
Moderní management cystické fibrózy a jeho vliv na zdravotní stav a přežívání českých nemocných
Modern Management of Cystic Fibrosis and Its Influence upon General Health and Survival of Czech Patients
Cystic fibrosis is a severe multisystemic autosomal recessively inherited disease. The disease becomes manifest with repeating respiratory tract infections, failure to thrive, high concentrations of salt in the sweat, and other complications. Up to 98% of adult men suffering from cystic fibrosis are infertile.
Management of cystic fibrosis must be complex, systematic, and should be managed by an experienced team of specialists. The expensive treatment the authors describe wasn’t fully accessible to patients until the year 1989. Comparing the status of patients on different therapeutic regimes allows assessing the importance of modern treatment and its influence upon nutritional status, lung function, and patient survival.
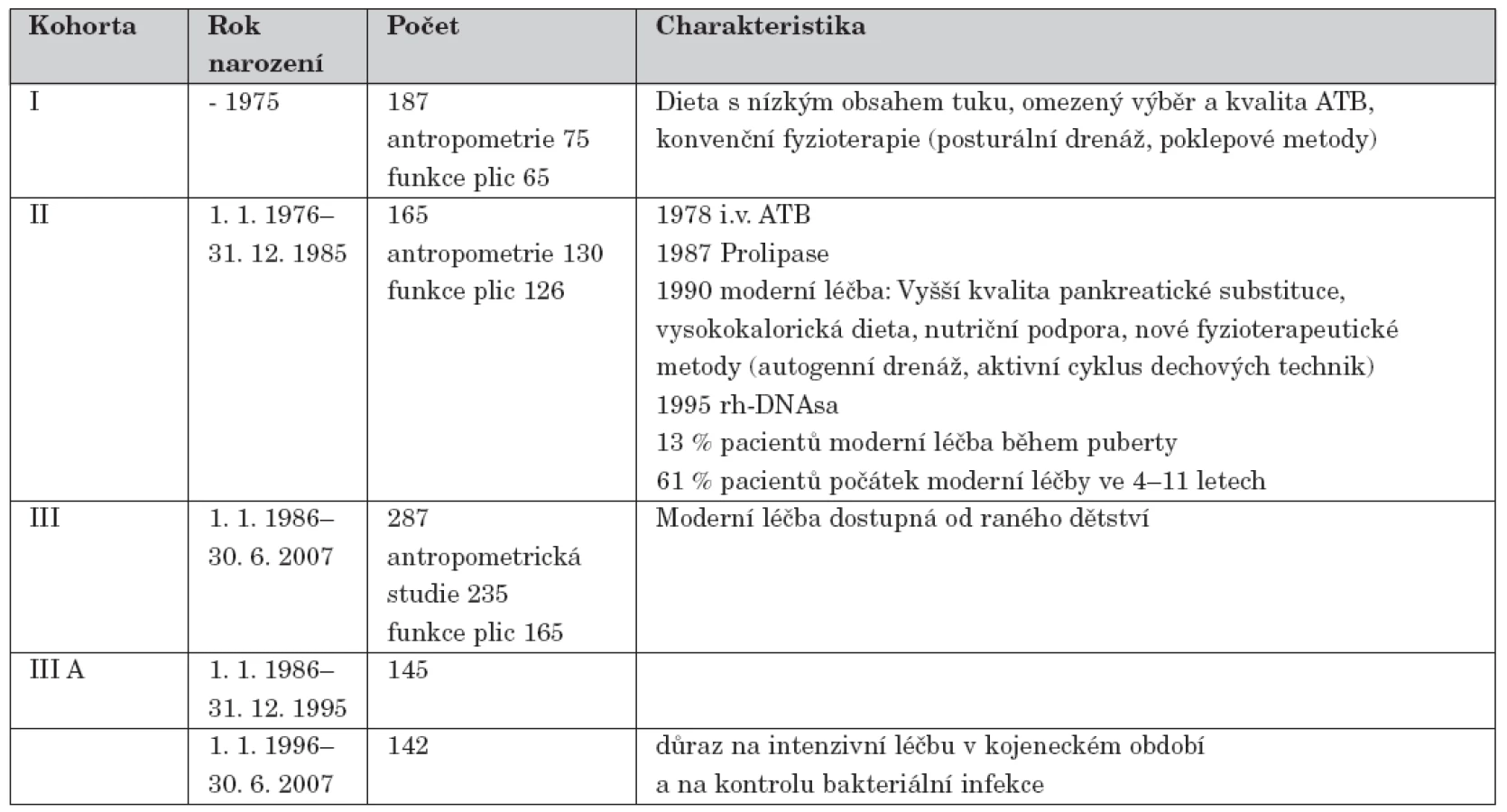
A group of 639 patients diagnosed between 1960 and June 6, 2007 at the CF Centre of Faculty Hospital Motol, Prague, was divided into three cohorts according to year of birth. Cohort I comprised 187 patients born before the year 1975. In cohort II are included 165 patients born between January 1, 1976 and December 31, 1985. Cohort III consists of 287 patients born after January 1, 1986. Cohort III is further divided into two subgroups.
Authors compared semi-longitudinal anthropometric and spirometric data in each category using the t-test. The length of survival in each group was analysed using Kaplan-Mayer’s function of survival and the cohorts were compared using the Long Rank test.
High mortality rate, poor nutritional status, which especially worsened during puberty, and growth retardation were characteristic for the oldest cohort. Modern treatment methods applied in cohort II led to improvement of nutritional status, central airway obstruction, and increased median of survival from 16.3 to 25.3 years. Modern treatment from early childhood in cohort III led to improvement of peripheral airway obstruction and a higher age of survival. Intensive treatment from early childhood lowers mortality during childhood to a minimum.
Authors have proven that modern management of CF improves nutritional status, lung function, and age of survival.
Key words:
cystic fibrosis, complex management, longitudinal follow-up, survival, lung function, nutritional status
Autoři:
D. Zemková 1; V. Skalická 1; J. Bartošová 1; L. Smolíková 2; J. Brázová 1; T. Fischerová 1; K. Böhmová 1; L. Pelikán 1; S. Koloušková 1; H. Chladová 1; P. Dřevínek 1; M. Macek ml. 3; P. Pohunek 1
Působiště autorů:
Pediatrická klinika UK 2. LF a FN Motol, Praha
přednosta prof. MUDr. J. Lebl, CSc.
1; Klinika rehabilitace UK 2. LF a FN Motol, Praha
přednosta doc. PaedDr. P. Kolář
2; Ústav biologie a lékařské genetiky UK 2. LF a FN Motol, Praha
přednosta prof. MUDr. M. Macek ml., DrSc.
3
Vyšlo v časopise:
Čes-slov Pediat 2008; 63 (2): 76-82.
Souhrn
Cystická fibróza je závažné multisystémové autozomálně recesivně přenášené dědičné onemocnění, projevující se opakovanými infekcemi dýchacích cest, neprospíváním, vysokým obsahem solí v potu a dalšími komplikacemi a u 98 % dospělých mužů i neplodností.
Léčení CF musí být komplexní, soustavné, vedené zkušeným týmem. Nákladná léčba, jejíž principy autoři popisují, je pacientům v plné míře dostupná až po roce 1989. Porovnání stavu pacientů při různých terapeutických režimech umožňuje zhodnotit význam moderní léčby, její vliv na stav výživy, funkci plic a přežívání.
Soubor 639 nemocných diagnostikovaných od roku 1960 do 30. 6. 2007 v CF centru ve Fakultní nemocnici v Motole byl rozdělen do 3 kohort podle věku narození. Kohortu I tvoří 187 pacientů narozených do roku 1975, kohortu II 165 nemocných narozených mezi 1. 1. 1976 a 31. 12. 1985 a III. kohortu 287 pacientů narozených od 1. 1. 1986, kteří se dále dělí na 2 podskupiny.
Autoři porovnávali semilongitudinální antropometrická a spirometrická data v jednotlivých věkových kategoriích pomocí t-testu. Přežívání v jednotlivých kohortách bylo analyzováno pomocí Kaplan-Mayerovy funkce přežití a kohorty porovnány pomocí log rank testu.
Pro nejstarší kohortu byla charakteristická vysoká úmrtnost, nepříznivý stav výživy zhoršující se především v pubertě a růstová retardace. Nasazení moderní léčby již v kohortě II vedlo ke zlepšení stavu výživy, obstrukce centrálních dýchacích cest a zvýšení mediánu přežití z 16,4 na 25,3 roků. Moderní léčba od raného dětství v kohortě III vedla ke zlepšení obstrukce periferních dýchacích cest a dalšího prodloužení věku dožití. Intenzivní léčba v raném dětství pak dovoluje snížit na minimum úmrtnost v dětském věku.
Autoři prokázali zlepšení stavu výživy, funkce plic a věku dožití při moderní komplexní léčbě CF. Skutečného úspěchu však lze dosáhnout pouze tehdy, když je intenzivní léčba uplatněna od raného dětství.
Klíčová slova:
cystická fibróza, komplexní léčba, longitudinální sledování, přežívání, funkce plic, stav výživy
Úvod
Cystická fibróza (CF) je závažné multisystémové autozomálně recesivně přenášené dědičné onemocnění. V některých zemích je CF nazývána též mukoviscidózou (mucus – hlen, viscidus – vazký). Projevuje se opakovanými infekcemi dýchacích cest, neprospíváním, vysokým obsahem solí v potu a u 98 % dospělých mužů i neplodností. Nemoc je provázena řadou komplikací, které mohou její obraz měnit a ovlivňovat její průběh. Někdy mohou být komplikace dokonce prvním projevem choroby [1].
Cystická fibróza je známa jako klinická jednotka 65 let. Toto onemocnění bylo dříve považováno za infaustní, smrtící v prvních letech života [2, 3]. S postupujícím výzkumem se prognóza nemoci měnila. U málokterého onemocnění došlo v posledních 20 letech k tak velkému rozvoji poznání a ke změnám v nazírání na podstatu jeho projevů jako právě u cystické fibrózy. Začínáme rozumět tomu, jak se základní defekt CFTR (Cystic Fibrosis Transmembrane Regulator) genu a porucha transportu elektrolytů buněčnými membránami promítá do klinických projevů [4, 5]. Přesto je léčba CF stále symptomatická. Je zaměřena především na redukci obstrukce dýchacích cest, kontrolu respirační infekce a zlepšení stavu výživy. Tyto tři základní kameny byly v posledních letech rozšířeny o protizánětlivou léčbu [6, 7, 8]. Důležitý je i včasný záchyt a intenzivní léčení všech komplikací. Léčení CF musí být komplexní, soustavné, vedené týmem, který dobře zná všechny patofyziologické pochody a poruchy a moderní přístupy, jak je řešit [9].
Po stanovení diagnózy na základě klinických příznaků, vyšetření koncentrace chloridů v potu a molekulárně genetického vyšetření následuje důkladná edukace rodičů o povaze onemocnění, možnostech léčby, s důrazem kladeným na techniky udržování dobré průchodnosti dýchacích cest, na výživu a na hygienická pravidla. Další péče zahrnuje pravidelná vyšetření v ambulanci, v případě zhoršení stavu pacienty hospitalizujeme. Základní schéma péče o pacienty je uvedeno v tabulce 1. Zvláštní důraz klademe na včasnou a agresivní léčbu infekce dýchacích cest závažnými patogeny, jakými jsou Staphylococcus aureus a Haemophilus influenzae a zvláště Pseudomonas aeruginosa a bakterie komplexu Burkholderia cepacia [10]. U pacientů s infekcí Pseudomonas aeruginosa se snažíme o eradikaci infekce pravidelnými i.v. antibiotickými kúrami (4x ročně na 14 dní), dvojkombinací antibiotik dávkovanými nad horní hranicí běžného rozmezí. Soustředíme se i na včasný záchyt komplikací, zvláště na diabetes mellitus vázaný na CF, postižení jater a osteoporózu [11, 12, 13].

Nezbytná je komplexní podpora rodiny celým CF týmem tvořeným lékaři, sestrami, fyzioterapeuty, antropologem, specializovaným klinickým psychologem, sociálním pracovníkem, poradcem pro výživu, koordinátorem CF klubu. Nedílnou součástí komplexní péče je i genetické poradenství.
Ve vyspělých zemích se tyto metody péče uváděly do praxe a rozvíjely postupně od 70. let. Zvláštní význam mělo především zdokonalování antibiotické léčby a přechod z diety s restrikcí tuků na vysokokalorickou stravu bohatou na tuky, což bylo možné díky vývoji účinnějších preparátů pro pankreatickou substituci.
U nás při prohlubující se izolaci a stagnující ekonomice na tuto nákladnou léčbu nebyly uvolněny prostředky. Možnosti moderní léčby se plně otevřely až po roce 1989, a to ve velmi krátkém období. Tento „nechtěný experiment“ dokládá význam a účinnost výše popsaných terapeutických schémat.
Pacienti a metodika
Ve Fakultní nemocnici v Praze Motole (později CF centrum Praha) bylo od roku 1960 do 30. 6. 2007 diagnostikováno a vyšetřeno 639 nemocných. Z nich 274 zemřelo, dlouhodobě se zde léčí 275 pacientů, z toho 182 v dětské části na Pediatrické klinice a 93 v centru pro dospělé na klinice TRN. Zbývajících 90 nemocných pacientů je vyšetřováno superkonsiliárně nebo byli po určení diagnózy předáni do jiných center.
Pacienti byli diagnostikováni na základě klinických příznaků a zvýšené koncentrace chloridů v potu, od 90. let je u každého pacienta provedeno molekulárně genetické vyšetření CFTR genu. Do souboru bylo zařazeno i 11 dětí diagnostikovaných v letech 2005–2006 novorozeneckým screeningem [14, 15, 16].
U 440 nemocných máme k dispozici antropometrické údaje pro semilongitudinální studii, u 356 nad 8 let i výsledky spirometrického vyšetření.
U pacientů jsme hodnotili genotyp a věk diagnózy, postižení zevně sekretorické funkce pankreatu a bakteriální infekci dýchacích cest. Dále jsme sledovali antropometrické parametry: tělesnou výšku, hmotnost, BMI, obvod hlavy a paže (vše v SD skóre) a tloušťku kožních řas nad tricepsem a scapulou (v mm) a torakální index (sagitální průměr hrudníku x 100/transverzální průměr) a vyhodnocovali je ve věku 1 a 3 let a dále v 8, 10, 12, 14, 16 a v 18 letech. Funkci plic jsme sledovali pomocí vitální kapacity plic (FVC), jednovteřinové kapacity (FEV1), vrcholového výdechového průtoku (PEF) a podle maximální výdechové rychlosti v poloze 25 %, 50 % a 75 % vitální kapacity (MEF25, MEF50, MEF75), a to od 8 let věku ve dvouletých intervalech. Výsledky spirometrického vyšetření jsou vyjádřeny v % náležitých hodnot podle Zapletala.
Podle věku narození byli pacienti rozděleni do 3 kohort, jak ukazuje tabulka 2.
Přežívání v jednotlivých kohortách bylo analyzováno pomocí Kaplan-Mayerovy funkce přežití a kohorty porovnány pomocí log rank testu.
Antropometrické a spirometrické parametry ve výše zmíněných věkových obdobích byly porovnány pomocí t-testu.
Výsledky
První kohorta je charakteristická vysokou úmrtností. Z dodnes přežívajících 25 pacientů je pouze 6 (24 %) homozygotních pro majoritní těžkou mutaci F508del, naproti tomu 12 (48 %) nemocných má alespoň jednu mutaci 4.–5. třídy (tzv. mírnou) nebo mutaci neznámou.
Výsledky antropometrického a spirometrického vyšetření shrnují tabulka 3 a grafy 1-4. Spolehlivou diagnostiku zajišťovalo již tehdy vyšetření chloridů v potu po stimulaci pilokarpinovou iontoforézou. V prvních letech ovšem byly zachyceny zpravidla spíše těžší formy nemoci. Do roku 1975 bylo diagnostikováno pouze 129 nemocných ve středním věku 0,5 roků, celkově medián věku diagnózy této kohorty však je 3 roky. Zbývajících 30 % pacientů z této kohorty bylo diagnostikováno v pozdějších věkových obdobích.





Po zavedení moderní léčby došlo ke zlepšení stavu výživy ve věkových kategoriích od 10 let. Patrné však bylo i zlepšení obvodu hlavy a tloušťky kožních řas v mladších věkových kategoriích od 3 let. Z hodnocení funkce plic vyplývá, že vitální kapacita (FVC) se prakticky neměnila. Ani FEV1 v 8 letech se nezvýšila, spíše zde byla patrná tendence k poklesuPEFa MEF (přežívalo více pacientů s těžkou obstrukcí). K významnému zlepšení (p <0,01) došlo u FEV1, a to od věku 10 let výše. V některých věkových kategoriích nad 10 let došlo i ke zvýšení PEF a MEF75. Ke zlepšení MEF50 došlo pouze u 16letých a 18letých a u MEF25 byly změny nevýznamné.
Jak ukazují Kaplan-Mayerovy funkce přežití (graf 5), věk pacientů se významně prodloužil (p = 1,57). Zlepšila se i diagnostika. Častěji byly zachyceny mírnější formy nemoci, celkově medián záchytu dané kohorty klesl na 0,7 roku.

Ve třetí kohortě opět došlo k významnému zlepšení přežívání.
U pacientů léčených od raného dětství moderními metodami jsme zaznamenali větší obvod hlavy od 1 roku a další zlepšení růstu a tělesného složení. Ve věkových kategoriích od 10 do 16 let se významně zlepšil tvar hrudníku. I po roce 1995 příznivý vývoj pokračuje a týká se především tělesného složení a obvodu hlavy.
Funkce plic v 8 letech se zlepšila ve všech parametrech oproti kohortě II. Hodnoty FEV1 vykazují zlepšení i v dalších věkových kategoriích, které však většinou nedosahuje hranice statistické významnosti. Můžeme nicméně říct že od 8 do 16 let je pokles FEV1 velmi pomalý, ke zhoršení dochází mezi 16. a 18. rokem. U pacientů III. kohorty, dokumentovaných v 16 i 18 letech věku, se FEV1 zhoršila o 11 % n. h. (rozmezí -33 až +15 %). Významným rozdílem mezi kohortou III léčenou od dětství a kohortou I s nedostatečnou léčbou je zlepšení MEF25 a MEF50 v kategoriích od 10 do 16 let. Pokračování příznivého trendu ukazuje i skupina dětí narozených od roku 1995. V 10 letech se PEF a MEF75 zlepšily natolik, že dosahují již normálních hodnot (97,8, resp. 99 % n. h.).
Ovšem všechny změny, které se po roce 1989 odehrály, nebyly jen pozitivní. Medián věku diagnózy celé kohorty III je 0,5 roku. Ve věku nad 10 let bylo diagnostikováno pouze 3,2 % pacientů, řada lehčích forem nepochybně zůstává nediagnostikována.
Zaměříme-li se podrobně na pacienty zachycené v jednotlivých letech, je patrné, že od roku 1999 věk při diagnóze stoupl; v letech 1999–2005 byl medián 1,5 roků. Nejednalo se pouze o nositele tzv. mírnějších mutací IV. a V. třídy, ale často o pozdní záchyt těžkých forem onemocnění.
Zavedení screeningu v rámci pilotního grantového projektu a s tím spojená informační kampaň vedla ke snížení mediánu věku diagnózy na 0,4 roku.
Diskuse
Léčba CF je komplexní, ekonomicky velmi nákladná. U nás je po roce 1989 dostupná pro všechny pacienty, hrazená ze zdravotního pojištění. Považujeme to za samozřejmé, ale ve všech postkomunistických zemích tomu tak není. Na tom má paní docentka Vávrová nedocenitelnou zásluhu, protože po celý svůj profesní život trvale sledovala nejnovější poznatky a intenzivně dbala na to, aby se pacientům i přes všechna úskalí dostávalo v rámci možností potřebného sledování a léčby.
Účinnost moderní komplexní léčby se prokázala postupně jak ve vyspělých zemích, tak u nás. Vysokokalorická strava a agresivní antibiotická léčba posunuly ve světě střední věk dožití nad 30 let [17], dnes ve vyspělých zemích dosahuje 37 let.
I náš „nechtěný experiment“ prokázal zlepšení stavu výživy i funkce plic. Zavedení moderní léčby během dětství nebo alespoň v pubertě může zlepšit stav výživy a zmírnit progresi obstrukce dýchacích cest. Tyto změny posunuly věk dožití nad 20 let. Skutečného úspěchu však lze dosáhnout pouze tehdy, když je intenzivní léčba uplatněna od raného dětství. To jasně dokazuje jak zlepšení obstrukce periferních dýchacích cest a tvaru hrudníku, ale i křivky přežívání. Do 16 let již nedochází k výraznému poklesu FEV1. Ovšem v našem souboru ve třetí kohortě je zatím zlepšení v 18 letech nedostatečné, mezi 16. a 18. rokem dochází k výraznému zhoršení. Jednou z příčin je epidemické rozšíření infekce bakteriemi komplexu Burkholderia cepacia v kohortách II a III. Navíc tito pacienti byli narozeni mezi lety 1986–1989, nejsou zde tedy ještě zahrnuti pacienti léčení od prvního roku života, kteří do tohoto věku ještě nedospěli a jejichž dosavadní vývoj probíhá příznivěji.
Tyto výsledky znovu upozorňují na význam včasné diagnózy. Proto se jevilo alarmující zvýšení věku diagnózy na konci 90. let a nárůst počtu pozdě diagnostikovaných pacientů (skrývajících se pod diagnózami jako asthma bronchiale a jiné alergické projevy, sinobronchiální syndrom, poruchy imunity, celiakie, nesnášenlivost kravského nebo dokonce mateřského mléka, nesnášenlivost jiných složek potravy, hepatopatie nejasné etiologie, suspektní dědičná metabolická porucha). Řešením tohoto problému je novorozenecký screening, který jsme vyzkoušeli v rámci pilotní grantové studie. Než však bude na celém území republiky rutinně prováděn, je nezbytné opakovaně na CF upozorňovat odbornou i laickou veřejnost a připomínat její příznaky [18, 19, 20]. (tab. 4)

Závěr
Kaplan-Mayerovy funkce přežití pacientů narozených po roce 1995 ukazují velmi příznivý trend a našim nemocným dávají naději na život srovnatelný s pacienty ve vyspělých zemích. Pokud jsou pacienti intenzivně léčeni od raného dětství, je úmrtí do 18 let věku výjimkou. Pro pacienty s CF jsou kritickým obdobím první tři roky života. Komplexní diagnostická a léčebná péče zahájená v okamžiku stanovení diagnózy má zásadní vliv na jejich další vývoj. Léčení musí být soustavné, od stanovení diagnózy, vedené zkušeným týmem.
Naděje na to, že v budoucnu bude možno léčit základní defekt, ovlivnit transport elektrolytů buněčnými membránami, aktivovat mutantní CFTR, ovlivnit jeho bílkovinný produkt či zasáhnout přímo genovou terapií jsou velké. Čím lépe budou pacienti kompenzováni v době zavedení nové léčebné možnosti, tím větší užitek z ní budou mít.
Práce vznikla s podporou VZ MZ ČR 64203-6405.
RNDr. Daniela Zemková
Pediatrická klinika UK 2. LF
FN Motol
V Úvalu 84
150 06 Praha 5
e-mail: dana.zemkova@lfmotol.cuni.cz
Zdroje
1. Vávrová V, a kol. Cystická fibróza. Praha: Grada, 2006. 520 s. ISBN 80-247-0531-1.
2. Houštěk J, Vávrová V. K výskytu cystické fibrózy pankreatu v ČSSR. Čs. Pediat. 1962;17 : 445–451.
3. Houštěk J, Vávrová V. Výskyt a klinické formy mukoviscidosy. Prakt. Lék. 1969;49 : 219–222.
4. Hodson ME, Geddes DM. Cystic Fibrosis. 2nd ed. London: Arnold 2000. ISBN 0-340-74208-9.
5. Bush A, et al. Cystic Fibrosis in the 21st Century. Basel, S. Karger AG, 2006. ISBN 3-8055-7960-8, ISSN 1422-2140.
6. Doering G, et al. Early intervention and prevention of lung disease in cystic fibrosis: a European consensus. Journal of Cystic Fibrosis 2004;3(2): 67–91.
7. Canton R, et al. Antimicrobial therapy for pulmonary pathogenic colonisation and infection by Pseudomonas aeruginosa in cystic fibrosis patients. Clinical Microbiology and Infection 2005;11(9): 690–703.
8. Sinaasappel M, et al. Nutrition in patients with cystic fibrosis: a European consensus. J. Cystic Fibrosis 2002;1(2): 51–75.
9. Kerem E, Conway S, Elborn S, et al. Standards of care for patients with cystic fibrosis: a European consensus. J. Cystic Fibrosis 2005;4(1): 7–26.
10. Döring G, et al. Antibiotic therapy against Pseudomonas aeruginosa in cystic fibrosis: a European consensus. European Respiratory Journal 2000;16(4): 749–767.
11. Moran A, et al. Diagnosis, screening and management of cystic fibrosis related diabetes mellitus: a consensus conference report. Diabetes Research and Clinical Practice 1999;45(1): 61–73.
12. Koloušková S, Zemková D, Bartošová J, et al. Vývoj porušené glukózové tolerance a diabetu u pacientů s cystickou fibrózou. Čes.-slov. Pediat. 2003;5 : 270–273.
13. Boyle MP. Update on maintaining bone health in cystic fibrosis. Current Opinion in Pulmonary Medicine 2006;12(6): 453–458.
14. Vávrová V. Diagnostika cystické fibrózy – potní test. Čes.-slov. Pediat. 2005;4 : 235–236.
15. Vávrová V, Macek M Jr. Současný stav diagnostiky cystické fibrózy. Čes.-slov. Pediat. 1999;5 : 220–226.
16. Rosenstein BJ, et al. The diagnosis of cystic fibrosis: a consensus statement. Cystic Fibrosis Foundation Consensus Panel. Journal of Pediatrics 1998;132(4): 589–595.
17. Corey M, McLaughlin FJ, Williams M, et al. A comparison of survival, growth and pulmonary function in patients with cystic fibrosis in Boston and Toronto. J. Clin. Epidemiol. 1988;41 : 583–591.
18. Vospělová J, Zapletalová J, Kolek A. Hypotonická dehydratace jako první příznak cystické fibrózy. Čes.-slov. Pediat. 2002;11 : 631–635.
19. Vávrová V, Zemková D, Skalická V, et al. Problémy v diagnostice cystické fibrózy – potřeba novorozeneckého screeningu. Čes.-slov. Pediat. 2006;12 : 703–709.
20. Holubová A, Balaščaková M, Skalická V, et al. Novorozenecký screening cystické fibrózy v České republice: závěry pilotní studie. Čes.-slov. Pediat. 2007;4 : 197–195.
Štítky
Neonatologie Pediatrie Praktické lékařství pro děti a dorostČlánek vyšel v časopise
Česko-slovenská pediatrie

2008 Číslo 2
- Efektivita kartáčku Sonicare For Kids u dětí předškolního věku
- Inosin pranobex v léčbě HPV infekcí děložního čípku
- Cytomegalovirové infekce u novorozenců a dětí
Nejčtenější v tomto čísle
- Novorozenecký screening v České republice a v Evropě
- Současné metodické postupy a přehled preimplantační, prenatální a postnatální DNA diagnostiky cystické fibrózy v České republice
- Doc. Věra Vávrová a historie cystické fibrózy v České republice
- Diabetes mellitus vázaný na cystickou fibrózu: diagnostika a terapie
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy