
Etiologie a diagnostika poruchy růstu u dětí, které se narodily malé na svůj gestační věk (SGA) s přetrvávající malou výškou v dětství (SGA-SS)
Etiology and diagnostic approach of growth failure in children born small for gestational age (SGA) with persistent short stature in childhood (SGA-SS)
Approximately 3–5% of all neonates are born with a birth weight and/or birth length of below -2 SD of the gestational age-specific normative values. They are assigned as being small for gestational age (SGA). About 90% of them will experience catch-up growth within the first two years of life. The additional 10%, who fail to catch-up and remain short during childhood, are referred as “small for gestational age-short stature” (SGA-SS) and fulfil arbitrary criteria for growth hormone (GH) administration.
The etiology of SGA-SS is heterogeneous. Some children have specific facial and other phenotypic features that allow targeted genetic testing; in others, the genetic testing might be more challenging. Significant genetic causes of SGA-SS include abnormal methylation patterns leading to Silver-Russell syndrome, defects of the GH – IGF-1 – growth plate axis, defective paracrine regulation of the chondrocyte, and abnormal formation of growth plate extracellular matrix. Identification of the genetic defect allows understanding the underlying pathophysiological mechanism of short stature and its inheritance pattern, facilitates a targeted search for comorbidities, and partly predicts the effect of treatment with growth hormone.
Keywords:
Genetics – small for gestational age – SGA – SGA-SS – Etiology – diagnostic procedures
Autoři:
L. Toni; L. Plachý; S. A. Amaratunga; A. Kodýtková; Š. Průhová; J. Lebl
Působiště autorů:
Pediatrická klinika 2. lékařské fakulty UK a Fakultní nemocnice Motol, Praha
Vyšlo v časopise:
Čes-slov Pediat 2020; 75 (4): 240-248.
Kategorie:
Sympozium: poruchy růstu
Souhrn
Přibližně 3–5 % dětí se rodí s porodní hmotností a/nebo délkou nižší než -2 SD ve srovnání s normou pro daný gestační věk a pohlaví. Označují se jako malé na gestační věk (small for gestational age – SGA). U 90 % SGA dětí dochází v prvních dvou letech k růstovému výšvihu, u zbylých 10 % růstový výšvih nenastane. Tyto děti mají růstové selhání navazující na intrauterinní růstovou retardaci, anglicky se označují jako „small for gestational age-short stature“ (SGA-SS) a mohou být indikovány k léčbě růstovým hormonem (GH).
Skupina dětí SGA-SS je etiologicky heterogenní. Některé mají charakteristické faciální a další fenotypické rysy, které indikují cílené genetické vyšetření, u ostatních dětí je pátrání po etiologii složitější. Mezi významné genetické příčiny SGA patří metylační poruchy spojené se Silverovým-Russellovým syndromem, změny v genech vedoucích k poruše osy růstový hormon – IGF-1 – růstová ploténka, k ovlivnění parakrinní regulace chondrocytů, nebo k poruše tvorby extracelulární matrix růstové ploténky. Určení genetické etiologie přispívá k porozumění patofyziologie růstové poruchy a jejího dědičného přenosu, umožní cíleně hledat komorbidity a do jisté míry predikovat účinek léčby růstovým hormonem.
Klíčová slova:
malý na svůj gestační věk – SGA – SGA-SS – genetika – etiologie – diagnostika
ÚVOD
Přibližně 3–5 % dětí se rodí s porodní hmotností a/nebo délkou nižší než -2 SD ve srovnání s normou pro daný gestační věk. Tyto děti označujeme jako malé pro svůj gestační věk (small for gestational age – SGA) [1]. SGA je většinou spojen s intrauterinní růstovou restrikcí (IUGR). I když pojmy SGA a IUGR se někdy zaměňují či užívají jako synonyma, IUGR označuje v porodnictví fetální růstovou restrikci na základě ultrazvukového měření velikosti plodu a dynamiky jeho růstu, zatímco SGA je pojem primárně neonatologický, dokumentovaný přesným změřením porodní hmotnosti a délky a porovnáním s normou pro daný gestační věk a pohlaví.
Stav SGA může být způsoben řadou exogenních, maternálních, placentárních nebo endogenních (embryonálních a fetálních) faktorů [2]. U 90 % dětí SGA dochází během prvních dvou let života k růstovému výšvihu, u zbylých 10 % v této době růstový výšvih nenastává [3]. Tyto děti se označují jako „small for gestational age – short stature“ (SGA-SS) neboli růstové selhání navazující na intrauterinní růstovou restrikci. Je známo, že zůstanou malé po celé dětství a dosáhnou významně nižší dospělé tělesné výšky. Proto se na základě studií účinnosti a bezpečnosti stala skupina dětí narozených SGA s postnatálním růstovým selháním (SGA-SS) na základě rozhodnutí Evropské lékové agentury (EMA) od roku 2003 indikací pro léčbu růstovým hormonem (GH) [4].
Etiologie růstového selhání navazující na SGA není zcela objasněna a je zřejmě heterogenní. Díky novým možnostem genetické diagnostiky se v poslední době dostává do popředí výzkum genetické a epigenetické etiologie této poruchy. Vychází se přitom z premisy o genetické podmíněnosti lidského růstu s tím, že pokud porucha růstu začíná již in utero, může se jednat s vyšší pravděpodobností o významnou genetickou predispozici ve srovnání s postnatálním rozvojem růstové poruchy. Ke genetickému vyšetření se používá spektrum metod od stanovení karyotypu, přes komparativní genomovou hybridizaci (aCGH), metylační studie, až po metody sekvenování nové generace (NGS). Tato vyšetření dokážou objasnit příčinu malého vzrůstu navazující na SGA v mnoha případech. V tomto souborném sdělení přinášíme přehled významných geneticky podmíněných příčin SGA-SS – poruch imprintingu, metylačních poruch a přehled základních genových defektů spojených s regulací osy GH – IGF-1 – růstová ploténka.
PORUCHY IMPRINTINGU A METYLAČNÍ PORUCHY. SILVERŮV-RUSSELLŮV SYNDROM A PŘÍBUZNÉ SYNDROMY
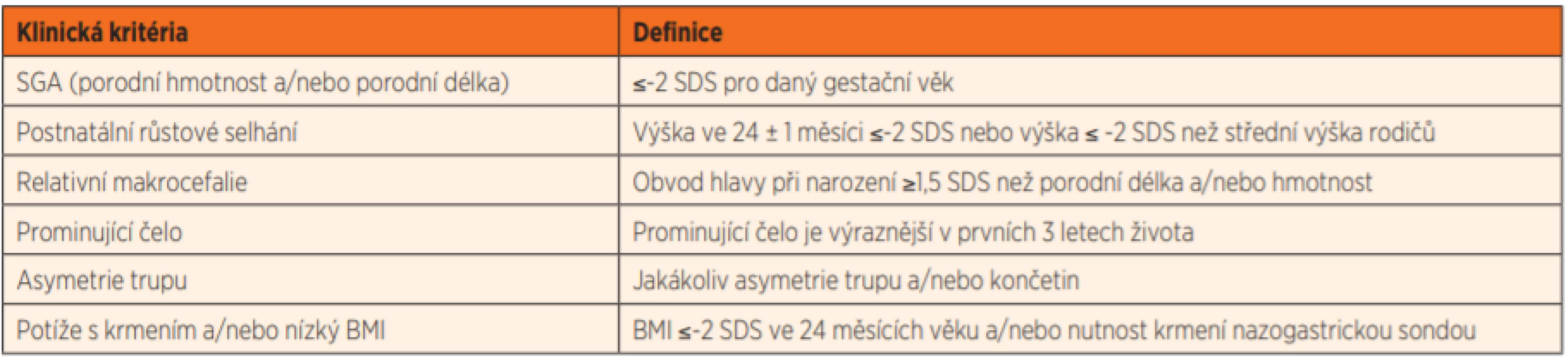
Pokud aktivita určitého genu závisí na maternálním či paternálním původu alely, hovoříme o genetickém imprintingu. Ve většině těchto případů se exprimuje (je aktivní) pouze maternální nebo paternální alela, zatímco druhá alela je umlčena. Jedna z častých imprintingových poruch spojená s fenotypem SGA-SS je Silverův-Russellův syndrom (SRS). Všechny děti se SRS se rodí malé na svůj gestační věk, zůstávají malé během dětství a mají typické dysmorfické rysy – relativní makrocefalii, trojúhelníkový obličej, prominující čelo, klinodaktylii malíčků a často také stranovou asymetrii včetně rozdílné délky končetin a plosek [5] (obr. 1a, b, c). Typické je obtížné krmení a neprospívání v kojeneckém věku [6]. SRS je primárně klinická diagnóza, k jejíž diagnostice používáme skórovací klinický systém dle Netchinové a Harbisona [7] (tab. 1).
Fig. 1a, b. A girl with Silver-Russell syndrome due to hypomethylation on chromosome 11 (11p15-SRS). She was born at 38th gestational week small-for-gestational-age – with birth weight 1700 g and birth length 42 cm. She failed to thrive and
to grow even postnatally (a – age 18 months, b – age 4 years). 1c. A boy with Silver-Russell syndrome due to maternal
uniparental disomy of short arm of 7th chromosome (UPD7). He was born at 36th gestational week with birth weight
1900 g and birth length 42 cm (SGA). Since early childhood, he failed to thrive and his weight was 4 kg when aged
1 year (fig.)

Genetickou etiologii lze při dnešní úrovni znalostí objasnit cca u 60 % dětí se SRS. Ve většině případů (téměř 50 % všech) je příčinou hypometylace v imprintingové kontrolní oblasti (imprinting control region – ICR) na paternální alele na chromosomu 11p15.5. V této oblasti jsou umístěny geny IGF2 a H19, které se podílejí na regulaci intrauterinního růstu [8]. Vedle toho byly popsány dvě vzácnější genetické odchylky ve stejné oblasti 11p15. Jedna z nich je spojena se zvýšenou expresí genu CDKN1C [9], což vede kromě intrauterinní růstové restrikce také k metafyzární dysplazii, kongenitální adrenální hypoplazii a malformaci genitálu u chlapců. Tento soubor příznaků se označuje jako IMAGe syndrom [10].
Příčinou 5–10 % případů SRS je uniparentální disomie (UPD) – přítomnost dvou homologních chromosomů nebo jejich částí, které pocházejí od jednoho rodiče. Nejčastěji se jedná o oblast s geny GRB10 a MEST na 7. chromosomu (UPD7) [11]. U dalších 40 % pacientů zůstává genetická etiologie SRS neobjasněna.
Dlouhodobé podávání růstového hormonu je u dětí SRS bezpečné a účinné, avšak během adrenarche a puberty může dojít k urychlení kostního zrání s vlivem na finální výšku [12].
Vedle UPD7 bylo popsáno několik dalších UPD syndromů spojených s malou výškou a typickým fenotypem (tab. 2). Jedním z nich je syndrom Templeové spojený s oblastí dlouhého raménka 14. chromosomu (14q). Může být vyvolán jak UPD 14. chromosomu, tak i paternální delecí, duplikací v oblasti 14q nebo hypometylací.
![Příklady imprintingových a metylačních poruch spojených s SGA. (Upraveno dle Finken MJJ, et al [2])](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image_pdf/600b230dfeeb2c41833059dfbe31f652.png)
Děti se syndromem Templeové se rodí SGA, jsou hypotonické, mají předčasnou pubertu a výrazně sníženou finální výšku [13]. Poruchy imprintingu nebo uniparentální disomie 15. chromosomu vedou ke vzniku syndromu Pradera-Williho, pro který je vedle dalších fenotypových projevů příznačná porodní hmotnost pod 10. percentilem a perzistující porucha růstu v dětství [14]. Také děti s uniparentální disomií 20. chromosomu patří mezi SGA-SS a mají potíže s krmením, na rozdíl od SRS dětí ale nemají typické dysmorfické rysy [15].
PORUCHY OSY GH – IGF-1 – RŮSTOVÁ PLOTÉNKA U DĚTÍ SGA-SS
Růstový hormon
I když růstový hormon hraje v prenatálním vývoji menší úlohu než IGF-1, je popsána řada stavů spojených s poruchou sekrece i působení (signalizace) růstového hormonu, které vedou k zpomalení intrauterinního růstu. U dětí s deficitem růstového hormonu je nižší průměrná porodní délka (-0,6 SD) i porodní hmotnost (-0,9 SD). Deficit růstového hormonu může být způsoben patogenními variantami řady různých genů, z nichž jsou nejčastější poruchy genů PROP1, GH1 a GHRH. Postnatální růstové selhání může být u dětí s deficitem růstového hormonu různě vyjádřené, efekt léčby růstovým hormonem bývá zpravidla příznivý [16].
Patogenní varianty genu kódující receptor pro růstový hormon (GHR) vedou ke vzniku necitlivosti na růstový hormon, tzv. Laronovu syndromu (obr. 2). Ten je charakterizován středně těžkou až těžkou poruchou růstu, poněkud disproporcionální postavou s kratšími končetinami a hypoplazií střední části obličeje, pro kterou je typický zejména vpáčený kořen nosu. Tyto děti mají koncentraci růstového hormonu v krvi normální nebo zvýšenou, zatímco koncentrace IGF-1 je velmi nízká. Léčebné podání růstového hormonu nemá u těchto jedinců žádný efekt, léčbou volby je podání rekombinantního lidského IGF-1 (rhIGF-1) [17].
Fig. 2. Phenotype of a boy with Laron syndrome due to a homozygous mutation of GHR gene encoding growth
hormone receptor. Besides of severe growth retardation, he has typical triangular face with prominent forehead and depressed nasal bridge (thanks to courtesy
of Prof. M. Savage).

Patogenní varianty v genech STAT5b, STAT3, IKBKB a IL2RG jsou též spojeny s necitlivostí na růstový hormon a současně s rozvojem závažného imunopatologického stavu [2]. Přehled základních genových defektů v ose GH – IGF-1 shrnuje tabulka 3.
![Genové defekty osy GH – IGF-1, spojené se SGA. (Upraveno dle Finken MJJ, et al. [2])](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image_pdf/ab9daddb9292245841e283591cd5af38.png)
IGF-1, IGF-2
Účinek růstového hormonu na periferní tkáně je zprostředkován především inzulinu podobnými růstovými faktory 1 a 2 (IGF-1, IGF-2). IGF-1 a IGF-2 jsou jednořetězcové polypeptidy, které vzájemně sdílejí přibližně 50 % své aminokyselinové sekvence [18]. IGF-1, ale zejména IGF-2, hrají významnou roli v časném fetálním vývoji prostřednictvím vazby na receptor IGF 1. typu (IGF1R), který je pro oba polypeptidy společný [19]. Po ukončení organogeneze se stává dominantním regulátorem růstu osa GH – IGF-1. Role IGF-2 v postnatálním vývoji není jasná. U myšího modelu vede inaktivace IGF-2 alely paternálního původu k těžké intrauterinní restrikci růstu, zatímco inaktivace alely maternálního původu nemá na růst žádný vliv [20]. Klíčová role IGF-1 v intrauterinním vývoji byla postupně dokumentována na příkladu čtyř jedinců z nepříbuzných rodin, kteří nesli patogenní varianty na obou alelách IGF1 genu. Všichni měli extrémně nízkou porodní hmotnost a délku i obvod hlavy a trpěli senzorineurální hluchotou (obr. 3) [21–23]. Dále byly popsány dvě rodiny s heterozygotními patogenními variantami IGF1, jejichž fenotyp byl mírnější [24, 25].
Fig. 3. Phenotype of the first diagnosed patient with biallelic IGF-I gene deletion. He suffered from severe
intrauterine growth restriction (birth weight -3.9 SD).
At age 15 years, he was microcephalic, intellectually
disabled, had hearing impairment and severely short
stature (body height -6.9 SD). IGF-I was undetectable,
IGFBP-3 5.8 mg/l, fasting insulin 27.3 mIU/l and stimulated growth hormone 61 μg/l (122 mIU/l) (photos and
clinical information overtaken from [21]).
![Fenotyp prvního diagnostikovaného pacienta s bialelickou delecí genu pro IGF-I. Chlapec prodělal těžkou
intrauterinní růstovou retardaci (porodní hmotnost
-3,9 SD). V 15 letech měl mikrocefalii, mentální retardaci, poruchu sluchu a závažnou růstovou poruchu (tělesná výška -6,9 SD). IGF-I bylo nedetekovatelné, IGF-BP3
5,8 mg/l, hladina inzulinu nalačno 27,3 mIU/l a hladina
růstového hormonu po stimulaci 61 μg/l (122 mIU/l)
(snímky a kazuistické informace převzaty z [21]).<br>
Fig. 3. Phenotype of the first diagnosed patient with biallelic IGF-I gene deletion. He suffered from severe
intrauterine growth restriction (birth weight -3.9 SD).
At age 15 years, he was microcephalic, intellectually
disabled, had hearing impairment and severely short
stature (body height -6.9 SD). IGF-I was undetectable,
IGFBP-3 5.8 mg/l, fasting insulin 27.3 mIU/l and stimulated growth hormone 61 μg/l (122 mIU/l) (photos and
clinical information overtaken from [21]).](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image_pdf/b78b81c0da1e47eb24769244fa7c46f6.png)
IGF1R
Gen IGF1R kóduje IGF receptor 1. typu (IGF1R), který má podobnou strukturu jako inzulinový receptor [26]. V literatuře najdeme řadu kazuistik popisujících různé varianty v IGF1R genu. Zajímavý je jejich značně variabilní fenotyp. Porodní hmotnost se pohybuje v rozmezí od -3,5 do -1,5 SD, porodní délka od -5,0 do +0,3 SD, výška při první návštěvě endokrinologa od -5 SD do -2,1 SD. Další fenotypické znaky zahrnují klinodaktylii, pectus excavatum, trojúhelníkový obličej, brachycefalii, ale také obtíže s krmením, opoždění psychomotorického vývoje a porušenou glukózovou toleranci. Odpověď na léčbu růstovým hormonem je u dětí s patogenními variantami IGF1R genu horší než u dětí SGA-SS bez genového defektu [2].
Vazebné proteiny pro IGF (IGF binding proteins; IGFBP)
Vazebné proteiny pro IGF (IGFBP-1 až IGFBP-6) prodlužují poločas IGF v cirkulující krvi a tím modulují jeho biologickou dostupnost a účinnost. Z šesti známých IGFBP je pro vazbu IGF-1 nejdůležitější IGFBP-3, v menší míře také IGFBP-5. Všechny IGFBP mají větší afinitu k IGF-1 než k receptoru IGF1R – vazba IGF-1 na IGFBP je tedy kompetitivní s vazbou IGF-1 na IGF1R. Zatím nebyly popsány žádné patogenní varianty v genech pro jednotlivé IGFBP [2].
Acidolabilní podjednotka (acid-labile subunit; ALS)
IGF-1 cirkuluje v krvi jako součást ternárního komplexu, který se skládá z IGFBP-3 a acidolabilní podjednotky (ALS). Zatímco poločas volně cirkulujícího IGF-1 je pouhých 10 minut, binární komplex IGF-1–IGFBP-3 přetrvává v krvi 30–90 minut a ternární komplex s ALS prodlužuje poločas na více než 12 hodin [27]. Studie u 312 dětí SGA-SS ukázala mírně nižší koncentraci ALS v krvi ve srovnání s běžnou dětskou populací (cca -0,5 SD), hladiny IGF-1 a IGFBP-3 byly ale ve srovnání se zdravou populací snížené podstatně významněji (<-1 SD) [28]. ALS je kódována genem IGFALS. Patogenní varianty genu IGFALS způsobují růstovou poruchu, která je mnohem závažnější v případě homozygotních mutací, delecí nebo duplikací než u heterozygotů nebo složených heterozygotů [29].
PAPP-A2 (pregnancy associated plasma protein A2)
PAPP-A2 je metaloproteináza, která štěpí vazbu mezi IGF-1 a IGFBP-3 (resp. IGFBP-5) a tím zvyšuje biologickou dostupnost IGF-1 v cílových tkáních. Doposud bylo popsáno 5 homozygotních mutací v genu PAPPA2, dvě z těchto pěti dětí byly SGA-SS. Všech pět dětí mělo zvýšené hladiny IGF-1, IGF-2, IGFBP-3, IGFBP-5 i ALS. Podle hypotézy autorů chybějící aktivita metaloproteinázy znemožňuje uvolnit IGF-1 a IGF-2 z ternárních komplexů. To sice zvyšuje jejich koncentraci v krvi prodloužením jejich poločasu, ale snižuje jejich biologickou dostupnost v cílových tkáních [30].
RŮSTOVÁ PLOTÉNKA A JEJÍ REGULACE
Poruchy parakrinní regulace
Genové defekty spojené s parakrinní signalizací v oblasti růstové ploténky vedou zpravidla k určité formě skeletální dysplazie. Většina těchto genů se exprimuje již in utero, proto bývá snížena porodní délka – směrodatná odchylka (SD) porodní délky vztažená ke gestačnímu věku je nižší než SD porodní hmotnosti. Těžší formy skeletálních dysplazií lze rozpoznat prenatálně či hned po narození; mírnější poruchy zůstávají nediagnostikovány a tyto děti se klasifikují jako SGA či v případě dominantní dědičnosti jako děti s familiárním malým vzrůstem (FSS). Při pátrání po možné skeleteální dysplazii u dětí s nižší porodní délkou je vhodné hodnotit opakovaně proporcionalitu: většina skeletálních dysplazií je postupně „asymetrická“ – končetiny jsou relativně zkrácené proti trupu, používá se označení „short-limbed dysplasias“ [31]. Asymetrie se ale zvýrazňuje s věkem a v prvních letech života nemusí být patrna.
Úplný výčet parakrinních faktorů v růstové ploténce přesahuje možnosti tohoto sdělení, proto zmíníme jen častější a/nebo klinicky významné poruchy. Fibroblastové růstové faktory (FGFs) hrají v regulaci růstové ploténky důležitou úlohu. Patogenní varianty v genu FGFR3, který kóduje třetí typ receptoru pro fibroblastový růstový faktor, ovlivňují růst kostí do délky a vedou k celé šíři fenotypů – od achondroplazie přes hypochondroplazii až k symetrické poruše růstu. I když u dětí s achondroplazií nejsou porodní hmotnost a délka výrazně sníženy (průměr -1 SD, resp. -0,7 SD), jejich nápadný fenotyp umožní okamžitou diagnózu (obr. 4). Děti s hypochondroplazií mají rhizomelické zkrácení končetin a relativní makrocefalii. Porodní délka a hmotnost může rovněž být snížena [2].
Fig. 4. Boy 5-year old with achondroplasia due to an FGFR3
gene mutation: disproportional stature, short limbs
(micromelia), brachycephalic skull, broadly prominent
forehead and mandible.

Dalším důležitým parakrinním regulátorem je natriuretický peptid typu C (CNP), který se váže na receptor natriuretického peptidu typu B (NPR-B) kódovaný genem NPR2. Homozygotní mutace genu NPR2 působí těžkou akromezomelickou dysplazii typu Maroteaux. Růstová porucha nositelů patogenní heterozygotní varianty v tomto genu je mírnější. Fenotyp bývá nenápadný (klinická diagnóza idiopatického/familiárního malého vzrůstu), v některých případech byl popsán obraz kostní dysplazie podobný Lériho-Weilovu syndromu (typický pro jedince s deficitem SHOX) se zkrácením předloktí a bérců (mezomelie). U NPR2 variant ale chybí charakteristická Madelungova deformita [32]. Heterozygotní varianty NPR2 zodpovídají zřejmě za 6 % případů dětí SGA-SS a familiárně malého vzrůstu [33].
IHH (Indian HedgeHog) hraje důležitou úlohu v embryonální morfogenezi, je exprimován v chondrocytech a reguluje jejich diferenciaci. Heterozygotní mutace genu IHH vedou k brachydaktylii typu A2, ale nedávno byly heterozygotní patogenní varianty nalezeny i u dětí s mírně disproporcionálním malým vzrůstem bez specifických skeletálních abnormit. Ve většině případů se tyto děti rodily malé na svůj gestační věk v parametru porodní délky a 50 % z nich mělo skeletální abnormitu na rentgenu ruky (například kratší střední článek III. a V. prstu s kónickým tvarem epifýzy) [34].
Poruchy tvorby extracelulární matrix
Jednou z příčin poruchy tvorby extracelulární matrix růstové ploténky jsou patogenní varianty genu ACAN, který kóduje bílkovinnou frakci proteoglykanu agrekan. Fenotypicky se může projevit mírná skeletální dysplazie, spondyloepifyzální dysplazie typu Kimberley nebo pouze malý vzrůst bez skeletálních abnormit, ale často s urychleným kostním věkem. 30–40 % dětí, které nesou patogenní variantu ACAN, se rodí SGA. Nositelé patogenních variant ACAN mohou v dospělosti trpět časně nastupující osteoartrózou a/nebo disekující osteochondritidou [35]. Mezi další příčiny stavu SGA-SS s narušenou strukturou extracelulární matrix patří patogenní varianty genů pro některé typy kolagenu, např. COL11A1, COL1A2, COL2A1 a jiné [36].
Poruchy intracelulární regulace
Relativně častou příčinou malého vzrůstu u dětí je aberace v genu SHOX, který kóduje SHOX protein (Short stature HOmeoboX-containing gene). Tento gen, nacházející se v pseudoautosomální oblasti obou pohlavních chromosomů X a Y, kóduje transkripční faktor s významnou úlohou v růstu dlouhých kostí. SHOX gen nepodléhá X-inaktivaci a za fyziologických okolností je přítomen ve dvou funkčních kopiích [37]. Bialelické inaktivační mutace způsobují závažnou Langerovou mezomelickou dysplazii. Heterozygotní mutace, delece či duplikace způsobují mírnější Lériho-Weilův syndrom (často s klasickou Madelungovou deformitou předloktí) anebo pouze SGA, resp. idiopatický či familiární malý vzrůst bez známek kostní dysplazie [38]. Chybění jedné kopie (haploinsuficience) SHOX je příčinou malého vzrůstu u dívek s Turnerovým syndromem.
Dalším významným intracelulárním regulátorem proliferace a diferenciace chondrocytů růstové ploténky je signalizační kaskáda Ras/MAPK. Úkolem Ras/MAPK signalizační kaskády je přenášet informaci mezi membránovým receptorem pro růstové faktory a buněčným jádrem. V buněčném jádře následně reguluje transkripci několika genů zodpovědných za buněčnou proliferaci, migraci, diferenciaci a apoptózu. Abnormální aktivace této kaskády vede k několika překrývajícím se syndromům se společným označením „RASopatie“, mezi které patří syndrom Noonanové, LEOPARD syndrom, Costellův syndrom, Legius syndrom a další. Hlavními příznaky RASopatií jsou malý vzrůst, typický faciální fenotyp, deformita hrudníku a kardiovaskulární abnormity [39].
Geny ovlivňující další buněčné procesy
Patogenní varianty genů, které kódují proteiny významné pro buněčné a mezibuněčné procesy v růstové ploténce a současně v dalších tkáních a orgánech, vedou ke skupině růstových poruch označovaných jako „primordiální trpaslictví (dwarfism)“. Tito jedinci mají zpravidla významně narušený jak intrauterinní, tak postnatální růst. Do této skupiny řadíme např. Floating Harbor syndrom, Meierův-Gorlinův syndrom, 3-M syndrom, syndrom Cornelia de Lange a další. Nositelé těchto syndromů mají příznačné fenotypické rysy, které mohou zkušeného klinického genetika nasměrovat k cílenému genetickému testování. U některých nemusí být fenotyp nápadný a jediná patrná patologie je SGA-SS [2].
CHROMOSOMÁLNÍ ABERACE A VARIABILITA POČTU KOPIÍ
Turnerův syndrom může být diagnostikován již prenatálně nebo hned po narození na základě typických dysmorfických známek – lymfedémů nártů nohou a hřbetů rukou, pterygia colli a dalších. V mnoha případech je diagnóza stanovena až později v dětství, zejména u dívek s mírnějším fenotypem při chromosomální mozaice. Téměř třetina dívek s Turnerovým syndromem se narodí SGA. Jejich prenatální i postnatální růstové selhání je způsobeno haploinsuficiencí SHOX [40].
Děti se všemi klasickými trisomickými chromosomálními aberacemi autosomů se rodí SGA. Vzhledem ke komplexní patologii těchto stavů zůstává otázka jejich tělesného růstu na okraji klinické významnosti.
Díky novým metodám genetické diagnostiky se dostala do popředí zájmu tzv. variabilita počtu kopií (CNV – copy number variation) lidského genomu. CNV je formou strukturální variability v podobě delecí a duplikací určitých úseků genomu, které jsou četné a mohou mít významný dopad na fenotyp. Z 671 pacientů vyšetřených pro malý vzrůst v rámci SGA-SS nejasné etiologie bylo pomocí aCGH objeveno několik patologických CNV u 87 pacientů (13 %) [41].
ALGORITMUS GENETICKÉHO VYŠETŘENÍ U DĚTÍ SGA-SS
Finken a spolupracovníci [2] navrhují zahájit vyšetření pečlivou anamnézou, fyzikálním vyšetřením, analýzou antropologických parametrů včetně růstových grafů, RTG levé ruky a předloktí a screeningovým laboratorním vyšetřením majícím za cíl vyloučit nehormonální příčiny poruchy růstu. Snímek ruky a předloktí doporučují provádět jak k určení kostního věku, tak k identifikaci skeletálních abnormit příznačných pro některé syndromy a genové defekty (SHOX, NPR2, ACAN, IHH a další).
U dívek s výškou nižší oproti cílové výšce o více než 1,6 SD doporučují autoři vyloučit Turnerův syndrom pomocí karyotypu a FISH. Po vyloučení Turnerova syndromu, patologického nálezu CNV a uniparentální disomie může být dalším krokem sekvenování nové generace (NGS) s analýzou panelu genů ovlivňujících růst. Podobný diagnostický postup volíme i u chlapců.
Pokud některá fenotypická známka vyvolává silné podezření na určitý syndrom (např. Madelungova deformita u defektů SHOX), volí se varianta přímého sekvenování kandidátního genu.
Ve specifických případech, kde máme podezření na přítomnost patogenní varianty v dosud neznámém genu, lze provést celoexomové sekvenování u tripletu (probanda a obou rodičů) [2]. Vzhledem k tomu, že genetická etiologie růstového selhání je u SGA dětí velmi různorodá, přináší pátrání ve větších kohortách dětí SGA-SS šanci na rozšíření biomedicínského poznání v této oblasti.
LÉČBA RŮSTOVÝM HORMONEM U DĚTÍ SGA-SS
Skupina dětí narozených SGA s postnatálním růstovým selháním (SGA-SS) se stala na základě rozhodnutí Evropské lékové agentury (EMA) od roku 2003 indikací pro léčbu růstovým hormonem. Nejnižší přípustný věk při zahájení terapie jsou v Evropě čtyři roky, v USA se terapie růstovým hormonem v této indikaci může zahájit již ve dvou letech věku. V případě těžké poruchy růstu již v kojeneckém věku a průkazu jasné genetické etiologie s nepříznivou prognózou dalšího růstu (například Silverův-Russellův syndrom) lze terapii GH zahájit i dříve. Ve většině evropských států je úvodní dávka GH 0,033 mg/kg/den.
V dosud zkoumaných kohortách dětí SGA-SS byla odezva na podávání GH poměrně různorodá. Tento jev zřejmě souvisí s heterogenní genetickou etiologií. Medián zlepšení finální výšky po léčbě oproti predikci činí 1,25 SD [42]. Puberta u dětí SGA-SS nastupuje v průměru ve stejném věku jako u ostatních vrstevníků [2]. Kromě pozitivního vlivu na růst má terapie růstovým hormonem poměrně dobře dokumentované lipolytické a anabolické působení, tedy celkově příznivý efekt na kardiovaskulární systém [43].
ZÁVĚR
Příčiny růstové poruchy navazující na SGA (SGA-SS) jsou různorodé, ale pro dítě je zapotřebí pokusit se je správně identifikovat. Po vyloučení sekundární poruchy růstu způsobené chronickým onemocněním (cystická fibróza, idiopatické střevní záněty, dlouhodobá kortikoterapie a další) přichází v úvahu primární porucha růstu, která může mít genetickou příčinu. Pokud má dítě SGA-SS určité fenotypické rysy, je namístě konzultace s klinickým genetikem a zahájení genetického vyšetření. Stanovení genetické diagnózy pomůže při cíleném hledání komorbidit, dovoluje určit diagnózu u případných dalších členů rodiny a upřesnit růstovou prognózu, a perspektivně také odhadnout šanci na úspěšnou léčbu růstovým hormonem. Postupně začínáme rozumět tomu, že u některých dětí může být tato léčba málo účinná, zatímco u jiných naopak výrazně zlepší růstovou rychlost a finální výšku.
Poznámka: Obrázky jsou publikovány se souhlasem zákonných zástupců dětí.
Problematiku genetické etiologie SGA-SS řeší autoři s podporou grantu AZV č. 18-07-00283 a GAUK č. 408120.
MUDr. Ledjona Toni
Pediatrická klinika 2. lékařské fakulty UK
a FN Motol
V Úvalu 84
150 06 Praha 5
e-mail: ledjonatoni@gmail.com
Zdroje
1. Lee PA, Chernausek SD, Hokken-Koelega AC, et al. International Small for Gestational Age Advisory Board consensus development conference statement: management of short children born small for gestational age, April 24–October 1, 2001. Pediatrics 2003;111 (6 Pt 1): 1253–1261.
2. Finken MJ, van der Steen M, Smeets CC, et al. Children born small for gestational age: Differential diagnosis, molecular genetic evaluation, and implications. Endocr Rev 2018; 39 (6): 851–894.
3. Hokken-Koelega AC, De Ridder MA, Lemmen RJ, et al. Children born small for gestational age: do they catch up? Pediatr Res 1995; 38 (2): 267–271.
4. Clayton P, Cianfarani S, Czernichow G, et al. Management of the child born small for gestational age through to adulthood: a consensus statement of the International Societies of Pediatric Endocrinology and the Growth Hormone Research Society. J Clin Endocrinol Metab 2007; 92 (3): 804–810.
5. Wakeling EL, Brioude F, Lokulo-Sodipe O, et al. Diagnosis and management of Silver–Russell syndrome: first international consensus statement. Nat Rev Endocrinol 2017; 13 (2): 105–124.
6. Marsaud C, Rossignol S, Tounian P, et al. Prevalence and management of gastrointestinal manifestations in Silver-Russell syndrome. Arch Dis Child 2015; 100 (4): 353–358.
7. Azzi S, Salem J, Thibaud N, et al. A prospective study validating a clinical scoring system and demonstrating phenotypical-genotypical correlations in Silver-Russell syndrome. J Med Genet 2015; 52 (7): 446–453.
8. Netchine I, Rossignol S, Dufourg MN, et al. 11p15 Imprinting center region 1 loss of methylation is a common and specific cause of typical Russell-Silver syndrome: clinical scoring system and epigenetic-phenotypic correlations. J Clin Endocrinol Metab 2007; 92 (8): 3148–3154.
9. Brioude F, Oliver-Petit I, Blaise A, et al. CDKN1C mutation affecting the PCNA-binding domain as a cause of familial Russell Silver syndrome. J Med Genet 2013; 50 (12): 823–830.
10. Arboleda VA, Lee H, Parnaik R, et al. Mutations in the PCNA-binding domain of CDKN1C cause IMAGe syndrome. Nat Genet 2012; 44 (7): 788–792.
11. Hitchins MP, Stanier P, Preece MA, et al. Silver-Russell syndrome: a dissection of the genetic aetiology and candidate chromosomal regions. J Med Genet 2001; 38 : 810–819.
12. Binder G, Liebl M, Woelfle J, et al. Adult height and epigenotype in children with Silver-Russell syndrome treated with GH. Horm Res Paediatr 2013; 80 (3): 193–200.
13. Ioannides Y, Lokulo-Sodipe K, Mackay DJ, et al. Temple syndrome: improving the recognition of an underdiagnosed chromosome 14 imprinting disorder: an analysis of 51 published cases. J Med Genet 2014; 51 (8): 495–501.
14. Lionti T, Reid SM, White SM, et al. A population-based profile of 160 Australians with Prader-Willi syndrome: trends in diagnosis, birth prevalence and birth characteristics. Am J Med Genet A 2015; 167 (2): 371–378.
15. Mulchandani S, Bhoj EJ, Luo M, et al. Maternal uniparental disomy of chromosome 20: a novel imprinting disorder of growth failure. Genet Med 2016; 18 (4): 309–315.
16. Mehta A, Hindmarsh PC, Stanhope RG, et al. The role of growth hormone in determining birth size and early postnatal growth, using congenital growth hormone deficiency (GHD) as a model. Clin Endocrinol (Oxf) 2005; 63 (2): 223–231.
17. Laron, Z. Laron syndrome (Primary growth hormone resistance or insensitivity): The personal experience 1958–2003. J Clin Endocrinol Metab 2004; 89 (3): 1031 – 1044.
18. Le Roith D, Bondy C, Yakar S, et al. The somatomedin hypothesis: 2001. Endocr Rev 2001; 22 (1): 53–74.
19. Constância M, Hemberger M, Hughes J, et al. Placental-specific IGF-II is a major modulator of placental and fetal growth. Nature 2002; 417 (6892): 945–948.
20. DeChiara TM, Efstratiadis A, Robertson EJ. A growth-deficiency phenotype in heterozygous mice carrying an insulin-like growth factor II gene disrupted by targeting. Nature 1990; 345 (6270): 78–80.
21. Woods KA, Camacho-Hübner C, Savage MO, et al. Intrauterine growth retardation and postnatal growth failure associated with deletion of the insulin-like growth factor I gene. N Engl J Med 1996; 335 (18): 1363–1367.
22. Walenkamp MJ, Karperien M, Pereira AM, et al. Homozygous and heterozygous ex - pression of a novel insulin-like growth factor-I mutation. J Clin Endocrinol Metab 2005; 90 (5): 2855–2864.
23. Netchine I, Azzi S, Houang M, et al. Partial primary deficiency of insulin-like growth factor (IGF)-I activity associated with IGF1 mutation demonstrates its critical role in growth and brain development. J Clin Endocrinol Metab 2009; 94 (10): 3913–3921.
24. van Duyvenvoorde HA, van Setten PA, Walenkamp MJ, et al. Short stature associated with a novel heterozygous mutation in the insulin-like growth factor 1 gene. J Clin Endocrinol Metab 2010; 95 (11): 363–367.
25. Fuqua JS, Derr M, Rosenfeld RG, et al. Identification of a novel heterozygous IGF1 splicing mutation in a large kindred with familial short stature. Horm Res Paediatr 2012; 78 (1): 59–66.
26. Francke U, Yang-Feng TL, Brissenden JE, et al. Chromosomal mapping of genes involved in growth control. Cold Spring Harb Symp Quant Biol 1986; 51 (2): 855–866.
27. Boisclair YR, Rhoads RP, Ueki I, et al. The acid-labile subunit (ALS) of the 150 kDa IGF - binding protein complex: an important but forgotten component of the circulating IGF system. J Endocrinol 2001; 170 (1): 63–70.
28. Renes JS, van Doorn J, Breukhoven PE, et al. Acid-labile subunit levels and the association with response to growth hormone treatment in short children born small for gestational age. Horm Res Paediatr 2014; 8 (2): 126–132.
29. Renes JS, van Doorn J, Hokken-Koelega A, et al. Current insights into the role of the growth hormone-insulin-like growth factor system in short children born small for gestational age. Horm Res Paediatr 2019; 92 : 15–27.
30. Dauber A, Muñoz-Calvo MT, Barrios V, et al. Mutations in pregnancy-associated plasma protein A2 cause short stature due to low IGF-I availability. EMBO Mol Med 2016; 8 (4): 363–374.
31. Bonafe L, Cormier-Daire V, Hall C, et al. Nosology and classification of genetic skeletal disorders: 2015 revision. Am J Med Genet A 2015; 167 (12): 2869–2892.
32. Bartels CF, Bükülmez H, Padayatti P, et al. Mutations in the transmembrane natriuretic peptide receptor NPR-B impair skeletal growth and cause acromesomelic dysplasia, type Maroteaux. Am J Hum Genet 2004; 75 (1): 27–34.
33. Olney RC, Bükülmez H, Bartels CF, et al. Heterozygous mutations in natriuretic peptide receptor-B (NPR2) are associated with short stature. J Clin Endocrinol Metab 2006; 91 (4): 1229–1232.
34. Vasques GA, Funari MFA, Ferreira FM, et al. IHH gene mutations causing short stature with nonspecific skeletal abnormalities and response to growth hormone therapy. J Clin Endocrinol Metab 2018; 103 (2): 604–614.
35. Gkourogianni A, Andrew M, Tyzinski L, et al. Clinical characterization of patients with autosomal dominant short stature due to aggrecan mutations. J Clin Endocrinol Metab 2017; 102 (2): 460–469.
36. Plachy L, Strakova V, Elblova L, et al. High prevalence of growth plate gene variants in children with familial short stature treated with growth hormone. J Clin Endocrinol Metab 2019; 104 (10): 4273–4281.
37. Rao E, Weiss B, Fukami M, et al. Pseudoautosomal deletions encopassing a novel homeobox gene cause growth failure in idiopathic short stature and Turner syndrome. Nat Genet 1997; 16 : 54–63.
38. Benito-Sanz S, Barroso E, Heine-Suñer D, et al. Clinical and molecular evaluation of SHOX/PAR1 duplications in Leri-Weill dyschondrosteosis (LWD) and idiopathic short stature (ISS). J Clin Endocrinol Metab 2011; 96 (2): 404–412.
39. Lebl J, Koloušková S, Toni L, et al. Syndrom Noonanové a další RASopatie: Etiologie, diagnostika a terapie. Čes-slov Pediat 2020; 75 (4): 219–226.
40. Ranke MB. Turner and Noonan syndromes: dinase – specific growth and growth-promoting therapies. In: Kelnar CJ, Savage MO, Saenger P, Cowell CT (Eds). Growth Disorders. 2nd ed. London, UK: Hodder Arnold; 2007 : 512–525.
41. Homma TK, Krepischi ACV, Furuya TK, et al. Recurrent copy number variants associated with syndromic short stature of unknown cause. Horm Res Paediatr 2018; 89 (1): 13–21.
42. Maiorana A, Cianfarani S. Impact of growth hormone therapy on adult height of children born small for gestational age. Pediatrics 2009; 124 (3): 519–531.
43. Lebl J, Kolouskova S, Steensberg A, et al. Metabolic impact of growth hormone treatment in short children born small for gestational age. Horm Res Paediatr 2011; 76 (4): 254–261.
Štítky
Neonatologie Pediatrie Praktické lékařství pro děti a dorostČlánek vyšel v časopise
Česko-slovenská pediatrie

2020 Číslo 4
- Efektivita kartáčku Sonicare For Kids u dětí předškolního věku
- Inosin pranobex v léčbě HPV infekcí děložního čípku
- Cytomegalovirové infekce u novorozenců a dětí
Nejčtenější v tomto čísle
- Syndrom Noonanové a další RASopatie: Etiologie, diagnostika a terapie
- Etiologie a diagnostika poruchy růstu u dětí, které se narodily malé na svůj gestační věk (SGA) s přetrvávající malou výškou v dětství (SGA-SS)
- Syndrom Noonanové z pohledu dětského kardiologa
- Prenatální fenotyp RASopatií
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy