
Posledních 25 let v diagnostice a léčbě myelodysplastického syndromu
Authors:
J. Čermák
Authors‘ workplace:
Ústav hematologie a krevní transfuze, Praha
Published in:
Transfuze Hematol. dnes,25, 2019, No. 1, p. 108-117.
Category:
Review/Educational Papers
Overview
Přehledné sdělení o vývoji diagnostiky a léčby myelodysplastického syndromu během posledních 25 let se shrnutím nových prognostických faktorů a terapeutických možností.
Klíčová slova:
myelodysplastický syndrom – diagnostika
ÚVOD
Již v padesátých letech minulého století byly publikovány články ukazující, že část akutních leukemií vzniká až po různě dlouhém předchorobí, jež je charakterizováno cytopenií, která je do různé míry vyjádřena v jednotlivých krevních řadách, a dále postupným nárůstem nezralých myeloidních buněk v kostní dřeni a v periferni krvi [1]. Klinické příznaky těchto stavů jsou odvislé od stupně postižení jednotlivých řad a zahrnují únavu, dušnost, opakované infekce a krvácení. Tyto stavy byly v minulosti nazývány preleukemií či doutnající leukemií [2]. V roce 1982 dostaly tyto choroby společný název myelodysplastické syndromy (MDS) a byla vytvořena jejich první mezinárodní klasifikace (tzv. FAB klasifikace) [3]. Základní podskupiny vytvořené FAB klasifikací jsou uvedeny v tabulce 1. FAB klasifikace jako první oddělila formy bez nadbytku blastů v kostní dřeni (RA, RARS), jež jsou spojeny s příznivější prognózou od prognosticky nepříznivých forem s nadbytkem blastů (RAEB, RAEB-T) a vytvořila základní prognostické parametry a podklady pro léčbu nemocných. V tabulce 2 a na obrázku 1 je uvedena délka přežití neléčených nemocných z registru ÚHKT rozdělených podle FAB klasifikace.
![FAB klasifikace myelodysplastického syndromu [3]](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image_pdf/ea9906751c468584c7cb33ffd832ea5b.png)

[data ÚHKT]
![Délka přežití neléčených nemocných s různými podtypy MDS podle FAB klasifikace
[data ÚHKT]](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image_pdf/5913d902b236e1161bdb52a0f5b7c8ef.png)
Počátkem osmdesátých let minulého století vznikla pod vedením doc. Neuwirtové skupina zabývající se MDS v Československu a začal vznikat registr nemocných s tímto onemocněním, který spolupracoval i se slovenskou MDS školou v rámci Československa (prof. Dieška, prof. Hrubiško, dr. Šteruská, dr. Mociková).
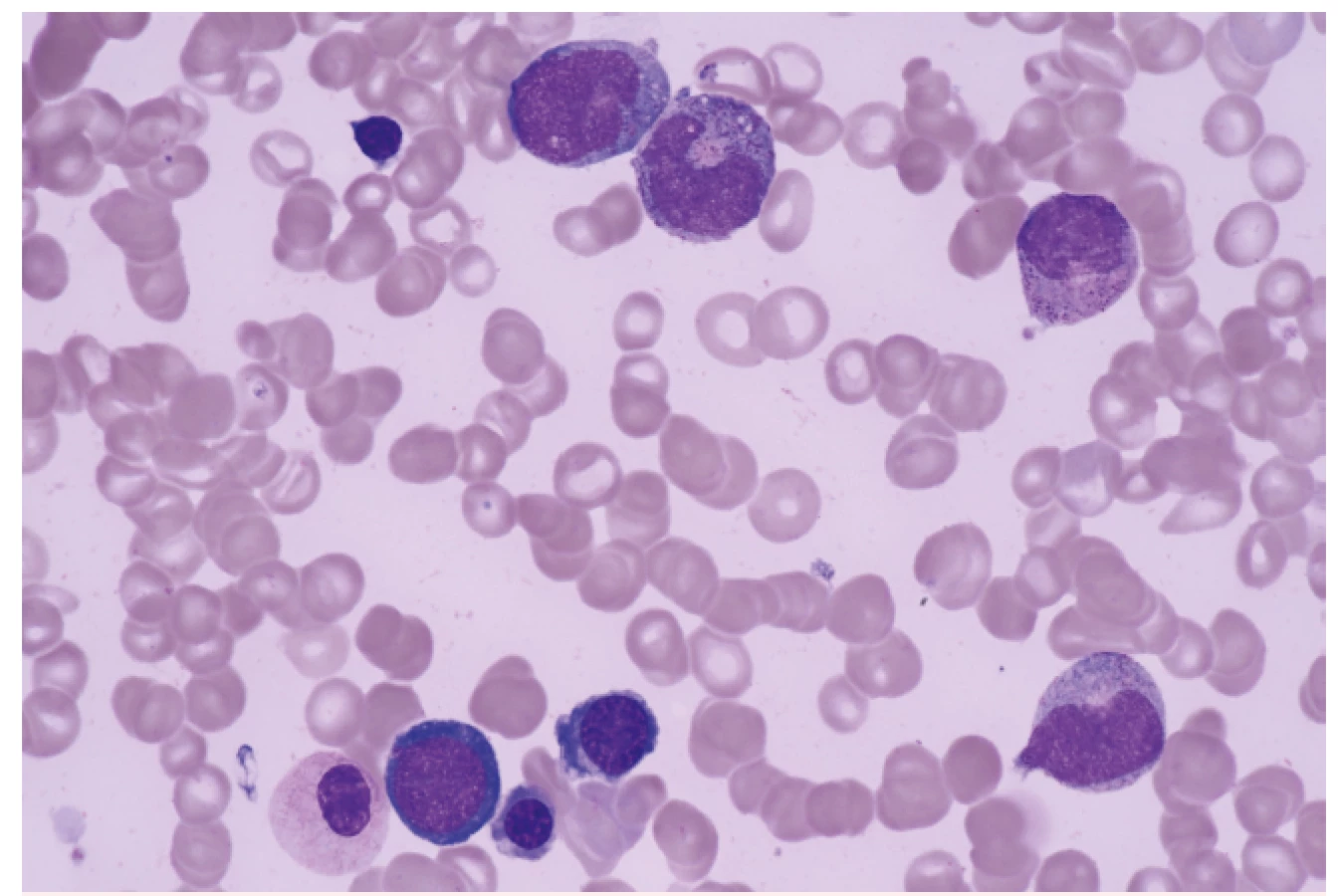
V této době byly k dispozici poměrně omezené diagnostické možnosti. Zásadní roli hrálo morfologické a cytochemické vyšetření aspirátu kostní dřeně a morfologické vyšetření trepanobioptického vzorku kostní dřeně (obr. 2). K vyšetření změn karyotypu sloužilo vyšetření mitotických chromozomů barvených tzv. G-pruhováním podle Wrighta. Vyšetření pomocí průtokové cytometrie nebylo standardním vyšetřením a molekulárně genetická analýza byla teprve v počátcích. Poměrně omezené byly i léčebné možnosti. I když FAB klasifikace provedla základní rozdělení podtypů MDS na prognosticky příznivé a nepříznivé, chybělo jednotné schéma prognostických parametrů, i když bylo vytvořeno několik systémů v různých zemích (Velká Británie – Bournemouth, SRN – Düsseldorf, Francie – Lille, Španělsko – Valencia). V léčbě MDS se v 80. letech minulého století významně uplatňovala podpůrná léčba – podávání transfuzí červených krvinek a krevních destiček, i když příprava koncentrátů na separátorech krevních buněk nebyla ještě standardním postupem. Recidivující infekce byly léčeny podáváním širokospektrých antibiotik a později i antimykotik. Velmi rozšířené bylo podávání kortikosteroidů za účelem zvýšení vyplavení krevních elementů do periferní krve. Dávky byly často vysoké a efekt nebyl úměrný vyvolané imunosupresi. Podrobnější analýza naší skupiny ukázala, že efekt kortikosteroidů a jejich kombinace s dalšími imunosupresivy (např. cyklosporinem A) může být přítomen zejména tam, kde se na obraze cytopenie podílí tzv. imunitně podmíněné selhání kostní dřeně [4]. Léčba nemocných s pokročilými stadii MDS měla špatné výsledky (tab. 3). Kombinovaná chemoterapie sice byla schopna navodit remisi u části nemocných, ale bez další léčby došlo u všech nemocných k relapsu. Překvapivě dlouhodobý efekt mělo cca u 10–15 % nemocných podání nízkých dávek (10–40 mg s. c. denně) cytosin arabinosidu (viz tab. 3).

[data UHKT]
Během devadesátých let minulého století se ukázalo, že pro prognózu nemocných a pro zvolení optimální léčby mají význam i další faktory, nikoli pouze orientační rozdělení na nemocné s příznivou a nepříznivou prognózou podle FAB klasifikace. V roce 1997 byl publikován tzv. Mezinárodní prognostický skórovací systém (IPSS) vycházející z analýzy 816 neléčených nemocných s primárním MDS [5]. Tento systém stanovil jako základní prognostické parametry počet blastů v kostní dřeni, počet cytopenií v periferní krvi a počet a typ změn karyotypu (obr. 3). Toto rozdělení se ukázalo významné jak pro délku přežití, tak pro riziko leukemické transformace choroby (obr. 4). Retrospektivní analýza dat nemocných s MDS [6] ale současně ukázala, že ani skupina nemocných s časnou formou MDS není prognosticky jednotná. Nálezy naší skupiny [7] a dalších týmů sdružených v tzv. MDS Foundation byly podnětem pro revizi klasifikace MDS vydanou WHO v roce 2001 [8] (tab. 4). Hlavním přínosem této klasifikace bylo vyčlenění prognosticky méně příznivé skupiny MDS s dysplazií ve více řadách za skupiny refrakterních anémií na jedné straně a nemocných s izolovanou del(5q) s relativně lepší prognózou na straně druhé. Nemocní s > 19 % blastů ve dřeni byli zařazeni již do skupiny akutních myeloidních leukemií s multilineární dysplazií a nemocní s chronickou myelomonocytární leukemií (CMML) byli přeřazeni do skupiny smíšených myelodysplasticko-myeloproliferativních syndromů. Na obrázku 5 je ukázána délka přežití neléčených nemocných z registru ÚHKT, která potvrzuje oprávněnost WHO klasifikace z roku 2001. Aktivita České MDS skupiny byla oceněna i přidělením organizace 5. světového sympozia o myelodysplastickém syndromu, jež se konalo v Praze v dubnu roku 1999. WPSS je prognostický systém kombinující WHO klasifikaci rizikové cytogenetické podskupiny podle IPSS a závislost na transfuzích [9, 10] (obr. 6).
![IPSS – Mezinárodní prognostický skórovací systém [5]](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image_pdf/31fbf2a052d80d3cbe17c54b549bfeb3.jpeg)
![WHO klasifikace MDS z roku 2001 [8]](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image_pdf/09722424a690b3e9a2ebd892fde40f9f.png)
![Délka přežití a riziko leukemické transformace u nemocných v jednotlivých
rizikových skupinách podle IPSS [5]](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image_pdf/8748d4142c53d465d0d611ccfa8c8c1d.png)
[data ÚHKT]
![Délka přežití neléčených nemocných s různými podtypy
MDS podle WHO klasifikace z roku 2001<br>
[data ÚHKT]](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image_pdf/576f9b4a4f38f8ed1c2b92aafcd96405.png)

Na přelomu minulého a současného století byly představeny nové diagnostické metody. Fluorescenční hybridizace in situ (FISH) umožnila diagnostiku chromozomálních změn na interfázních jádrech [11] (obr. 7), nicméně předpokládaný přínos této metody pro zpřesnění klasifikace nemocných podle IPSS se neukázal jako zásadní, i když upřesnil detekci některých prognosticky významných aberací karyotypu (zejména na 5. a 7. chromozomu) [12]. Vyšetření karyotypu pomocí FISH je na místě tam, kde se nedaří získat mitózy pro vyšetření karyotypu pomocí G-pruhování. Další metodou umožňující zpřesnění detekce změn karyotypu je vyšetření polymorfismu jednotlivých nukleotidů užitím vyšetření polymorfismu jednotlivých nukleotidů tzv. SNP (single nucleotide polymorphism assay), tuto metodu lze výhodně kombinovat s vyšetřením buněk v metafázi [13]. Vyšetření abnormit exprese povrchových antigenů pomocí průtokové cytometrie prokázalo určité změny charakteristické pro diagnostiku nemocných s MDS [14] a v poslední době i pro jejich prognózu, nicméně, průtoková cytometrie je stále pokládána jen za fakultativní metodu pro diagnostiku MDS.

Zavedení transplantace krvetvorných buněk (SCT) do léčby MDS vedlo k zásadní změně prognózy nemocných. Tato metoda je dodnes jediným kurativním přístupem k nemocným s MDS. Na obrázku 8 je porovnávající tříleté přežití u transplantovaných a netransplantovaných nemocných podle jednotlivých rizikových skupin podle IPSS [5, 15]. Z výsledků vyplývá, že nemocní s nízkým a středním-1 rizikem jsou indikování spíše ke konzervativní léčbě, kdežto u nemocných se středním-2 a vysokým rizikem je transplantace metodou volby, pokud to dovoluje stav nemocného. Názor na podání kombinované chemoterapie před SCT u nemocných s nadbytkem blastů není zcela jednotný. Naše studie opakovaně potvrdily příznivý efekt redukce počtu blastů pod 10 % před SCT na incidenci relapsů i přežití [16]. U nemocných s časnými formami MDS (refrakterní anémie a refrakterní cytopenie s dysplazií ve více řadách podle WHO 2001 klasifikace) je transplantace indikována u nemocných se středním-2 rizikem, tj. s hlubokou pancytopenií a nepříznivými změnami karyotypu. Nicméně příznivý efekt na dlouhodobé přežití se u nemocných s časnými formami choroby projevuje ve srovnání s pouhou podpůrnou léčbou až po více než 4 letech [16], příčinou je stále relativně vysoké procento komplikací v časné době po transplantaci na jedné straně a pozdní mortalita (často na onemocnění nesouvisející přímo s MDS) u netransplantovaných nemocných. Tento fakt byl také důvodem pro hledání nových léčebných přístupů, jež by zlepšily dlouhodobé přežití nemocných s časnými formami MDS, kteří nejsou indikováni k transplantaci.
![Srovnání přežití transplantovaných a netransplantovaných
nemocných v jednotlivých rizikových skupinách podle IPSS
TRM – mortalita spojená s transplantací, 3rDFS – tříleté přežití bez
známek choroby [5, 15].](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image_pdf/8bee7a2fefc872568288277764483f35.jpeg)
Kombinovaná imunosuprese podle identických schémat užívaných u aplastické anémie je podávána zejména u nemocných s hypoplastickou formou MDS [17]. Efekt byl popsán u třetiny nemocných, nicméně do 5 let po podání antithymocytárního globulinu (ATG) došlo k relapsu či k progresi směrem k AML u poloviny nemocných [16, 17]. Z růstových faktorů jsou podávány zejména erytropoezu stimulující látky (ESA), nejčastěji erytropoetin alfa (EPO) či darbopoetin alfa. Efekt těchto látek je závislý na přítomnosti alespoň určitého procenta funkční erytropoezy a absolutního či relativního deficitu endogenního erytropoetinu. Tyto faktory odráží i schéma predikce odpovědi na ESA [18], u nemocných s hladinou endogenního EPO v séru < 500 IU/l a závislostí na ≤ 2 TU erytrocytů měsíčně je pozorována odpověď u cca 70 % nemocných, u nemocných s hladinou EPO v séru < 100 IU/l pak dokonce u více než 80 % nemocných. Celkově lze říci, že efekt erytropoezu stimulujících látek je přítomen cca u třetiny nemocných s časnými stadii MDS. Přínos podávání agonistů trombopoetinového receptoru u časných forem MDS zatím nebyl definitivně zhodnocen vzhledem k jejich možnému stimulačnímu účinku na produkci časných myeloidních prekursorů patologického klonu, jenž byl pozorován u některých nemocných.
Přetížení železem vzniká u nemocných s MDS jednak díky opakovanému přívodu transfuzemi, jednak díky jeho zvýšenému vstřebávání v důsledku přítomné anémie s hypoxií, která tlumí tvorbu hepcidinu. Důsledkem může být orgánové přetížení železem, jež je závažné zejména při postižení srdce a jater. Na přelomu tisíciletí byly do léčby přetížení železem zavedeny perorální chelátory, jež jsou užívány zejména u nemocných s časným MDS s dysplazií vyjádřenou především v erytropoéze, která vede k opakovanému podávání transfuzí [10, 19]. Z perorálních látek je užíván především deferasirox, deferiprone je méně výhodný jednak pro horší snášenlivost, jednak pro riziko vzniku agranulocytózy a je vyhrazen pro nemocné, kde nelze použít deferasirox (renální selhání, intolerance deferasiroxu) [20].
První dvě desetiletí nového tisíciletí jsou spojena jednak s rozvojem molekulárně genetických metod v diagnostice MDS a v hledání významu mutací některých genů pro prognózu nemocných, jednak s dalším rozvojem transplantačních metod a s rozšířením léčebných možností díky novým lékům. Metoda transplantace krvetvorných buněk s užitím redukovaného přípravného režimu sice umožnila provést transplantaci i u starších nemocných díky snížení peritransplantační mortality, na druhé straně se díky použití nemyeloablativních přípravných režimů zvýšilo procento potransplantačních relapsů, takže rozdíl v dlouhodobém přežití nemocných nebyl při srovnání se standardně provedenou transplantací významný [21]. Význam SCT od haploidentického dárce u MDS zatím čeká na zhodnocení na rozsáhlejší skupině nemocných. Z řady metodik snažících se snížit riziko potransplantačních relapsů včetně některých nových léků zatím žádná nevedla k zásadnímu přínosu pro nemocné.
Dvěma klíčovými skupinami léků, jež během posledních 15 let ovlivnily prognózu nemocných s MDS, jsou lenalidomid a hypometylační látky (HMA). Lenalidomid působí na několika úrovních, indukuje selektivní degradaci buněk s delecí 5q indukcí jejich apoptózy, stimuluje erytropoézu ovlivněním aktivity p53 genu a má i imunomodulační účinek. Efekt lenalidomidu je tudíž nejprůkaznější u nemocných s izolovanou del(5q) [22]. Analýza našich nemocných léčených lenalidomidem ukázala dosažení nezávislosti na transfuzích po léčbě u více než 90 % nemocných s vymizením cytogenetické aberace u více než poloviny pacientů [23]. Zavedení HMA do léčby MDS významně zlepšilo prognózu zejména starších nemocných s pokročilými formami MDS, kteří nemohou být transplantováni. HMA (azacytidin, decitabin) jsou schopny ovlivněním stupně metylace genů uplatňujících se v diferenciaci časných prekursorů obnovit jejich funkci, a navodit tak vyhasínající diferenciaci mladých buněk. Výsledkem je snížení počtu blastů ve dřeni a stabilizace krevního obrazu [24]. Léčba 162 nemocných léčených azacytidinem v rámci České MDS skupiny byla efektivní u více než 53 % nemocných s průměrnou dobou přežití ukázala průměrnou dobu přežití léčených nemocných 16,9 měsíce ve srovnání se 7,5 měsíců u nemocných léčených kombinovanou chemoterapií či podpůrnou léčbou, jeden rok přežívalo 60,6 % nemocných léčených azacytidinem vs. 29,9 % nemocných léčených jiným typem léčby [25]. Nicméně, naše retrospektivní analýza ukázala, že 3 roky přežívá jen 26 % a 5 let 3,8 % nemocných léčených azacytidinem oproti 49 % transplantovaných nemocných [16] – obrázek 9). Podávání HMA tedy výrazně zlepšilo krátkodobou prognózu nemocných s pokročilým MDS, kteří nemohou být transplantováni, ale výrazněji neovlivnilo jejich dlouhodobé přežití. V současné době jsou studovány další možnosti využití HMA, jednak jako alternativy kombinované chemoterapie za účelem redukce počtu blastů před SCT, jednak jako udržovací léčby po SCT či po kombinované chemoterapii za účelem prevence relapsu. Z léčiv studovaných v posledních době se jako nadějný přípravek jeví luspatercept, jenž ovlivňuje stupeň inefektivní erytropoézy u časných forem MDS a tím zvyšuje hladinu hemoglobinu a snižuje závislost na transfuzích [26]. Rigosertib, efektivní blokátor proteinů produkovaným RAS genem, je schopen navodit efekt u části nemocných, u nichž selhala hypometylační léčba.

V letech 2009–2011 proběhla nová retrospektivní analýza neléčených nemocných s MDS za účelem revize IPSS [27]. Bylo hodnoceno 7 012 nemocných z 11 zemí, včetně nemocných z České republiky. Analýza však pouze potvrdila prognostický význam počtu blastů ve dřeni, počtu a hloubky cytopenií v periferní krvi a změn karyotypu a provedla podrobnější prognostické rozdělení změn karyotypu a hloubky cytopenie v periferní krvi. Revidovaný Mezinárodní prognostický systém (IPSS-R) je uveden v tabulce 5. IPSS-R potvrdil, že nemocní s velmi nízkým a nízkým rizikem jsou indikováni ke konzervativní léčbě, zatímco nemocní s vysokým a velmi vysokým rizikem jsou indikováni k transplantaci. Stále není jednotný názor na léčbu nemocných se středním rizikem, mladší nemocní by měli být indikováni k transplantaci, u ostatních se očekává možný prognostický přínos nových, zejména molekulárně genetických faktorů. V roce 2013 byly Evropskou pracovní skupinou pro MDS vydány Doporučené diagnostické a léčebné postupy pro nemocné s MDS [28], tato pravidla se stala i základem pro Doporučené postupy u MDS vydané Českou pracovní skupinou pro MDS [29].
![IPSS-R – revidovaný prognostický skórovací systém pro nemocné s MDS [27]](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image_pdf/17d083ba66626f553d2b4bb66b4adebf.png)

Molekulárně genetické studie se naplno rozvinuly během posledních 10 let díky zavedení nových technik sekvenovaní nové generace a celogenomového sekvenování. Analýza ukázala prognostický význam přítomnosti a počtu tzv. „driver“ mutací, tj. mutací genů hrajících důležitou roli v regulaci buněčné proliferace a diferenciace, a to jak u časných stadií MDS (EZH2, nRAS, ASXL1, RUNX1, TP53) [30], tak u všech skupin nemocných (ASXL1, RUNX1, TP53 a EZH2) [31]. Přítomnost určitých mutací typických pro MDS stoupá s věkem [32] a může mít i diagnostický význam při jinak nevysvětlitelné cytopenii zejména u starších nemocných. V roce 2016 byla publikována zatím poslední revize WHO klasifikace MDS [33] (tab. 6), ve které je již jedním z diagnostických kritérií pro MDS s prsténčitými sideroblasty přítomnost mutace sestřihového genu SF3B1. V současné době probíhá rozsáhlá analýza molekulárně genetických dat nemocných hodnocených při tvorbě IPSS-R, jež by měla přispět k zařazení některých mutací do prognostických schémat. Obecná shoda zatím panuje o prognosticky nepříznivém významu přítomnosti mutace TP53 genu [34].
![WHO klasifikace MDS z roku 2016 [33]](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image_pdf/57284d182c4c5e02a93f27758e0b2116.png)
V současné době je pro další rozvoj léčby nemocných s MDS očekáváno zhodnocení přínosu vyšetření mutací genů hrajících zásadní úlohu v kontrole růstu a diferenciace buňky a také změn jejich exprese. Tyto faktory by měly přispět ke zpřesnění charakteristiky mladších nemocných indikovaných k časné SCT i bez nálezu zmnožení blastů v kostní dřeni a blíže identifikovat i prognosticky nepříznivou podskupinu nemocných se středním rizikem podle IPSS-R, kteří by měli být rovněž transplantováni. V léčebných přístupech k nemocným s MDS zůstává několik nedořešených otázek. Zejména se jedná o přístup k nemocným s pokročilými stadii MDS s nadbytkem blastů, komplexními změnami karyotypu a přítomností mutace TP53 genu, kde i po SCT přežívá 3 roky méně než 10 % nemocných [15]. Další otázkou je jakými potransplantačními přístupy snížit incidenci relapsů po SCT a zvýšit efektivitu jejich léčby. Rovněž nevyřešenou otázkou zůstává efektivní léčba nemocných, u nichž selhává léčba hypometylačními látkami. I přes tyto problémy je ale nutno konstatovat, že za posledních 25 let došlo díky zavedení nových léčebných přístupů k zásadní změně prognózy nemocných s MDS, jak dokumentuje obrázek 10.

ZKRATKY
CMML – chronická myelomonocytární leukemie
EPO – erytropoetin alfa
ESA – erytropoézu stimulující agens
FAB – francouzsko-americko-britská
FISH – fluorescenční in situ hybdridizace
HMA – hypometylační látky
IPSS – mezinárodní prognostický skórovací systém
IPSS-R – revidovaný mezinárodní prognostický skórovací systém
MDS – myelodysplastický syndrom
RA – refrakterní anémie
RAEB – refrakterní anémie s nadbytkem blastů
RAEB-T – refrakterní anémie s nadbytkem blastů v transformaci
RARS – refrakterní anémie se zmnožením prsténčitých sideroblastů
SCT – transplantace krvetvorných buněk
WHO – Světová zdravotnická organizace
WPSS – Prognostický skórovací systém na bázi WHO klasifikace
Čestné prohlášení
Autor práce prohlašuje, že v souvislosti s tématem, vznikem a publikací tohoto článku není ve střetu zájmů a vznik ani publikace článku nebyly podpořeny žádnou farmaceutickou firmou.
Doručeno do redakce dne 11. 11. 2018.
Přijato po recenzi dne 28. 11. 2018.
prof. MUDr. Jaroslav Čermák, CSc.
Ústav hematologie a krevní transfuze
U Nemocnice 1
128 00 Praha 2
e-mail: cermak@uhkt.cz
Sources
1. Block M, Jacobson LO. Preleukemic acute leukemia. JAMA 1953;152 : 1018–1029.
2. Rheingold JJ, Kaufman R, Adelson E, Lear A. Smoldering acute leukemia N Engl J Med 1963;268 : 812–815.
3. Bennett JM, Catovsky D, Daniel MT, et al. Proposals for the classification of the myelodysplastic syndromes. Br J Haematol 1982;51 : 189–199.
4. Jonasova A, Neuwirtova R, Cermak J, et al. Cyclosporin A therapy in hypoplastic MDS patients and certain refractory anaemias without hypoplastic bone marrow. Br J Haematol 1998;100 : 304–309.
5. Greenberg P, Cox C, LeBeau MM, et al. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood 1997;89 : 2079–2088.
6. Germing U, Gattermann N, Strupp C, et al. Validation of the WHO proposals for a new classification of primary myelodysplastic syndromes: a retrospective analysis of 1600 patients. Leuk Res 2000;24 : 983–992.
7. Cermák J, Michalova K, Brezinova J, Zemanova Z. A prognostic impact of separation of refractory cytopenia with multilineage dysplasia and 5q - syndrome from refractory anemia in primary myelodysplastic syndrome. Leuk Res 2003;27 : 221–229.
8. Vardiman JW, Harris NL, Brunning RD. The World Health Organization (WHO) classification of the myeloid neoplasms. Blood 2002;100 : 2292–2302.
9. Malcovati L, Della Porta MG, Strupp C, et al. Impact of the degree of anemia on the outcome of patients with myelodysplastic syndrome and its integration into the WHO classification-based Prognostic Scoring System (WPSS). Haematologica 2011;96 : 1433–1440.
10. Cermak J, Kacirkova P, Mikulenkova D, Michalova K. Impact of transfusion dependency on survival in patients with early myelodysplastic syndrome without excess of blasts. Leuk Res 2009;33 : 1469–1474.
11. Ransdorfová Š, Březinová J, Šárová I, et al. Využití interfázní fluorescenční in situ hybridizace pro analýzu CD34+ buněk v periferní krvi u nemocných s myelodysplastickými syndromy. Transfuze Hematol dnes 2016;21 : 90–96.
12.
Rigolin GM, Bigoni R, Milani R, et al. Clinical importance of interphase cytogenetics detecting occult chromosome lesions in myelodysplastic syndromes with normal karyotype. Leukemia 2001;15 : 1841–1847.
13. Tiu RV, Gondek LP, O’Keefe CL, et al. Prognostic impact of SNP array karyotyping in myelodysplastic syndromes and related myeloid malignancies. Blood 2011;117 : 4552–4560.
14. Westers TM, Ireland R, Kern W, et al. Standardization of flow cytometry in myelodysplastic syndromes: a report from an international consortium and the European LeukemiaNet Working Group. Leukemia 2012;26 : 1730–1741.
15. Degg HJ, Guardiola P. Allogeneic hemopoietic stem cell transplantation in patient with myelodysplastic syndrome or myelofibrosis. Int J Hematol 2002;76(suppl.2):29–34.
16. Cermak J, Vitek A, Mikulenkova D, et al. An analysis of real life data obtained from 30 years follow-up of primary myelodysplastic syndromes (MDS) patients confirms a leading role of stem cell transplantation (SCT) for achievement of prolonged survival. Blood 2018;132 : 4372.
17. Molldrem JJ, Caples M, Mavroudis D, et al. Antithymocyte globulin for patients with myelodysplastic syndrome. Br J Haematol 1997;99 : 699–705.
18. Hellström-Lindberg E, Gulbrandsen N, Lindberg G, et al. Scandinavian MDS Group. A validated decision model for treating the anaemia of myelodysplastic syndromes with erythropoietin+granulocyte colony-stimulating factor: significant effects on quality of life. Br J Haematol 2003;120(6):1037–1046.
19. List AF, Baer MR, Steensma DP, et al. Deferasirox reduces serum ferritin and labile plasma iron in RBC transfusion-dependent patients with myelodysplastic syndrome. J Clin Oncol 2012;30 : 2134–2139.
20. Cermak J, Jonasova A, Vondrakova J, et al. A comparative study of deferasirox and deferiprone in the treatment of iron overload in patients with myelodysplastic syndromes. Leuk Res 2013;37 : 1612–1615.
21. Martino R, Iacobelli S, Brand R, et al. Myelodysplastic Syndrome subcom-
mittee of the Chronic Leukemia Working Party of the European Blood and Marrow Transplantation Group. Retrospective comparison of reducedintensity highdose conditioning for allogeneic hematopoietic stem cell transplantation using HLA-identical sibling donors in myelodysplastic syndromes. Blood 2006;108 : 836–846.
22. Fenaux P, Giagounidis A, Selleslag D, et al. MDS-004 Lenalidomide del5q Study Group. A randomized phase 3 study of lenalidomideversus placebo in RBC transfusion-dependent patients with Low-/Intermediate-1-risk myelodysplastic syndromes with del5q. Blood 2011;118 : 3765–3776.
23. Neuwirtová R, Jonášová A, Čermák J, et al. Analýza nemocných s myelodysplastickým syndromem (MDS) s delecí dlouhého ramene 5.chomozomu (del5q)), sledovaných Českou MDS pracovní skupinou: význam pro diagnostické zařazení a určení prognózy. Transfuze Hematol dnes 2009;15 : 204–209.
24. Fenaux P, Mufti GJ, Hellstrom-Lindberg E, et al. International Vidaza High-Risk MDS Survival Study Group. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: a randomised, open-label, phase III study. Lancet Oncol 2009;10 : 223–232.
25. Jonášová A, Čermák J, Červínek L, et al. První zkušenosti České MDS skupiny s terapií 5-azacytidinem u nemocných s myelodysplastickým syndromem s vyšším rizikem (IPSS střední 2 a vysoké riziko), akutní myeloidní leukémií do 30 % myeloblastů a chronickou myelomonocytární leukémií II. Transfuze Hematol dnes 2013;19 : 125–133.
26. Platzbecker U, Germing U, Götze KS, et al. Luspatercept for the treatment of anaemia in patients with lower-risk myelodysplastic syndromes (PACE-MDS): a multicentre, open-label phase 2 dose-finding study with long-term extension study. Lancet Oncol 2017;10 : 1338–1347.
27. Greenberg PL, Tuechler H, Schanz J, et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood 2012;120 : 2454–2465.
28. Malcovati L, Hellström-Lindberg E, Bowen D, et al. Diagnosis and treatment of primary myelodysplastic syndromes in adults: recommendations from the European LeukemiaNet. Blood 2013;122 : 2943–2964.
29. Čermák J, Jonášová A. Myelodysplastický syndrom. Transfuze Hematol dnes 2010;16 : 42–46.
30. Bejar R, Stevenson KE, Caughey BA, et al. Validation of a prognostic model and the impact of mutations in patients with lower-risk myelodysplastic syndromes. J Clin Oncol 2012;30 : 3376–3382.
31. Bejar R, Levinw R, Ebert BE. Unraveling the molecular pathophysiology of myelodysplastic syndromes. J Clin Oncol 2011;29 : 504–515.
32. Malcovati L, Galli A, Travaglino E, et al. Clinical significance of somatic mutation in unexplained blood cytopenia. Blood 2017;129 : 3371–3378.
33. Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision of the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016;127 : 2391–2405.
34. Bejar R. Implications of molecular genetic diversity in myelodysplastic syndromes. Curr Opin Hematol 2017;24 : 73–78.
Labels
Haematology Internal medicine Clinical oncologyArticle was published in
Transfusion and Haematology Today

2019 Issue 1
Most read in this issue
- Hodgkinův lymfom – nekončící příběh
- Non-Hodgkinův lymfom v České republice
- Vývoj transfuzní služby v České republice po roce 1990
- Chronická lymfocytární leukemie – současné využití moderních prognostických a prediktivních faktorů v diagnostice