused by mutations in the COL1A1 and COL1A2 genes encoding the subunits of collagen I. Recently, mutations in the CRTAP and LEPRE1 genes were found to cause some rare forms of human OI. Many OI cases exist where the causative mutation has not yet been found. We investigated Dachshunds with an autosomal recessive form of OI. Genotyping only five affected dogs on the 50 k canine SNP chip allowed us to localize the causative mutation to a 5.82 Mb interval on chromosome 21 by homozygosity mapping. Haplotype analysis of five additional carriers narrowed the interval further down to 4.74 Mb. The SERPINH1 gene is located within this interval and encodes an essential chaperone involved in the correct folding of the collagen triple helix. Therefore, we considered SERPINH1 a positional and functional candidate gene and performed mutation analysis in affected and control Dachshunds. A missense mutation (c.977C>T, p.L326P) located in an evolutionary conserved domain was perfectly associated with the OI phenotype. We thus have identified a candidate causative mutation for OI in Dachshunds and identified a fifth OI gene.
Introduction
Collagen I is the most abundant protein in the human body and its highly ordered fibril structure is responsible for its special mechanical properties. Together with inorganic hydroxylapatite it is the main component of bones and gives them elasticity while the hydroxylapatite alone would be very brittle. Defects in the structure of the highly ordered collagen I triple helix lead to osteogenesis imperfecta (OI), a disease characterized by extremely fragile bones and teeth. OI is sometimes also accompanied by blue sclera, hearing loss, dwarfism, dentinogenesis imperfecta, and other complications. Seven subtypes of human OI are distinguished based on the underlying genetic defects and phenotypic severity [1]. OI affects an estimated 6 to 7 per 100,000 people worldwide [http://ghr.nlm.nih.gov/condition=osteogenesisimperfecta/]. Approximately 85–90% of the human OI cases are caused by mutations in the COL1A1 or COL1A2 genes encoding the two different subunits of collagen I. More than 800 distinct mutations in these two genes have been described and most of them lead to autosomal dominant forms of OI [2]. The maturation and correct folding of collagens is a complicated process, which involves a large number of accessory proteins and chaperones. Recently mutations in two of these accessory proteins were found in patients with autosomal recessive forms of OI [3]–[7]. Both of these proteins are involved in the 3-hydroxylation of a specific proline residue in collagen I. One represents the enzymatically active prolyl-3-hydroxylase 1 itself and is encoded by the LEPRE1 gene [3]. The other is called cartilage-associated protein (CRTAP) and forms a complex with the prolyl-3-hydroxylase [4]. For some human OI cases the underlying mutation has not yet been found.
OI also occurs in dogs and the dog may represent a better model for human OI than genetically engineered mice because of its larger body size and the resulting similarity of mechanical forces that act on the skeleton. OI in dogs has been described in Golden Retrievers, Beagles, Collies, Poodles, Norwegian Elkhounds, and Bedlington Terriers [8]–[12]. In Golden Retrievers a COL1A1 mutation and in Beagles a COL1A2 mutation has been reported to cause OI [8],[9]. For other canine OI cases the underlying genetic defect has not been elucidated. We have observed a severe form of OI in rough-coated Dachshunds that is inherited as a monogenic autosomal recessive trait [13]. In our initial analysis of the OI Dachshunds we did not find any mutations in the COL1A1 or COL1A2 genes. Therefore, we hypothesized that a mutation in a novel OI gene may be responsible for the observed bone defects in Dachshunds. Consequently, we started a positional cloning approach to identify this mutation.
Results
Collection of informative families and exclusion of COL1A1 and COL1A2
We collected samples from six Dachshund families segregating for congenital OI (Figure 1; Video S1). The parents of all available cases were healthy (Figure S1). The pedigrees were consistent with a monogenic autosomal recessive inheritance although the ratio of affected Dachshunds from the presumed carrier x carrier matings was slightly higher than expected with 14 out of 36 total pups affected instead of the expected 9/36. The available pedigree records indicate that the affected dogs from the German Dachshund breeding population share common ancestors and most likely trace back to a single common founder. As the OI phenotype in Dachshunds shows striking clinical similarities to human OI forms, we initially hypothesized that mutations in COL1A1 or COL1A2 might cause the canine disease. In order to validate whether a mutation in one of these genes might be responsible for OI, we genotyped three gene associated microsatellite markers derived from the surrounding genome sequence of COL1A1 (located on chromosome 9 (CFA 9) at 29.5 Mb) and COL1A2 (located on CFA 14 at 22.8 Mb), respectively. Two-point linkage analysis in the six available families clearly excluded the COL1A2 gene but indicated a suggestive linkage of OI to the region of COL1A1 with a positive LOD score of 1.5 (Table S1). However, the re-sequencing of COL1A1 using DNA of four affected and four healthy Dachshunds did not reveal any disease associated sequence polymorphism within the 51 coding exons and flanking intron regions of the COL1A1 gene. Furthermore, haplotype analysis revealed five different CFA 9 microsatellite marker haplotypes in affected dogs, which was not compatible with our assumption of a single founder mutation in all OI affected dogs.

Mapping of the causative mutation
Based on the pedigrees of our samples we hypothesized that the affected Dachshunds most likely were inbred to one single founder animal. Under this scenario the affected Dachshunds were expected to be identical by descent (IBD) for the causative mutation and flanking chromosomal segments. Therefore, we decided to apply a homozygosity mapping approach to determine the position of the mutation in the canine genome. We genotyped approximately 50,000 evenly spaced SNPs from five affected dogs and five obligate carriers. We analyzed the cases for extended regions of homozygosity with simultaneous allele sharing. Only one genome region fulfilled our search criteria (Table S2). On CFA 21 all five affected genotyped dogs were homozygous and shared identical alleles over 102 SNP markers corresponding to a 5.82 Mb interval from 23.58–29.40 Mb (Figure 2). We then examined the five obligate carriers for the mutation and reconstructed one copy of the disease-associated haplotype in each dog. One of the carriers showed a recombination event, which allowed us to narrow down the critical interval harboring the causative mutation to 4.74 Mb from 24.66–29.40 Mb (Figure 2).

Identification of a functional candidate gene and mutation analysis
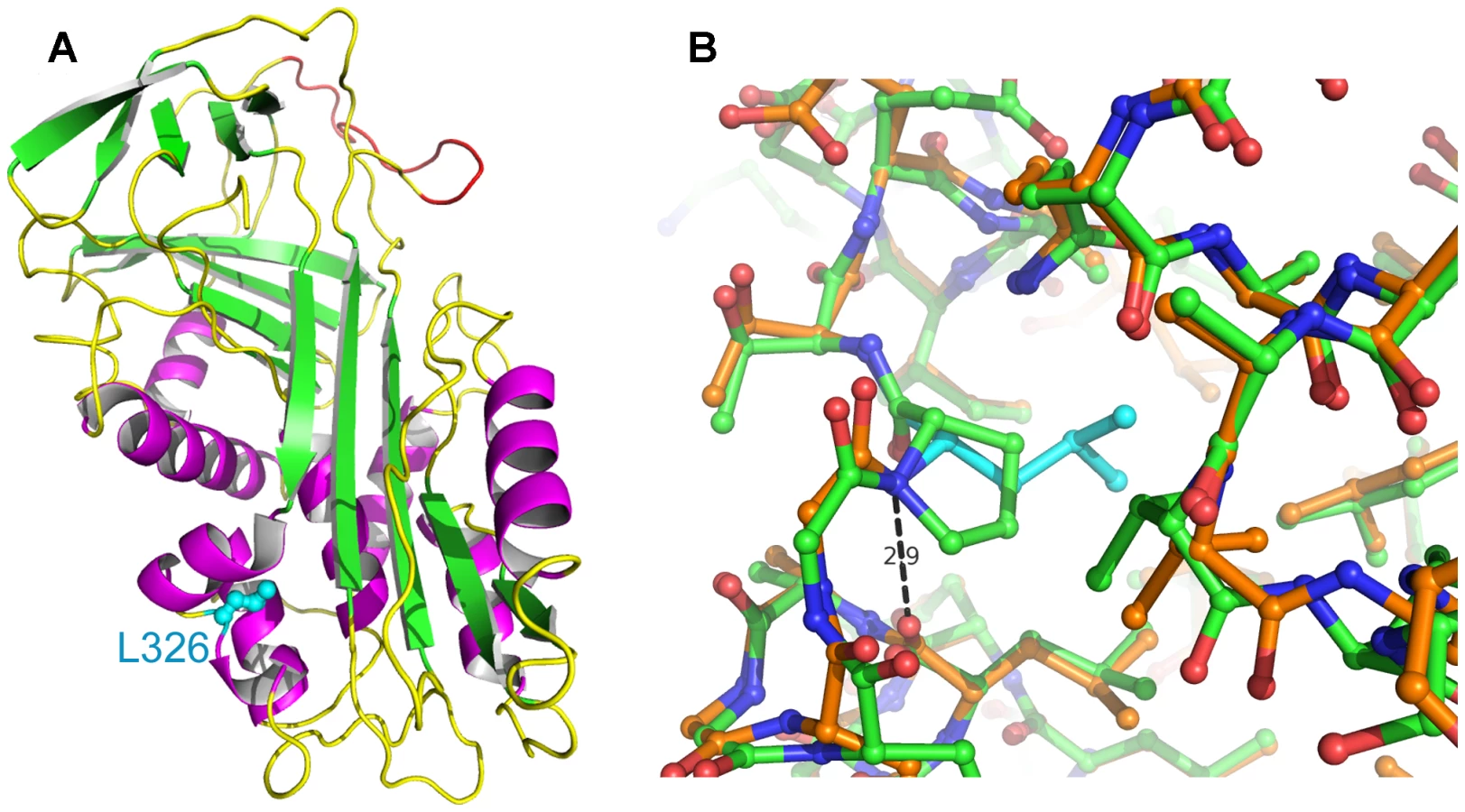
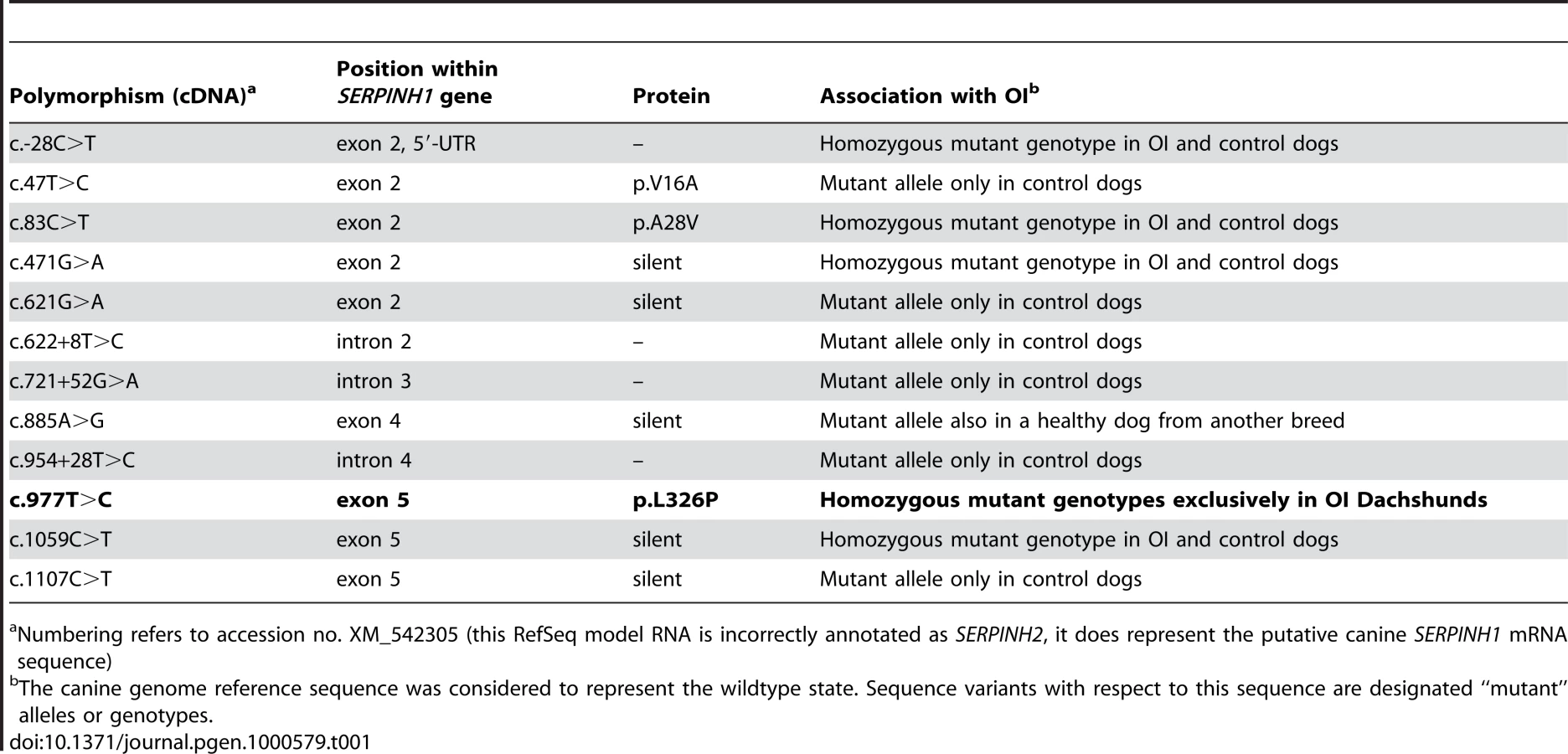
As the quality of dog genome annotation is still far from perfect, we inferred the gene annotation of the mapped interval from the corresponding human interval. The dog OI interval corresponds to two human segments from 3.59–3.84 Mb and from 71.30–76.48 Mb on HSA 11. The two human intervals contain 98 annotated genes including 8 annotated pseudogenes (NCBI MapViewer, build 36.3). A careful inspection of these genes and database searches of their presumed function revealed SERPINH1 as a functional candidate gene within the critical interval at 26.0 Mb on CFA 21. SERPINH1 encodes a serine protease inhibitor, also called heat shock protein 47 (HSP47) or collagen binding protein 1. Serpinh1 deficient mice die at around day 11 of development due to defective collagen synthesis [14] and it was shown that Serpinh1−/− fibroblasts produce abnormally thin and branched collagen type I fibres [15]. In order to further validate SERPINH1 as positional candidate gene for OI, we genotyped two gene associated microsatellite markers derived from the surrounding genome sequence of CFA 21 (Table S1). The obtained LOD score of 4.1 conclusively confirmed the linkage of OI to the candidate gene region in the Dachshund families. All OI affected dogs showed homozygosity at both tested microsatellites and all genotyped parents had one copy of the disease associated haplotype. We therefore investigated whether mutations in the canine SERPINH1 gene might be responsible for the OI phenotype. We designed PCR primers for the amplification of the four coding exons and determined the genomic sequence of two affected and two control dogs. This analysis revealed twelve polymorphisms including three non-synonymous substitutions (Table 1). Of these polymorphisms only a single SNP located in SERPINH1 exon 5 (c.977T>C; Figure 3) showed perfect association to the OI phenotype (Table 2, Figure S1). All 11 affected dogs were homozygous C/C and all 13 known carriers were heterozygous C/T. One grandmother and 16 out of 22 healthy full - and half-sibs of OI affected dogs were also heterozygous C/T. None of 66 unrelated healthy Dachshunds had the homozygous C/C genotype, but twelve of them were also presumed carriers with the C/T genotype. Thus the allele frequency of the deleterious C-allele within the unrelated Dachshunds was 18%. The mutation was encountered in wire-haired and short-haired Dachshunds. The mutant C-allele was absent from 79 control dogs from 75 diverse dog breeds (Table S2). RT-PCR on bone cDNA confirmed that the SERPINH1 RNA is normally spliced in an affected dog. We sequenced the cDNA from an affected dog and it did contain the mutant C-nucleotide at position +977 confirming that the mutant mRNA is expressed at normal levels. The c.977T>C substitution is predicted to result in an exchange of a highly conserved leucine to a proline in the SERPINH1 protein sequence (p.L326P, Figure 4). We modeled the wildtype and mutant SERPINH1 protein structures based on experimentally determined structures of serpins and found that the p.L326P mutation indeed affects the three-dimensional structure of SERPINH1 (Figure 5).



Discussion
We have applied an efficient SNP-based homozygosity mapping strategy to map the causative gene for OI in Dachshunds using only five affected dogs and five obligate carriers. The special population structure of purebred dog breeds with a limited amount of inbreeding on the one hand increases the occurrence of recessive phenotypes and on the other hand provides ideal prerequisites to map the underlying genes for these traits [16]. In this study we did not have enough high-quality DNA samples of unrelated Dachshunds for a genome-wide association study. However, the homozygosity mapping approach for this recessive trait basically required only samples from the five affected dogs to map the causative gene to one unique chromosome segment of 5.82 Mb. Adding the five obligate carriers further reduced this interval to 4.74 Mb. Thus the use of genome-wide canine SNP genotyping data enables very efficient positional cloning projects of Mendelian traits even if only very few samples are available.
The mapped OI interval contains a very good functional candidate gene, SERPINH1. We found a non-synonymous mutation in this gene, which is perfectly associated with the OI phenotype in Dachshunds, and confirmed the presence of this mutation on the genomic DNA and mRNA level. Although we cannot provide functional proof of the causality of the mutation at this time, the wealth of functional data, which are available for the SERPINH1 gene, strongly supports the hypothesis that p.L326P is indeed the causative mutation. SERPINH1 or HSP47 is a molecular chaperone of the serpin family. It promotes the correct folding of the collagen I triple helix [17]. This triple-helical structure would normally not be stable at temperatures above 35 °C. SERPINH1 is present in high concentration in the endoplasmic reticulum and specifically binds to and stabilizes the triple helices of nascent collagens [18]–[20]. Apparently the complete absence of SERPINH1 leads to embryonic lethality due to deficiencies in several types of collagen [14]. It is an evolutionary conserved protein with 97% identity between human and dog and 64% identity between human and zebrafish. The p.L326P mutation lies within the conserved serpin domain and the wildtype leucine is conserved across all SERPINH1 sequences while in other, more distantly related serpins like antitrypsin or ovalbumin, it is conservatively replaced by isoleucine, valine or methionine. It is located at the interface of helices hB, hC and hI (Figure 5A) [21]. Leu326 has backbone dihedral angles Φ/Ψ of about −90/+88 degrees. Proline has a Φ-angle restricted to about −60 degrees due to its five-membered ring and most frequently Ψ-angles of −45 or +135 degrees. Therefore, it is likely that the mutation L326P results in an increased strain. Furthermore, in the wildtype Leu326 donates a main-chain H-bond to Leu321 that is not possible with the imino acid proline (Figure 5B). Thus it is conceivable that this mutation affects the proper folding and stability of the native conformation, possibly reducing the protein level significantly. Additionally, this non-conservative amino acid substitution could affect the ability of SERPINH1 to bind and stabilize collagen triple helices. We speculate that the p.L326P mutation in OI affected dogs probably does not represent a complete null allele but has some residual activity, which results in live-born dogs with a severe form of OI instead of the embryonic lethality seen in Serpinh1 knock-out mice. The phenotype of OI affected dogs primarily indicates a deficiency in collagen I, the most abundant collagen, whereas basal membranes, which contain collagen IV, do not seem to be severely altered [13].
Our finding of a SERPINH1 p.L326P mutation in dogs with OI provides a valuable model for human medicine and identifies SERPINH1 as a fifth OI gene in addition to COL1A1, COL1A2, CRTAP, and LEPRE1. It has already been shown that a functional SNP in the promoter of the human SERPINH1 gene is associated in African American women with an increased risk for preterm premature rupture of membranes [22]. Our study indicates that coding mutations of the SERPINH1 gene might be responsible for recessive forms of human OI, where no mutation in the four known OI genes has been found. It has been proposed to develop SERPINH1 binding molecules as drugs against fibrosis [23]. The findings of our study emphasize that such a therapeutic strategy will have to be very carefully adjusted in order not to have adverse effects on the physiological production of collagen.
In conclusion, we have identified the p.L326P mutation in the canine SERPINH1 gene as the candidate causative mutation for OI in Dachshunds. This result allows genetic testing and eradication of a lethal disease from the Dachshund breeding population. Our study also provides a defined animal model and a novel genetic mechanism for a lethal or severely debilitating human hereditary disease.
Materials and Methods
Animals
We collected samples from OI affected rough-coated Dachshunds (n = 11), their healthy littermates (n = 22), sires (n = 4), dams (n = 7), and one grandmother. We performed parentage verification to confirm the pedigree documentation from the breeders (Figure S1). In addition, we collected two Dachshunds recorded as sires of OI affected puppies. Parents of affected offspring were classified as obligate carriers (n = 13). We also collected 66 unrelated healthy Dachshunds resulting in a total of 113 samples from the Dachshund breed. Furthermore, we sampled 79 control dogs from 75 different breeds for the re-sequencing of SERPINH1 exon 5 (Table S3).
DNA and RNA extraction
Genomic DNA was isolated from blood or tissue using the Nucleon Bacc2 kit (GE Healthcare). Total RNA was isolated from bone or skin using Trizol reagent according to the manufacturer's instructions (Invitrogen).
Linkage analysis in candidate genes
Microsatellite markers were amplified using the Multiplex PCR Kit (Qiagen) and fragment size analyses were determined on an ABI 3730 capillary sequencer (Applied Biosystems) and analyzed with the GeneMapper 4.0 software (Applied Biosystems). Twopoint parametric linkage analysis under the assumption of OI segregating as a biallelic autosomal recessive trait with complete penetrance was performed with Merlin software version 1.1.2 [24]. The frequency of the mutant allele in the considered population was unknown and there were no data available that would have made it possible to estimate the frequency in a reliable manner. For the calculations a frequency of 0.001 for the mutant allele was assumed. The LOD score test statistic was used to estimate the proportion of linked families and the corresponding maximum heterogeneity LOD score. Within the available families, a maximum LOD score of 5.573 would have been possible. To reconstruct the most likely haplotypes, we applied the ‘best’ option of the Merlin software.
Mapping of the OI mutation
Genomic DNA from five affected Dachshunds and five carriers was genotyped on the canine Affymetrix version 2 SNP genotyping microarray (49,663 SNPs). The results were analyzed with PLINK [http://pngu.mgh.harvard.edu/~purcell/plink/]. To identify extended homozygous regions with allele sharing across all five affected animals the options –homozyg-group and –homozyg-match were applied. All given positions correspond to the build 2.1 dog genome assembly [http://www.ncbi.nlm.nih.gov/projects/mapview/map_search.cgi?taxid=9615].
Mutation analysis
Primers for the amplification of each of the four SERPINH1 coding exons with flanking regions were designed with the software Primer3 [http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi] after masking repetitive sequences with RepeatMasker (Smit, A.F.A and Green, P. [http://repeatmasker.genome.washington.edu/]). The sequences of the primers are listed in Table S4. For the mutation analysis PCR products were amplified of two affected and two unrelated healthy dogs using TopTaq polymerase (Qiagen). The subsequent re-sequencing of the PCR products was performed after rAPid alkaline phosphatase (Roche) and exonuclease I (New England Biolabs) treatment using both PCR primers with the ABI BigDye Terminator Sequencing Kit 3.1 (Applied Biosystems) on an ABI 3730. Sequence data were analyzed with Sequencher 4.8 (GeneCodes).
RT–PCR
Aliquots of 1 µg total RNA were reverse transcribed into cDNA using 20 pmol (T)24V primer and Omniscript reverse transcriptase (Qiagen). Two microliters of the cDNA were used as a template in PCR. PCR reactions were performed as described above and primer sequences are given in Table S5. The canine SERPINH1 cDNA sequence was deposited under accession FN395288 in the EMBL nucleotide database.
Homology modeling
Models of wildtype and mutant SERPINH1were produced employing 3D-JIGSAW [25],[26] with template PDB entries1OO8 and 1OPH [27]. Figures were prepared using PyMOL [http://www.pymol.org].
Supporting Information
Zdroje
1. RauF
GlorieuxF
2004 Osteogenesis imperfecta. Lancet 363 1377 1385
2. MariniJC
ForlinoA
CabralWA
BarnesAM
San AntonioJD
2007 Consortium for osteogenesis imperfecta mutations in the helical domain of type I collagen: regions rich in lethal mutations align with collagen binding sites for integrins and proteoglycans. Hum Mutat 28 209 221
3. CabralWA
ChangW
BarnesAM
WeisM
ScottMA
2007 Prolyl 3-hydroxylase 1 deficiency causes a recessive metabolic bone disorder resembling lethal/severe osteogenesis imperfecta. Nat Genet 39 359 365
4. MorelloR
BertinTK
ChenY
HicksJ
TonachiniL
2006 CRTAP is required for prolyl 3 - hydroxylation and mutations cause recessive osteogenesis imperfecta. Cell 127 291 304
5. BarnesAM
ChangW
MorelloR
CabralWA
WeisM
2006 Deficiency of cartilage-associated protein in recessive lethal osteogenesis imperfecta. N Engl J Med 355 2757 2764
6. BaldridgeD
SchwarzeU
MorelloR
LenningtonJ
BertinTK
2008 CRTAP and LEPRE1 mutations in recessive osteogenesis imperfecta. Hum Mutat 29 1435 1442
7. BodianDL
ChanTF
PoonA
SchwarzeU
YangK
2009 Mutation and polymorphism spectrum in osteogenesis imperfecta type II: implications for genotype-phenotype relationships. Hum Mol Genet 18 463 471
8. CampbellBG
WoottonJA
MacLeodJN
MinorRR
2000 Sequence of normal canine COL1A1 cDNA and identification of a heterozygous alpha1(I) collagen Gly208Ala mutation in a severe case of canine osteogenesis imperfecta. Arch Biochem Biophys 384 37 46
9. CampbellBG
WoottonJA
MacleodJN
MinorRR
2001 Canine COL1A2 mutation resulting in C-terminal truncation of pro-alpha2(I) and severe osteogenesis imperfecta. J Bone Miner Res 16 1147 1153
10. HolmesJR
PriceCHG
1957 Multiple fractures in a collie: Osteogenesis imperfecta. Vet Rec 69 1047 1050
11. LettowE
DammrichK
1960 Clinic and pathology of osteogenesis imperfecta in young dogs. Zbl Vet Med 7 936 966
12. SchmidtV
1967 Osteogenesis imperfecta in 2 collie litter siblings. Wien Tierärztl Monatsschr 54 92 100
13. SeeligerF
LeebT
PetersM
BrugmannM
FehrM
2003 Osteogenesis imperfecta in two litters of dachshunds. Vet Pathol 40 530 539
14. NagaiN
HosokawaM
ItoharaS
AdachiE
MatsushitaT
2000 Embryonic lethality of molecular chaperone hsp47 knockout mice is associated with defects in collagen biosynthesis. J Cell Biol 150 1499 1506
15. IshidaY
KubotaH
YamamotoA
KitamuraA
BächingerHP
2006 Type I collagen in Hsp47-null cells is aggregated in endoplasmic reticulum and deficient in N-propeptide processing and fibrillogenesis. Mol Biol Cell 17 2346 2355
16. KarlssonEK
Lindblad-TohK
2008 Leader of the pack: gene mapping in dogs and other model organisms. Nat Rev Genet 9 713 725
17. MakareevaE
LeikinS
2007 Procollagen triple helix assembly: an unconventional chaperone-assisted folding paradigm. PLoS ONE 2 e1029 doi:10.1371/journal.pone.0001029
18. KoideT
AsadaS
TakaharaY
NishikawaY
NagataK
2006 Specific recognition of the collagen triple helix by chaperone HSP47: minimal structural requirement and spatial molecular orientation. J Biol Chem 281 3432 3438
19. KoideT
NishikawaY
AsadaS
YamazakiCM
TakaharaY
2006 Specific recognition of the collagen triple helix by chaperone HSP47. II. The HSP47-binding structural motif in collagens and related proteins. J Biol Chem 281 11177 11185
20. ThomsonCA
TenniR
AnanthanarayananVS
2003 Mapping Hsp47 binding site(s) using CNBr peptides derived from type I and type II collagen. Protein Sci 12 1792 1800
21. HuberR
CarrellRW
1989 Implications of the three-dimensional structure of alpha 1-antitrypsin for structure and function of serpins. Biochemistry 28 8951 8966
22. WangH
ParryS
MaconesG
SammelMD
KuivaniemiH
2006 A functional SNP in the promoter of the SERPINH1 gene increases risk of preterm premature rupture of membranes in African Americans. Proc Natl Acad Sci USA 103 13463 13467
23. ThomsonCA
AtkinsonHM
AnanthanarayananVS
2005 Identification of small molecule chemical inhibitors of the collagen-specific chaperone Hsp47. J Med Chem 48 1680 1684
24. AbecasisGR
ChernySS
CooksonWO
CardonLR
2002 Merlin - rapid analysis of dense genetic maps using sparse gene flow trees. Nature Genet 30 97 101
25. BatesPA
KelleyLA
MacCallumRM
SternbergMJ
2001 Enhancement of protein modeling by human intervention in applying the automatic programs 3D-JIGSAW and 3D-PSSM. Proteins, Suppl 5 39 46
26. BatesPA
2002 Domain Fishing: a first step in protein comparative modelling. Bioinformatics 18 1141 1142
27. DementievA
SimonovicM
VolzK
GettinsPG
2003 Canonical inhibitor-like interactions explain reactivity of alpha1-proteinase inhibitor Pittsburgh and antithrombin with proteinases. J Biol Chem 278 37881 37887
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2009 Číslo 7
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- A Missense Mutation in the Gene in Dachshunds with Osteogenesis Imperfecta
- Scientists←Editors←Scientists: The Past, Present, and Future of
- Prediction and Interaction in Complex Disease Genetics: Experience in Type 1 Diabetes
- PCH'ing Together an Understanding of Crossover Control
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy