
Ocular Manifestations of Granulomatosis with Polyangiitis
Authors:
E. Říhová; P. Svozílková; M. Brichová; A. Klímová; P. Kuthan; P. Diblík
Authors‘ workplace:
Přednosta: prof. MUDr. Jarmila Heissingerová, Ph. D., MBA
; Oční klinika, 1. lékařská fakulta, Univerzita Karlova a Všeobecná fakultní nemocnice v Praze
Published in:
Čes. a slov. Oftal., 74, 2018, No. 5, p. 167-174
Category:
doi:
https://doi.org/10.31348/2018/5/1
Overview
Granulomatosis with polyangiitis (GPA), formerly known as Wegener’s granulomatosis, is an autoimmune vasculitis of small vessels, presenting as necrotizing granulomatous inflammation especially of the upper and lower respiratory tract and necrotizing glomerulonephritis. GPA affects more often Caucasians in northern states, predominantly is affected the age-range group of 50 – 60 years. GPA may affect any organ; the eye symptoms are stated in the range of 16–78 %. The eye symptoms are very variable, and in up to 27 % they are the first sign of undiagnosed GPA. The etiology of GPA was not until now explained. Anti-neutrophil cytoplasmic antibodies (ANCA) play important role in the pathogenesis of this disease. GPA is ranked among ANCA associated vasculitis. The GPA is diagnosed on the basis of clinical signs and symptoms of systemic vasculitis, laboratory and histological tests and imaging studies. Immunomodulative therapy made a contribution to the improvement of GPA prognosis in the last decades; biological treatment reaches the prominence of the GPA treatment procedures. Good collaboration with other specialties is necessary for the early diagnosis and treatment of this life and vision threating disease. The ophthalmologist in the collaboration with specialists of other medical branches may take an important part in the GPA diagnostics, monitoring of the disease’s course, or adverse affects of the medication.
This paper pays attention to the eye symptoms of the GPA; the literature is supplemented with own photographs of GPA eye symptoms in patients followed up at the Department of Ophthalmology, First medical faculty, Charles University and General Faculty Hospital in Prague, Czech Republic, E.U.
Key words:
Granulomatosis with polyangiitis (GPA), orbit, scleritis, peripheral ulcerative keratitis (PUK), immunomodulation
INTRODUCTION
Granulomatosis with polyangiitis (GPA) is a life-threatening disease characterised in its classic form by necrotising vasculitis of the small blood vessels, above all inflicting the respiratory tracts and kidneys. GPA may affect any organ, ocular manifestations are stated in 50% of cases (2, 17). The disease was first described in 1936 by the German pathologist Wegener, after whom it was originally named – Wegener's granulomatosis. At present the recommended nomenclature is granulomatosis with polyangiitis, GPA is classified among ANCA associated vasculitis (16, 51). GPA may occur in any age group, but most frequently between the ages of 50 and 60. In children and adolescents (up to 19 years) the incidence is stated at between 8-15% of cases (22, 25). GPA afflicts individuals of Caucasian race from northern states more frequently, with the highest incidence recorded in northern Europe and New Zealand. The prevalence is approximately 30 per 1 million of the population. The incidence of GPA is 13-21 per 1 million of the population. In Norway it is estimated at 12 per 1 million, and in England 10.3 per 1 million of the population. The incidence in the Czech Republic is not precisely known. A minimal difference between the sexes is stated (2, 16, 17, 29, 32, 38). To date the etiology has not been entirely clarified, although current theories assume a possible contribution of hypersensitivity to certain microbial agents or genetic predispositions. Affliction with the disease is linked with the formation of anti-neutrophil cytoplasmic antibodies (ANCA) targeted against antigens contained in the primary granules of neutrophils, most commonly against proteinase 3 (PR3) and less often against myeloperoxidase (MPO) (27, 29).
Classification: According to the affected organs, GPA is manifested in generalised or limited form, i.e. without affecting the kidneys. GPA is characterised by necrotising granulomas of the upper and/or lower respiratory tracts, systemic vasculitis and necrotising glomerulonephritis. In the initial stages, even generalised GPA may begin as the organ-limited form, after which the manifestations of the affliction of further organs progressively increase. The classification criteria of GPA according to the American College of Rheumatology 1990 (24, 27) are as follows: 1. Inflammation in the area of the oral and nasal cavity (ulceration in oral cavity, purulent or haemorrhagic rhinitis), 2. Abnormal finding on X-ray of the chest with finding of nodules, infiltrates or cavities; 3. Abnormal urinary sediment (microscopic haematuria with or without the presence of leukocyturia), 4. Granulomatous inflammation of the vascular walls or perivascular area, demonstrated by biopsy. The presence of two or more criteria confirms the diagnosis with 88% sensitivity and 92% specificity. According to the more recent criteria the clinical course has two typical parts. The initial phase is characterised by granulomatous, ulcerative and antibiotic-resistant rhinitis, sinusitis or mastoiditis. In the second phase there is a presence of pulmonary manifestation and necrotising glomerulonephritis (29, 32).
Clinical picture: In approximately 30-50% of patients, initial non-specific manifestations of systemic pathologies are stated, such as high temperature, fatigue, weakness, torpidity and weight loss. Frequent manifestations of GPA are nosebleeds, chronic cold and chronic sinusitis. Affliction of the upper and lower respiratory tracts is stated in 90% of cases, and may also be manifested as non-specific acute or chronic inflammation. Inflammations are of a necrotising character with a tendency towards ulceration, and may lead to the destruction of nasal cartilage with the onset of a “saddle nose”. Recurring bilateral inflammations of the middle ear and transitory hearing malfunction in middle age are in 90% a manifestation of GPA, and affliction of the larynx is also possible (30, 49). Cough and chest pain may be manifested also in haemoptysis (12, 29). In as many as 75% of patients there is affliction of the kidneys during the course of the disease. The beginnings may be asymptomatic, later with manifestations of microscopic haematurias to rapidly progressing glomerulonephritis with an acutely developing picture of renal insufficiency (10, 11, 27, 32). Arthralgia and myalgia are common, arthritis affects primarily large joints without subsequent deformations. Upon affliction of the skin, maculopapular exanthema and subcutaneous nodules to ulcers are evident. Skin manifestations of GPA are stated in 50% of cases. Pericarditis, dilated cardiomyopathy, arrhythmia, changes on heart valves, affliction of intramural or epicardial coronary arteries are rare conditions. Diabetes insipidus is a manifestation of granulomatous affliction of the pituitary gland. Rarer conditions are neurological manifestations of GPA, from mononeuritis multiplex, polyneuropathy, paresis of the cranial nerves to strokes, in rare cases with the formation of granulomas and abscesses (10, 11, 16, 19, 32, 43).
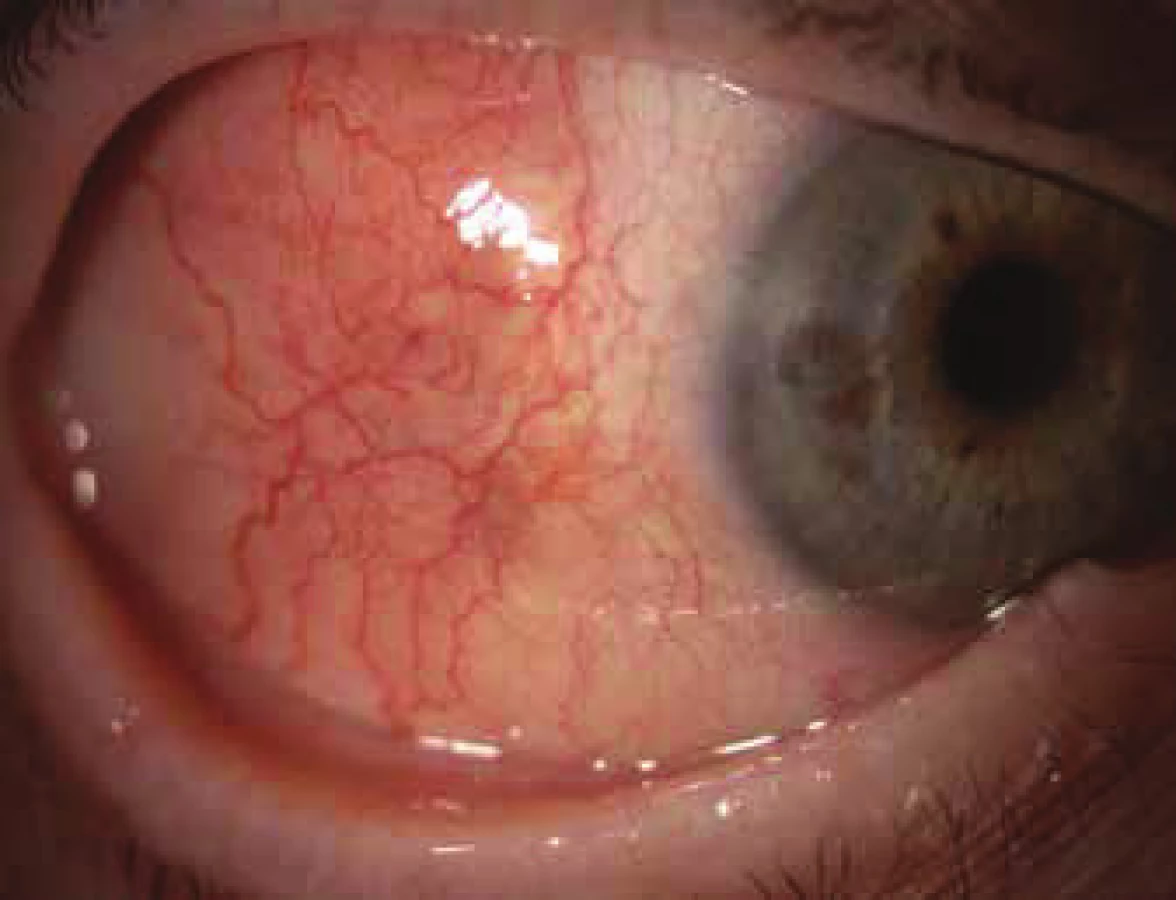
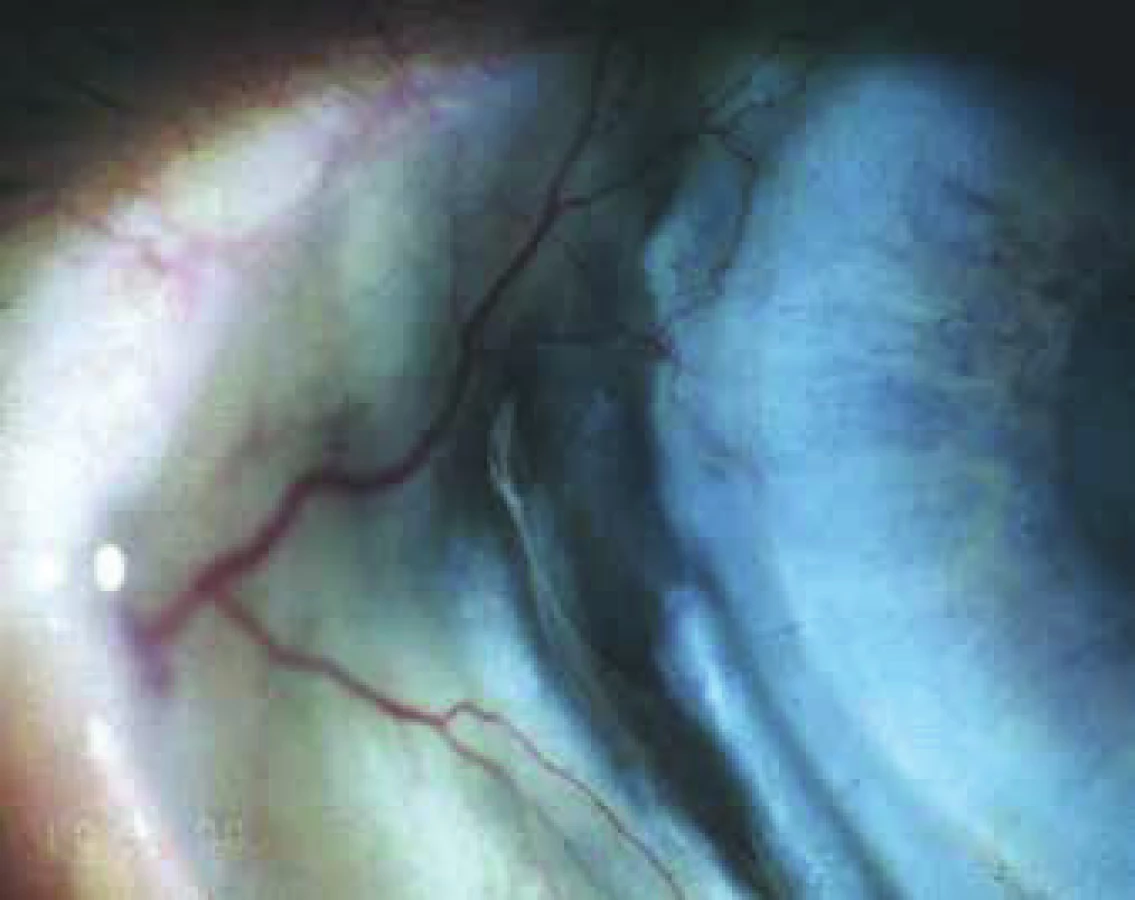
Ocular manifestation of GPA is stated in the literature in 50% (16-78%) of cases. Ocular manifestations of GPA may be a component of a systemic pathology, in 29% of cases the first symptoms of a hitherto undiagnosed disease, but also the only organ manifestation of the limited form of GPA in up to 30% of cases (17). Serious ocular manifestations of GPA contribute to deterioration of vision to blindness in 8-37% of cases (51, 53). Clinical symptoms of inflammation are manifested in various intensity of pain, reddening or edema of the eyelids, diplopia, protrusion of the eyeballs, epiphora, red eye, efferent pupillary defect, deterioration of visual acuity or blind spots in the visual field (Fig. 1). Subjective complaints and clinical findings are dependent upon the intensity of the affliction of any part of the eye, though the most frequent and most serious manifestations of GPA are sclerokeratitis and inflammatory infiltration of the eye socket (43, 46, 51, 54). Affliction of the orbit is stated in 15-60% of cases (24, 34). Granulomatous inflammation of the soft tissues of the orbit may take place primarily, more often it is linked with inflammation of the paranasal cavities (19, 43). In 14-30% of cases bilateral affliction of the orbits is stated (Fig. 2), pain is an accompanying symptom in only ¼ of patients (34). Pseudotumour has been described in children as the first symptom of GPA (27, 49). Infiltrations of the soft tissues in the orbit are manifested in saturation and swelling of the eyelids, protrusion, restriction of motility of the eye, diplopia and malfunctions of vision. Retrobulbar granulomatous masses compress the optic nerve and vascular bundle, and may lead to atrophy of the optic nerve with irreversible deterioration of vision. Extensive masses may cause destruction of the bone structures of the orbit (34, 59). Vasculitis of the posterior ciliary vessels or central retinal artery may be the cause of a deterioration of vision as a consequence of ischemic neuropathy (Fig. 3) (19, 43). Fibrosis of tissues as a consequence of chronic pseudotumour of the orbit leads in approximately 1/3 of patients with GPA to ringed fibrosis of the tissues of the optic behind the eyeball, fibrous remodelling of the afflicted extraocular muscles with subsequent motility disorder, retraction, chronic pain and enophthalmos resistant to immunosuppresive therapy (10, 43, 48, 53, 55). Pronounced fibrosis in the area of the lamina cribrosa and surroundings of the optic nerve in connection with atrophy with compression of the nerve fibres has been documented also histopathologically (10). Neuritis of the optic nerve is not a frequent manifestation of GPA, and may leave permanent changes with pronounced deterioration of vision. Inflammations of the extraocular muscles (Fig. 4) – orbital myositis - are manifested in sudden pain with a malfunction of motility of the eye, and may affect any extraocular muscle, with the rectus muscles more commonly afflicted bilaterally (34, 48). Dacryoadenitis may be a component of inflammatory infiltration of the orbit, and subjective complaints – edema of the eyelid, pain upon movement of the eye – are obscured. Affliction of the eyelids is not common, and manifestations are very diverse. Granulomatous inflammation may destroy the eyelids (Fig. 5), subsequent deformation is the cause of ectropion or entropion connected with trichiasis (47). Obstruction of the ductus nasolacrimalis is mostly linked with inflammations of the sinonasal cavities, and affects approximately 7% of patients with GPA, in whom it is usually a later manifestation (5, 12, 40, 34). Affliction of the anterior segment of the eye is common upon GPA. Conjunctivitis with petechiae, granulomas, ulcerations, dry eye syndrome with variously severe manifestations on the conjunctiva or cornea (Fig. 6) are stated in approximately 16% (12). The conjunctiva is an easily accesssible sample for histological examination. Affliction of the conjunctiva is often linked with inflammation of the subglottis (49). Scleritis is a frequent manifestation of systemic pathologies, and after rheumatoid arthritis GPA is stated as the second most common connected systemic pathology (51). Scleritis is stated in 12% as the first symptom indicating GPA. It is manifested clinically in pronounced pain and reddening. Scleritis is mostly bilateral, manifested more often as anterior than posterior inflammation of the sclera, more frequently nodular (Fig. 7) than diffuse and in men in up to 67% in the necrotising form of scleritis in GPA (Fig. 8) (17, 21). The surface of necrotising scleritis is avascular, ischemic, with scleral constriction and frequently also exposed choroidea. In severe, insufficiently treated forms it may progress to a devastating form of inflammation perforating ocular structures (Fig. 9), with subsequent secondary infection or phthisis of the eye (5, 8, 47, 56). Spontaneous perforations are rare, but may be the result also of even minimal trauma. English authors (41) drew attention to necrotising scleritis caused by eye surgery (cataract, glaucoma, pars plana vitrectomy). In up to 35% of patients with scleritis, perilimbal minor infiltrations of the cornea (Fig. 10) are evident, which indicate severe form of systemic vasculitis (21, 35). Peripheral ulcerous keratitis (Fig. 11) is a very frequent manifestation of GPA. In its beginnings it is manifested in perilimbal infiltrations of the cornea, and progresses around the periphery with spreading ulceration. Occlusively necrotising vasculitis of the arterior ciliaris anterior leads to peripheral ulcerous keratitis, frequently bilateral, and due to the common vascular supply is also linked with scleritis. Constriction to perforation of the affected cornea (Fig. 12) are complications of insufficiently treated basic pathology (24, 35). Anterior uveitis in GPA is rare (Fig. 13) and is frequently linked with keratoscleritis (12, 24). The incidence of manifestations of GPA on the posterior segment of the eye is stated in 16%. Intermedial or posterior granulomatous uveitis is stated in approximately 2.5%. Sclero-choroidal granulomas connected with choroidal stripes, effusion, exudative amotio or acute posterior multifocal pigment epitheliopathy have been described as individual ocular manifestations in patients with GPA (7, 18, 20, 33). Vasculitis is of lesser intensity. Retinal vasculopathy, retinal occlusion with haemorrhage into the vitreous body are stated more frequently than vasculitis (23, 28, 30). Retinopathy – cotton wool spots and intraretinal haemorrhage are rare in GPA, and may be linked also with nephropathy and hypertension accompanying this disease. Central retinal vein occlusions have been published also in relation to young patients with GPA without the presence of vasculitis in the angiographic picture (30, 57).











Diagnosis of GPA is determined on the basis of a correlation of the clinical picture with laboratory findings, results of imaging methods and if applicable also tissue biopsy of the affected organ. Examination by a specialist in the field according to the afflicted organ, with targeted questioning for GPA is essential. All the symptoms of granulomatosis need not necessarily be expressed simultaneously at the time of diagnosis. The diversity of the manifestations, which are sometimes non-specific for GPA, causes a delay in correct diagnosis by two years on average. Limited forms of GPA affect in particular the respiratory tract and the eyes. A component of the diagnostic balance is also thorough differential diagnostics (6, 8, 9, 17, 43). The value of sedimentation of erythrocytes is constantly increased from the laboratory examinations. Anaemia is present in approximately 50%. There is usually a raised level of C-reactive proteins (CRP), gamma globulins (primarily IgA) and circulating immune complexes (CIK). There may be a presence of erythrocyturia or proteinuria. In the case of renal damage, there is an increase in the level of urea and creatinine and a decrease in glomerular filtration. A significant diagnostic criterion is anti-neutrophil cytoplasmic antibodies – ANCA. A finding of ANCA andibodies in plasma, usually of subtype c-ANCA against PR3, occurs in the initial phase in approximately 50% of patients, in the phase of generalisation around 90%. Sensitivity and specificity of the PR3-ANCA examination is up to 90%, fluctuating with the activity of the pathology, and is one of the criteria for assessing the effectiveness of treatment. However, fluctuations in the level of c-ANCA need not necessarily be connected with the activity of ocular manifestations of GPA. In the isolated ocular form, titres of ANCA may be negative (18, 19, 29, 32). Imaging methods are an essential accompaniment to diagnosis in the case of affliction of the upper respiratory tracts, paranasal cavities, orbit and neurological forms of GPA. Radiological findings may demonstrate infiltration of the orbit, thickening or fibrosis of the optic nerve and extraocular muscles, erosion of the orbital wall, and are undoubtedly of benefit for the diagnosis of GPA, but are not specific (9, 43). As a result, histological examination of the granuloma or infiltrated tissue, not only of the orbit but any affected organ, may support the diagnosis of GPA (22, 39). In the diagnostic balance of GPA it is necessary to consider also other pathologies such as ANCA-associated vasculitis, Graves orbitopathy, orbital cellulitis, tumorous infiltration, sarcoidosis, polyarteritis nodosa, systemic lupus erythematosus, rheumatoid arthritis, IgG4-related diseases. Ocular inflammations of an infectious and non-infectious origin and certain post-medication reactions (masquerade syndromes) must be taken into account upon determining diagnosis or relapse of GPA (29, 43, 53, 60).
Treatment of GPA is governed by the severity of the affected organs, the activity of the pathology and the age of the patient. The standard procedure in the treatment of GPA is divided into induction and maintenance. The main aim of induction therapy is to induce a rapid remission, the purpose of the second phase is to maintain long-term remission. The generally recognised and recommended treatment is combined immunosuppression. Cyclophosphamide with corticoids are the drugs of first choice. The initial doses of prednisone 1 mg/kg/day and cyclophosphamide 2 mg/kg/day usually alleviate manifestations of GPA. In rapidly progressing or fulminant forms of GPA they are recommended in pulse intravenous form (1, 5, 29, 37, 54). At present the use of cyclophosphamide is minimised due to the carcinogenic risk. Neither non-steroid therapy nor monotherapy with corticoids usually has a sufficient influence on manifestations of GPA. Azathioprine may be an alternative to cyclophosphamide, but is less effective, especially in the acute phase of the disease. Methotrexate, cyclosporine A and mycophenolate mofetil are other pharmaceuticals which can maintain the remission of certain forms of GPA (42). Biological treatment (infliximab and rituximab are currently preparations which may induce and maintain remission of the more severe forms of GPA (14, 25, 55, 61). Rituximab administered with corticoids is highly effective in the acute phase of GPA and may induce long-term remission. According to the international literature, treatment with rituximab is comparable with cyclophosphamide, and has lesser side effects. Treatment with rituximab appears to be more effective upon manifestations of necrotising vasculitis resistant to treatment by cyclophosphamide than in the case of granulomatous manifestations of GPA (32). In the orbital form of GPA it is important to differentiate the active inflammatory disease of GPA from the no longer active fibrotic form, which will not respond to immunomodulation therapy. In the literature long-term treatment with infliximab on patients with bilateral infiltration of the orbit is positively evaluated (52), but no significant positive results are stated with treatment using etanercept (58). In the Czech Republic both preparations are off-label, but for patients with severe form of the pathology or upon intolerance of cyclophosphamide it is possible to administer them at specialised centres. It is necessary to continue long-term with the treatment according to the clinical picture and laboratory parameters. Remission of GPA means complete disappearance not only of the clinical manifestations of the disease, but also a return of laboratory examinations to the usual values. GPA with persistently increased values of d-ANCA and CRP usually has a worse prognosis, and relapses occur more often. Recurrence of GPA takes place usually upon a reduction of the doses of medications or following the cessation of their application. After adjustment of treatment, recurrence of the disease need not reach the previous level. Treatment is supervised by a specialist in the given field, and interdisciplinary consultation of the clinical manifestations and treatment is essential. Local therapy of ocular manifestations of GPA is conducted by an ophthalmologist. Local anti-inflammatory therapy may alleviate inflammatory manifestations of the anterior segment of the eye, but without general therapy its effect is limited. Lubricant is sufficient in the treatment of dry eye syndrome, but it is necessary to treat inflammation of the anterior segment intensively with corticoids in the form of drops or ointments. According to the clinical manifestations of necrotic scleritis or infiltration of the cornea we recommend antibiotic preparations, sometimes in combination with a corticoid or cyclosporine A in the form of drops (5, 35, 51). Complication of inflammation such as pronounced constriction or perforation of the cornea or sclera may be resolved by cyanoacrylate glue, covering by the conjunctiva, amnio, scleral implant or keratoplasty of the affected – constricted or perforated – tissue (13, 35, 45). The good postoperative effect of dacryocystorhinostomy on stenosis of the ductus nasolacrimalis may be complicated by postoperative necrosis of the scar or skin fistula (40). Orbital manifestations of GPA often progress also during immunosuppression therapy, decompression of the orbit may be effective in certain cases (49). Orbital infection, fistula or abscess have been described as serious complications of orbital manifestations (11, 13, 34). Cataract and secondary glaucoma are complications of ocular manifestations of GPA and long-term corticoid therapy, it is recommended to perform surgery in remission of GPA with an increase of general medication at least for a period of 1 month (13, 24). Depot corticoids applied periocularly increase the potency of necrotic processes of the sclera, and as a result are not recommended in the case of GPA. Patients with GPA are treated with immunosuppressive medications long-term, and as immunoincompetent individuals are at risk from ocular infections such as cytomegaloviral retinitis, herpes zoster keratitis (3, 6, 14) or orbital abscesses (11). Biological medications may paradoxically induce ocular changes similar to intraocular inflammation – for example snowballs in the vitreous body, maculopathy, vasculopathy similar to systemic lupus erythematosus or sarcoidosis (60). Some local medications reducing intraocular pressure such as brimonidine are not recommended upon acute manifestations of inflammation of the anterior segment of the eye.
The prognosis of GPA is dependent on the extent of the pathology – the frequency and severity of organic affliction. GPA is characterised by systemic vasculitis, but there are substantial differences between the limited and generalised forms of the disease. The prognosis of GPA without treatment is infaust, the patient dies within two years, mostly from kidney failure (56). Immunosuppressive therapy markedly improves the prognosis of this life and sight threatening pathology, as many as 80% of patients survive 10 years (34, 38, 39, 43). Patients with limited form of GPA are younger on average at the beginning of the illness than patients with generalised form, and are more often women. The isolated form often progresses into a chronic inflammation of the upper respiratory tracts, with signs of destruction, such as saddle nose. These patients have a better prognosis, with a good response to immunosuppressive therapy (40). Complete remission of GPA is stated in up to 70% of patients, but one half of them suffer a relapse upon reduction of the maintenance treatment or after the termination thereof, sometimes many years later. The renewal or increase of treatment alleviates the clinical manifestations of GPA, but remission may not be full (24, 29, 32). The treatment should always be the result of interdisciplinary co-operation. The prognosis of ocular manifestations of GPA and resulting vision are dependent on the localisation of the inflammation and the severity of affliction of the ocular tissues. The most common and most serious sight threatening manifestations of GPA are considered to be affliction of the orbit and sclerokeratitis. Affliction of the retina or disc of the optic nerve often leave irreversible damage also in remission of the general pathology. The risk of deterioration of vision is higher in patients in whom it is more difficult to induce remission, with a higher number of relapses and resistant type of GPA (17, 24). Recurrences of ocular manifestations of GPA may be the first sign of relapse of inflammations also of other organs affected by this disease, and an ophthalmologist in co-operation with specialists in the given fields adjusts general therapy according to the clinical manifestations of GPA and tolerance of the medication by the patient. For determination of a correct and timely diagnosis and the application of targeted treatment of GPA it is essential to ensure good interdisciplinary co-operation with a nephrologist, immunologist, rheumatologist, otorhinolaryngologist, dermatologist, gastroenterologist, radiologist and histopathologist (46).
Summary: GPA is a rare, life and sight threatening pathology. Timely diagnosis and correct, directed treatment may induce long-term remission of the disease, avoid complications connected with this pathology and improve the quality of life of patients. Ocular manifestations of GPA may be the first symptom of a previously not manifested or undiagnosed systemic disease, and thus enable the ophthalmologist to contribute to the diagnosis of this illness. Scleritis is the most frequent ocular manifestation of GPA, and inflammatory infiltration of the orbital tissue is the most severe sight-threatening manifestation of this systemic pathology.
We would like to thank Dr. J. Žabek, B. Braun-Avitum Prague and the team of doctors at the Department of Nephrology at the 1st Faculty of Medicine, Charles University and General University Hospital in Prague for their kind co-operation in the diagnosis and treatment of patients with GPA observed at the Department of Ophthalmology at the 1st Faculty of Medicine, Charles University and General University Hospital in Prague.
Parts of this study were presented at the symposium of the 17th South Bohemian Alois Timra Days in Hluboká nad Vltavou in May 2017 and at the Clinical Seminar of the Department of Ophthalmology at the 1st Faculty of Medicine, Charles University and General University Hospital in Prague in March 2018.
Department of Ophthalmology, 1st Faculty of Medicine, Charles University and General University Hospital Prague Chief Doctor: prof. MUDr. Jarmila Heissingerová,Ph.D., MBA
The authors of the study declare that no conflict of interest exists in the compilation, theme and subsequent publication of this professional communication, and that it is not supported by any pharmaceuticals company.
Received: 13. 6. 2018
Accepted: 30. 10. 2018
Available on-line: 27. 3. 2019
doc. MUDr. Eva Říhová, CSc.

Oční klinika 1. LF UK a VFN v Praze
U Nemocnice 2
128 08 Praha 2
Sources
1. Alloway, JA., Cupps, TR.: High–dose methylprednisolone for retro-orbital Wegener‘s granulomatosis. J Rheumatol. 20(4); 1993 : 752-754.
2. Banerjee, SH., Grayson, PC.: Vasculitis Around the World: Epidemiologic insights into causality and a need for global partnership. J Rheumatol. 44(2); 2017 : 136-139.
3. Bertelmann, E., Liekfeld, A., Pleyer, U. et al.: Cytomegalovirus retinitis in Wegener‘s granulomatosis: case report and review of the literature. Acta Ophthalmol Scand. 83(2); 2005 : 258-261.
4. Blaise, P., Robe-Collignon, N., Andris, C. et al.: Wegener granulomatosis and posterior ischemic optic neuropathy. Eur J Intern Med. 18(4); 2007 : 326-327.
5. Charles, SJ., Meyer, PA., Watson, PG.: Diagnosis and management of systemic Wegener‘s granulomatosis presenting with anterior ocular inflammatory disease. Br J Ophthalmol. 75(4); 1991 : 201-207.
6. Charlier, C., Henegar, C., Launay, O. et al.: Risk factors for major infections in Wegener granulomatosis: analysis of 113 patients. Ann Rheum Dis. 68(5); 2009 : 658-663.
7. Chiquet, C., Lumbroso, L., Denis, P. et al.: Acute posterior multifocal placoid pigment epitheliopathy associated with Wegener‘s granulomatosis. Retina 19(4); 1999 : 309-313.
8. Cocho, L., Gonzales-Gonzales, LA., Molina-Prat, N. et al.: Scleritis in patients with granulomatosis with polyangiitis (Wegener). Br J Ophthalm. 100(8); 2016 : 1062-1065.
9. Courcoutsakis, NA., Langford, CA., Sneller, MC. et al.: Orbital involvement in Wegener‘s granulomatosis: MR findings in 12 patients. J Comput Assist Tomogr. 21(3); 1997 : 452-458.
10. Cutler, WM., Blatt, IM.: The ocular manifestations of lethal midline granuloma (Wegener‘s granulomatosis). Am J Ophthalmol. 42(1); 1956 : 21-35.
11. de Silva, DJ., Cole, C., Luthert, P. et al.: Masked orbital abscess in Wegener‘s granulomatosis. Eye 21(2); 2007 : 246-248.
12. Fortney, AC., Chodosh, J.: Conjunctival ulceration in recurrent Wegener granulomatosis. Cornea 21(6); 2002 : 623-624.
13. Foster, CS.: Peripheral ulcerative keratitis in Melki SA., Fava LA.: Cornea and refractery atlas of clinical wisdom, chapter 17, 2011, SLACK corp, print, USA.
14. Freeman, WR., Stern, WH., Gross, JG. et al.: Pathologic observations made by retinal biopsy. Retina. 10(3); 1990 : 195-204.
15. Freidlin, J., Wong, I.G., Acharya, N.: Rituximab treatment for peripheral ulcerative keratitis associated with Wegener‘s granulomatosis. Br J Ophthalmol. 91(10); 2007 : 1414-1416.
16. Greco, A., Marinelli, C., Fusconi, M. et al.: Clinic manifestations in granulomatosis with polyangiitis. Int J Immunopath and Pharmacol. 29(2); 2016 : 151-159.
17. Harper, SL., Letko, E., Samson, CM. et al.: Wegener‘s granulomatosis: the relationship between ocular and systemic disease. J Rheumatol. 28(5); 2001 : 1025-1032.
18. Hejcmanová, D., Koblížek, V., Ruta, J.: Wegenerova granulomatóza - kazuistické sdělení, Cesk Slov Oftalmol, 63(1); 2007 : 55-62.
19. Hoffman, GS., Kerr, GS., Leavitt., RY. et al.: Wegener‘s granulomatosis: an analysis of 158 patients. Ann Intern Med. 116(6); 1992 : 488-498.
20. Huong du, LT., Tran, TH., Piette, JC.: Granulomatous uveitis revealing Wegener‘s granulomatosis. J Rheumatol. 33(6); 2006 : 1209-1210.
21. Jabs, DA., Mudun A., Dunn J.P. et al.: Episcleritis and scleritis: clinical features and treatment results. Am J Ophthalmol. 130(4); 2000 : 469–476.
22. Kalina, PH., Lie, JT., Campbell. RJ. et al.: Diagnostic value and limitations or orbital biopsy in Wegener‘s granulomatosis. Ophthalmology 99(1); 1992 : 120-124.
23. Kamei, M., Yasuhara, T., Tei, M. et al.: Vitreous hemorrhage from a ciliary granuloma associated with Wegener granulomatosis. Am J Ophthalmol. 132(6); 2001 : 924-926.
24. Kubaisi, B., Abu Samra, K., Foster, CS.: Granulomatosis with polyangiitis: an apdated rewiev of ocular disease manifestation. Intractable Rare Dis Res. 5(2); 2016 : 61-69.
25. Lasave, AF., You, C., Ma, L. et al.: Long-term outcomes of rituximab therapy in patients with noninfectious posterior uveitis refractory to conventional immunosuppressive therapy. Retina 38(2); 2018 : 395-402.
26. Leavitt, RY., Fauci, AS., Bloch, DA. et al.: The American College of Rheumatology 1990 criteria for the diagnosis of Wegener‘s granulomatosis. Arthritis Rheum. 33(8); 1990 : 1101-1107.
27. Levi, M., Kodsi, SR., Rubin, SE. et al.: Ocular involvement as the initial manifestation of Wegener‘s granulomatosis in children. J AAPOS. 12(1); 2008 : 94-96.
28. Lida, T., Spaide, RF., Kantor, J.: Retinal and choroidal arterial occlusion in Wegener‘s granulomatosis. Am J Ophthalmol. 133(1); 2002 : 151-152.
29. Lošťáková, V., Kolek, V., Vašáková, M. et al.: Granulomatóza s polyangiitidou (Doporučený postup při diagnostice, terapii a sledování vývoje onemocnění) [KAP. 6.4] Sekce intersticiálních plicních procesů ČPFS – dostupné na www.pneumologie.cz
30. Lukáš, J., Hroboň, M., Rambousek, P. et al.: Lokalizovaná forma Wegenerovy granulomatozy s postižením ucha a oka. Otolaryng Foniat. 52(1); 2003 : 29-32.
31. Lutalo, MK., DCruz, DP.: Diagnosis and classification of granulomatosis with polyangiitis. J Autoimun. 48-49; 2014 : 94-98.
32. Martínková, K., Valkovský, I., Handlos P.: Granulomatóza s polyangiitidou. Interni Med. 16(5); 2014 : 199-201.
33. Masuda, T., Izumi, Y., Takeshita, H. et al.: Granulomatosis with polyangiitis presenting as a choroidal tumor. Case Rep Rheumatol. 2015; 2015 : 271823. doi: 10.1155/2015/271823.
34. McNab, AA.: The 2017 Doyne Lecture: the orbit as a window to systemic disease. Eye (Lond). 32(2); 2018 : 248-261.
35. Messme, EM., Foster, CS.: Vasculitic peripheral ulcerative keratitis. Surv Ophthalmol. 43(5); 1999 : 379-396.
36. Metee, A., Kimyon, S., Saygili, O. et al.: Bilateral acute angle-closure glaucoma as a first presentation of granulomatosis with polyangiitis (Wegener’s) Arq Bras Oftalmol. 79(5); 2016 : 336-338.
37. Meyer, PA., Watson, PG., Franks, W. et al.: Pulsed immunosuppressive therapy in the treatment of immunologically induced corneal and scleral disease. Eye (Lond) 1(4); 1987 : 487-495.
38. Mohammad, AJ., Jacobsson LT., Westman, KW. et al.: Incidence and survival rates in Wegener‘s granulomatosis, microscopic polyangiitis, Churg-Strauss syndrome and polyarteritis nodosa. Rheumatology (Oxford) 48(12); 2009 : 1560-1565.
39. Muller, K., Lin, JH.: Orbital granulomatisis with polyangiitis: clinical and pathological findings. Arch Pathol Lab Med. 138(8); 2014 : 1110-1114.
40. Eloy, P., Leruth, E., Bertrand, B. et al.: Successful endonasal dacryocystorhinostomy in a patient with Wegener‘s granulomatosis. Clin Ophthalmol. 3; 2009 : 651-656.
41. O‘Donoghue, E., Lightman, S., Tuft, S. et al.: Surgically induced necrotising sclerokeratitis (SINS) -precipitating factors and response to treatment. Br J Ophthalmol. 76(1); 1992 : 17-21.
42. Pagnoux, C., Mahr, A., Hamidou, MA. et al.: Azathioprine or methotrexat maintenance for ANCA-associated vasculitis. N Engl J Med. 359(26); 2008 : 2790-2803.
43. Pakalniskis, MG., Berg, AD., Policeni, BA.: The Many Faces of granulomatosis with polyangiitis: A review of the head and neck imaging manifestations. Am J Roentgenol. 205(6); 2015 : 619-629.
44. Phillip, R., Luqmani. R.: Mortality in systemic vasculitis: a systematic review. Clin Exp Rheumatol. 26(5); 2008 : 94-104.
45. Pakrou, N., Selva, D., Leibovitch, I.: Wegener‘s granulomatosis: ophthalmic manifestations and management. Semin Arthritis Rheum. 35(5); 2006 : 284-292.
46. Reinhold-Keller, E., Beuge, N., Latza, U. et An indesciplinary approach to the care of patients with Wegenerś granulomatosis. Long-Term Outcome in 155 Patients. Arthritis Rheum. 43(5); 2000 : 1021-1032.
47. Říhová, E., Havlíková, M., Michalová, K. et al.: Diagnóza a léčba Wegenerovy granulomatózy na podkladě očních změn. Cesk Slov Oftalmol. 53(4); 1997 : 223-228.
48. Salam, A., Meligonis, G., Malhotra, R.: Superior oblique myositis as an early feature of orbital Wegener‘s granulomatosis. Orbit 27(3); 2008 : 203-206.
49. Santiago, YM., Fay, A.: Wegener‘s granulomatosis of the orbit: a review of clinical features and updates in diagnosis and treatment. Semin Ophthalmol. 26(4-5); 2011 : 349-355.
50. Seo, P., Specks, U., Keogh, KA.: Efficacy of rituximab in limited Wegener‘s granulomatosis with refractory granulomatous manifestations. J Rheumatol. 35(10); 2008 : 2017-2023.
51. Soukiasian, SH.: Wegeners granulomatosis in Foster et Vitale: Diagnosis and treatment of uveitis. Saunders company, Philadelphia, 2011 : 661-672.
52. Svozílková, P., Říhová, E., Brichová, M. et al.: Infliximab v léčbě Wegenerovy granulomatózy. Cesk Slov Oftalmol, 62(4); 2006 : 280-286.
53. Talar-Williams, C., Sneller, M.C., Langford, C.A. et al.: Orbital socket contracture: a complication of inflammatory orbital disease in patients with Wegener‘s granulomatosis. Br J Ophthalmol. 89(4); 2005 : 493-497.
54. Tarabishy, AB., Schulte, M., Papliodis, GN. et al.: Wegeners granulomatosis: Clinical manifestations, differential diagnosis and management of ocular and systemic disease. Surv Ophthalmol. 55(5); 2010 : 429-443.
55. Taylor SR., Salama, AD., Joshi, L et al.: Rituximab is effective in the treatment of refractory ophthalmic Wegener‘s granulomatosis. Arthritis Rheum. 60(5); 2009 : 1540-47.
56. Vogiatzis, KV.: Bilateral blindness due to necrotizing scleritis in a case of Wegener‘s granulomatosis. Ann Ophthalmol. 15(2); 1983 : 185-188.
57. Wang, M., Khurana, RN., Sadda, SR.: Central retinal vein occlusion in Wegener‘s granulomatosis without retinal vasculitis. Br J Ophthalmol. 90(11); 2006 : 1435-1436.
58. WGET Research Group: Design of the Wegener‘s granulomatosis etanercept trial (WGET). Control Clin Trials. 23(4); 2002 : 450-468.
59. Woo, TL., Francis, IC., Wilcsek, GA. et al.: Australasian orbital and adnexal Wegener‘s granulomatosis. Ophthalmology 108(9); 2001 : 1535-43.
60. You, C., Sahawneh, HF., Ma, L., Kubaisi, B. et al.: A review and update on orphan drugs for the treatment of noninfectious uveitis. Clin Ophthalmol. 11; 2017 : 257-265.
61. You, C., Ma, L., Lasave, AF. et al.: Rituximab induction and maintenance treatment in patients with scleritis and granulomatosis with polyangiitis (Wegener‘s). Ocul Immunol Inflamm. 19; 2017 : 1-8.
Labels
OphthalmologyArticle was published in
Czech and Slovak Ophthalmology

2018 Issue 5
Most read in this issue
- Ocular Manifestations of Granulomatosis with Polyangiitis
- Merkel Cell Carcinoma of the Eyelids (Clinical-Histological Study)
- Pachychoroid Disease of the Macula – Case Report
- Pilot Results of Implantation of the New Hydrophobic Intraocular Lens Zeiss LUCIA 611P in the Czech Republic