
Generalizované makulopapuly s bělavým středem
Clinicopathological Correlation Generalized Maculopapules with Whitish Center
The authors present a case of a 66-year-old patient with skin manifestations of disseminated maculopapules with a porcelain-white center and a rim of ectatic capillaries on the periphery. Six months after the onset of skin manifestation, neurological impairment resembling a stroke occurred, with diplopia and left-sided hemiparesis. Skin biopsy and brain magnetic resonance imaging performed 16 months after the onset of skin manifestation were consistent with the diagnosis of malignant atrophic papulosis. Progressive neurologic involvement led to the patient’s death 18 months after the onset of skin lesions. The authors present a current overview of knowledge about this disease.
Keywords:
histopathology – differential diagnosis – malignant atrophic papulosis – pathognomonic clinical signs – neurologic symptoms
Autoři:
M. Důra 1,2; L. Řandová 1; M. Petráčková 1; J. Štork 1
Působiště autorů:
Dermatovenerologická klinika 1. LF UK a VFN
1; Bioptická laboratoř s. r. o.
2
Vyšlo v časopise:
Čes-slov Derm, 100, 2025, No. 6, p. 231-234
Kategorie:
Klinicko-patologické korelace
Souhrn
Autoři prezentují případ 66letého pacienta s kožními projevy charakteru diseminovaných makulopapul s porcelánově bílým středem a s lemem ektatických kapilár na periferii. Šest měsíců po vzniku kožních projevů došlo k neurologickému postižení charakteru cévní mozkové příhody, s diplopií a levostrannou hemiparézou. Kožní biopsie a magnetická rezonance mozku provedené 16 měsíců od vzniku kožních projevů odpovídaly diagnóze maligní atrofické papulózy. Progredující postižení CNS vedlo k úmrtí nemocného 18 měsíců od vzniku kožních projevů. Autoři uvádí současný přehled poznatků o tomto onemocnění.
Klíčová slova:
histopatologie – diferenciální diagnóza – maligní atrofická papulóza – patognomické klinické projevy – neurologické postižení
KLINICKÝ PŘÍPAD
Pacientem byl 66letý muž. Matka pacienta zemřela na karcinom slinivky, otec trpěl blíže nespecifikovanými osteopenickými obtížemi. Další členové rodiny (dvě sestry, dcera a syn) byli bez chronických chorob. Pacient asi před 15 lety prodělal boreliózu, rok poté prodělal dvakrát erysipel. Před dávnou dobou utrpěl sériovou zlomeninu žeber a zlomeninu obratle. Alergie pacient neudával, trvalou medikaci neužíval. Pacient byl ve starobním důchodu, dříve pracoval jako truhlář. Byl celoživotní nekuřák.
Před 10 měsíci pacient prodělal cévní mozkovou příhodu, projevující se náhlou poruchou vizu a diplopií při pohledu doleva. V obdobné době zaznamenal brnění v oblasti levé dolní končetiny a příznaky chabé parézy.
Klinicky byla zvažována polyneuropatie. Pacient byl ve sledování neurologa.
Před 16 měsíci (tedy půl roku před cévní mozkovou příhodou) pacient pozoroval výsev postupně přibývajících, mírně svědivých projevů na trupu a končetinách, které údajně začínaly jako puchýřek. Lokální a protrahovaná celková antibiotická terapie byla bez efektu. Lokální kortikosteroidy vykázaly pouze mírný efekt.
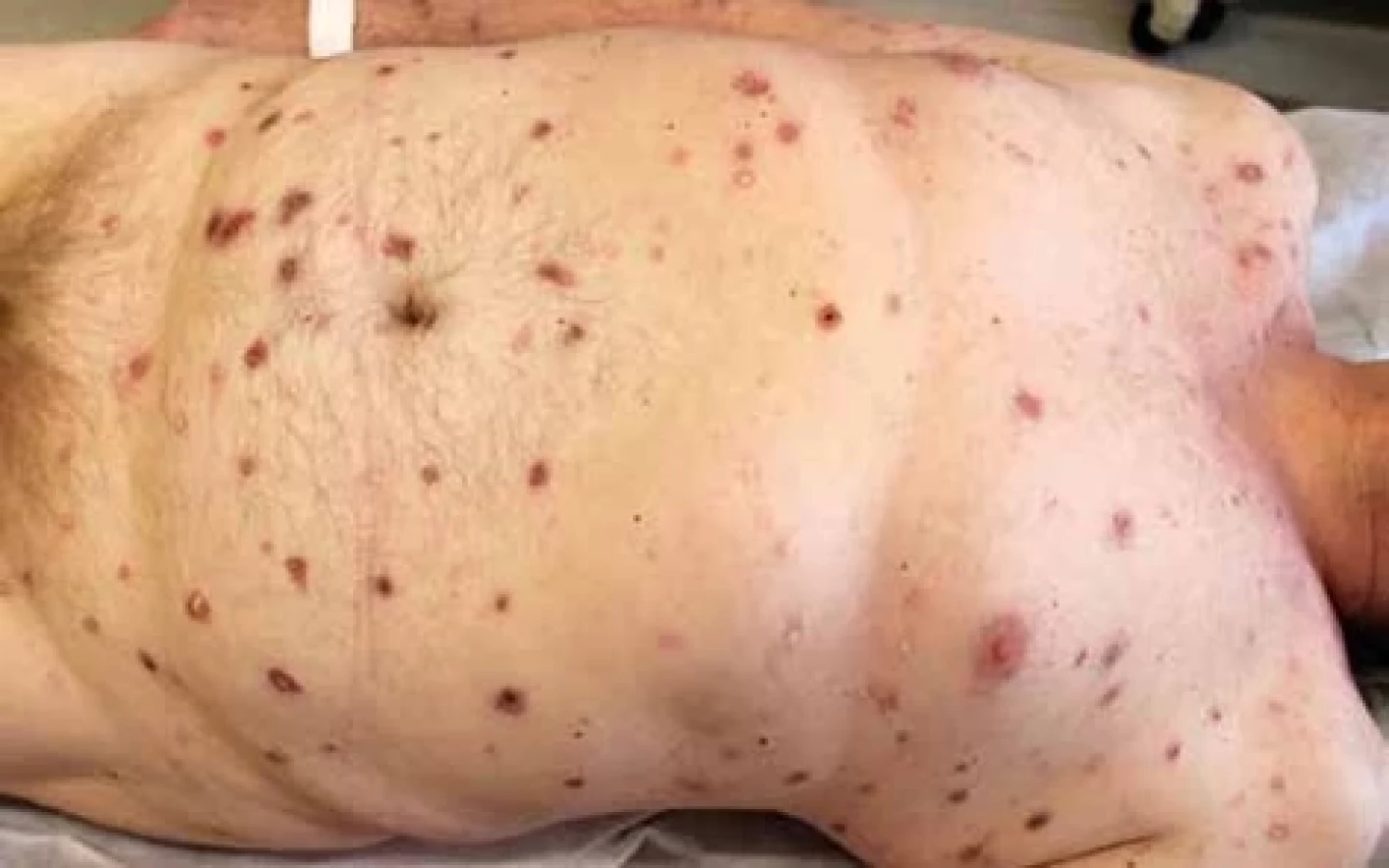
Při klinickém vyšetření byly patrné generalizované diseminované erytematózní makulopapuly, místy v centru s krustou či s tužším porcelánově bílým středem a s periferním lemem ektatických kapilár (obr. 1–3). Klinickému obrazu odpovídal i obraz dermatoskopický (obr. 3). Provedena byla excize jedné léze s bělavým centrem (obr. 4).



HISTOPATOLOGICKÝ NÁLEZ
Úsek mírně akantotické epidermis v centru excize chybí. Pod tímto úsekem je patrná klínovitá nekróza vaziva s intersticiálními depozity mucinu znázorňujícími se v alciánové modři s vrcholem dosahujícím středního koria, kde je přítomen perivaskulární lymfocytární infiltrát. Jednoznačné známky vaskulopatie či vaskulitidy nebyly ani v prokrojených řezech prokázány.
Závěr
Maligní atrofická papulóza (Köhlmeierova-Degosova choroba).
Průběh
U pacienta následně došlo k pocitům bušení srdce a zhoršenému dýchání. Zároveň pacient udával občasné průjmy. Další symptomy (bolesti, hubnutí, B příznaky) nebyly přítomny.
Kardiologické vyšetření prokázalo fibrilaci síní s rychlou odpovědí komor a drobnou poruchu intraventrikulárního vedení. Nastavena byla medikace apixaban 5 mg 2krát denně, metoprolol 50 mg 2krát denně, furosemid 40 mg denně a spironolakton 25 mg denně.
Neurologické vyšetření odhalilo frustní levostrannou hemiparézu, parézu n. abducens vlevo a reziduální polyneuropatický syndrom dolních končetin. Pravděpodobná byla kardioembolizační etiologie prodělané cévní mozkové příhody.
Magnetická rezonance mozku s angiografií vykázala postmalatická ložiska kortikosubkortikálně frontoparietálně vpravo, stenotické úseky a. cerebri media a a. cerebri anterior, zesílení leptomening a chronický subdurální hematom. Nález odpovídal diagnóze Degosovy choroby.
Přibližně za 2 měsíce byl pacient po pádu přijat na interní lůžko pro celkové zhoršení stavu, ztrátu soběstačnosti, imobilitu a dyspneu. Krátce nato pacient umírá.
DISKUSE A STRUČNÝ PŘEHLED
Generalizované makulopapuly s bělavým středem – maligní atrofická papulóza. Stručný přehled
Maligní atrofická papulóza (MAP), syn. Degosova či Köhlmeierova-Degosova choroba, je extrémně vzácné onemocnění postihující kůži i vnitřní orgány, řadící se mezi okluzivní vaskulopatie malých cév. Onemocnění bylo poprvé popsáno v roce 1941 Köhlmeierem a o rok později, v roce 1942, Degosem [3, 8]. O 6 let později, v roce 1948, Degos zavádí pro toto onemocnění pojem „papulose atrophiante maligne“ [9].
Onemocnění postihuje obě pohlaví, poněkud častěji ženy, poměr muži : ženy činil 1 : 1,6 [5]. Vyskytuje se především u mladých dospělých mezi druhou a čtvrtou dekádou života, zejména kavkazské rasy. Průměrný věk pacientů v době vzniku je 34 let [7]. Známy jsou případy familiárního výskytu, podle některých zdrojů byl familiární výskyt zaznamenán v 8,1 % analyzovaných případů [5]. Dosud bylo v literatuře popsáno řádově několik stovek případů.
Klinicky se MAP vyznačuje přítomností mnohočetných projevů, které mají v iniciální fázi charakter erytematózních makulopapul velikosti do 5 mm. V rozmezí 2–4 týdnů se v centru těchto projevů tvoří bělavá vtažená jizva, která nadále perzistuje jako porcelánově bílá tuhá makula s periferním lemem telangiektazií. Klinicky mohou léze připomínat projevy livedoidní vaskulopatie typu „atrophie blanche“. Projevy jsou lokalizovány na trupu a končetinách, obvykle s maximem v oblasti břicha a typicky vynechávají oblast dlaní a chodidel. Nejčastěji jsou asymptomatické [7]. Dermatoskopický obraz se odvíjí od stáří léze. V plně rozvinuté fázi má obraz „trnové koruny“ s bělavým jizevnatým centrem a periferním lemem lineárních či vlásenkovitých cév [1].
Kromě kůže se MAP manifestuje zejména postižením gastrointestinálního traktu a centrálního nervového systému. Postižení gastrointestinálního traktu je přítomno v 70 % případů [2]. Projevit se může náhlými bolestmi břicha, hematemézou či přítomností krve ve stolici. Postižení může vyústit až do perforace, nejčastěji tenkého střeva [10]. Právě perforace střeva s následným rozvojem peritonitidy je častou příčinou smrti. Postižení CNS je přítomno u 45 % pacientů, doprovázeno je bolestmi hlavy, mozkovou příhodou, epilepsií, diplopií, poruchami paměti či čití [2]. MAP může negativně ovlivnit i další orgány (oko, ledviny, srdce či játra).
Kožní postižení obvykle předchází vzniku systémového postižení. Robustní analýza 357 publikovaných případů prokázala průměrnou dobu od vzniku kožních lézí do rozvoje systémových příznaků 2 roky, pohybující se od 0 do 28 let. Mortalita činila 57 % [7].
Vedle klasické (maligní, systémové) formy byla prokázána i existence benigní (nesystémové) formy atrofické papulózy (tzv. benigní atrofická papulóza, BAP). Při ní onemocnění postihuje pouze kůži, bez postižení vnitřních orgánů [9]. Pravděpodobnost benigního průběhu onemocnění v době vzniku kožních lézí je dle některých prací udávána přibližně 70 %, po 7 letech bez systémového postižení tato pravděpodobnost dosahuje 97 % [11].
Etiologie MAP není dosud zcela ozřejměna. V současnosti je onemocnění považováno za formu okluzivní vaskulopatie. Zvažována je genetická příčina v genech pro složky komplementu či koagulační faktory. Některými autory je zvažována souvislost MAP s autoimunitními onemocněními charakteru lupus erythematosus či antifosfolipidového syndromu. Laboratorní markery, které by byly schopné upozornit na MAP či monitorovat její vývoj, dosud nebyly popsány. Provedena byla dosud pouze jedna studie hodnotící hladinu zánětlivých a trombo-okluzivních markerů u 6 případů benigní atrofické papulózy [12].
Histopatologický obraz kožních lézí se odvíjí od jejich stáří. V časné fázi je patrný obraz pouhé junkční (interface) dermatitidy a edém koria s přítomností mucinu. V plně rozvinuté fázi nález vykazuje přítomnost klínovité nekrózy se světle se barvícími snopci kolagenních vláken, doprovázenou řídkým perivaskulárním smíšeným zánětlivým infiltrátem na periferii nekrotického úseku. Při spodině nekrotického okrsku může být zastižen obraz trombotické vaskulopatie či lymfocytární vaskulitidy [6]. Povrchová epidermis je atrofická, kryta hyperkeratózou, případně též nekrotická. V pozdní, finální fázi jsou přítomny klínovité jizevnaté změny se sklerotizací postiženého koria a inkontinence melaninu [2]. Histopatologický obraz pozdní fáze může připomínat obraz lichen sclerosus [6].
V klinické diferenciální diagnóze zvažujeme ostatní onemocnění provázená okluzivní vaskulopatií, zejména antifosfolipidový syndrom, lupus erythematosus či dermatomyozitidu. Degos-like léze byly popsány u pacientů se SLE a některými autory je z tohoto důvodu zvažována souvislost či možný překryv MAP se systémovými onemocněními pojiva [4].
Terapeuticky nejlepšího efektu dosahují antiagregancia (kyselina acetylsalicylová) [7]. Experimentálně byl zaznamenán efekt treprostinilu (analogu prostacyklinu) či v ČR neregistrovaného dipyridamolu. Terapeutický efekt vykázala monoklonální protilátka eculizumab. Eculizumab je namířen proti C5 složce komplementu, čímž blokuje její rozštěpení C5 konvertázou na C5a a C5b. C5a je silný anafylatoxin s protrombotickými a proinflamatorními vlastnostmi. C5b je součástí terminálního komplexu komplementu C5b-9, jenž má obdobné biologické vlastnosti. Zaznamenány byly případy efektivní léčby kortikosteroidy, dalšími imunosupresivy, antimalariky či intravenózními imunoglobuliny [2]. V případech postižení gastrointestinálního traktu se uplatňuje chirurgická léčba.
Zdroje
- ANKER, J. P., KAMINSKA-WINCIOREK, G., LALLAS, A. et al. The dermoscopic variability of Degos disease at different stages of progression. Dermatol Pract Concept, 2014, 4(3), p. 59–61.
- BOLOGNIA, J. L., SCHAFFER, J. V., CERRONI, L. Dermatology. 5th Edition. Philadelphia: Elsevier/Saunders, 2024, 2 vol., p. 412–413.
- DEGOS, R., DELORT, J., TRICOT, R. Dermatite papulo-squameuse atrophiante. Bull Soc Fr Dermatol Syphiligr, 1942, 49, p. 148–150.
- JANG, M. S., PARK, J. B., YANG, M. H. et al. Degos-li-ke lesions associated with systemic lupus erythematosus. Ann Dermatol, 2017, 29(2), p. 215–218.
- KALETA, K. P., JARIENĖ, V., THEODORIDIS, A., NIKOLAKIS, G., ZOUBOULIS, C. C. Atrophic papulosis (Köhlmeier-Degos disease) revisited: a cross-sectional study on 105 patients. J Eur Acad Dermatol Venereol, 2022, 36(11), p. 2190–2194.
- KIM, E., MOTAPARTHI, K. Benign atrophic papulosis (Degos disease) with lymphocytic vasculitis and lichen sclerosus-like features. Am J Dermatopathol, 2018, 40(4), p. 272–274.
- KIM, P. J., LYTVYN, Y., KASHETSKY, N., BAGIT, A., MUFTI, A., YEUNG, J. Clinical manifestations and treatment outcomes in degos disease: a systematic review. J Eur Acad Dermatol Venereol, 2021, 35(8), p. 1655–1669.
- KÖHLMEIER, W. Multiple Hautnekrosen bei Thromboangiitis obliterans. Arch Dermatol Syphilol, 1941, 181, p. 792–793.
- POCK, L., ŠIMLOVÁ, J., MACHÁČKOVÁ, R., KOSKUBA, J., HERCOGOVÁ, J. Papulosis maligna atrophicans (morbus Degos). Čes-slov Derm, 2007, 82(4), p. 190–193.
- SATTLER, S. S., MAGRO, C. M., SHAPIRO, L. et al. Gastrointestinal Kohlmeier-Degos disease: a narrative review. Orphanet J Rare Dis, 2022, 17(1), p. 172.
- THEODORIDIS, A., KONSTANTINIDOU, A., MAKRANTONAKI, E., ZOUBOULIS, C. C. Malignant and benign forms of atrophic papulosis (Kohlmeier-Degos disease): systemic involvement determines the prognosis. Br J Dermatol, 2014, 170, p. 110–115.
- ZOUBOULIS, C. C., THEODORIDIS, A., MAKRANTONAKI, E. Inflammation and thrombo-occlusive vessel signalling in benign atrophic papulosis (Köhlmeier-Degos disease). J Eur Acad Dermatol Venereol, 2022, 36(11), p. 2195–2198.
Do redakce došlo dne 9. 12. 2025.
Adresa pro korespondenci:
MUDr. Miroslav Důra, Ph.D.
Dermatovenerologická klinika 1. LF UK a VFN
U Nemocnice 499/2 128 00 Praha 2
e-mail: miroslav.dura@vfn.cz
Štítky
Dermatologie Dětská dermatologieČlánek vyšel v časopise
Česko-slovenská dermatologie

2025 Číslo 6
- Nástroje k hodnocení závažnosti psoriázy v klinických studiích
- Systémová léčba atopické dermatitidy konečně i u dětí
- Inovace v hojení ran: krytí Zetuvit Plus Silicone Border pro optimální management exsudátu z ran
Nejčtenější v tomto čísle
- Kaposiho sarkom
- Využití mobilních aplikací v diagnostice kožních nádorů
- Generalizované makulopapuly s bělavým středem
- Spomienka na doc. MUDr. Klaudiu Kolibášovú, PhD., mim. prof.
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy