
OČNÍ PROJEVY GRANULOMATÓZY S POLYANGIITIDOU
Authors:
E. Říhová; P. Svozílková; M. Brichová; A. Klímová; P. Kuthan; P. Diblík
Authors‘ workplace:
Přednosta: prof. MUDr. Jarmila Heissingerová, Ph. D., MBA
; Oční klinika, 1. lékařská fakulta, Univerzita Karlova a Všeobecná fakultní nemocnice v Praze
Published in:
Čes. a slov. Oftal., 74, 2018, No. 5, p. 167-174
Category:
doi:
https://doi.org/10.31348/2018/5/1
Overview
Granulomatóza s polyangiitidou (GPA), dříve známá jako Wegenerova granulomatóza, je autoimunní vaskulitida malých cév, projevující se jako nekrotizující granulomatózní zánět především horních a dolních cest dýchacích, a nekrotizující glomerulonefritida. GPA postihuje častěji bělochy severských států, nejčastěji je postižena věková skupiny 50 - 60 let. GPA může postihnout kterýkoli orgán, oční projevy jsou uváděny v rozmezí 16 - 78 %. Oční projevy mohou být velmi různorodé a až v 27 % prvním projevem nediagnostikované GPA. Etiologie GPA nebyla dosud objasněna. Anti-neutrofilní cytoplazmatické protilátky (ANCA) hrají významnou roli v patogenezi této nemoci, GPA je řazena mezi ANCA asociované vaskulitidy. GPA je diagnostikována na základě klinických projevů systémové vaskulitidy, laboratorních, zobrazovacích a histologických vyšetření. Imunomodulační terapie přispěla ke zlepšení prognózy GPA v posledních desetiletích, biologická léčba se dostává do popředí léčebných postupů GPA. Dobrá mezioborová spolupráce je nezbytná pro včasnou diagnózu a léčbu této život a zrak ohrožující choroby. Oftalmolog ve spolupráci se specialisty jiných oborů se může významně podílet na diagnostice a léčbě GPA, monitorování průběhu choroby, nebo nežádoucích účinků léčiv.
Tato práce upozorňuje na oční projevy GPA, literární zdroje jsou doplněny vlastní fotodokumentací očních projevů GPA u pacientů sledovaných na Oční klinice 1.LF UK a VFN v Praze.
Klíčová slova:
Granulomatóza s polyangiitidou (GPA), orbita, skleritida, periferní ulcerózní keratitida (PUK), imunomodulace
ÚVOD
Granulomatóza s polyangiitidou (GPA) je život ohrožující choroba charakterizovaná ve své klasické podobě nekrotizující vaskulitidou malých cév postihující především dýchací cesty a ledviny. GPA může postihnout kterýkoli orgán, oční projevy jsou uváděny v 50 % [2,17]. Nemoc popsal v roce 1936 německý patolog Wegener, dle kterého byla původně pojmenována – Wegenerova granulomatóza. V současné době je doporučeno názvosloví granulomatóza s polyangiitidou, GPA je řazena mezi ANCA asociované vaskulitidy [16,51]. GPA se vyskytuje v kterékoli věkové skupině, nejčastěji však mezi 50 – 60 rokem života. U dětí a mladistvých (do 19 let) je výskyt uváděn mezi 8 - 15 % [22,25]. GPA postihuje častěji bílou rasu severských států, nejvyšší výskyt byl zaznamenán v severní Evropě a na Novém Zélandu. Prevalence je přibližně 30/1 milion obyvatel. Incidence GPA je 13–21/1 milion obyvatel. V Norsku je odhadována na 12/1 milion a v Anglii 10,3/1 milion obyvatel. Výskyt v naší republice není přesně znám. Je udáván minimální rozdíl mezi pohlavím [2,16,17,29,32,38]. Etiologie dosud nebyla zcela objasněna, současné teorie však předpokládají možný podíl hypersenzitivity na některá mikrobiální agens či genetické predispozice. Onemocnění souvisí s tvorbou protilátek proti cytoplasmě neutrofilů (ANCA) namířených proti antigenům obsaženým v primárních granulech neutrofilů, nejčastěji proti proteináze 3 (PR3) a méně často proti myeloperoxidáze (MPO) [27,29]. Klasifikace. Podle postižených orgánů se GPA projevuje ve formě generalizované nebo limitované, tj. bez postižení ledvin. GPA je charakterizovaná nekrotizujícími granulomy horních a/nebo dolních cest dýchacích, systémovou vaskulitidou a nekrotizující glomerulonefritida. V počátcích může i generalizovaná GPA začít jako orgánově limitovaná forma, a projevy postižení dalších orgánů jsou časově odstupňované. Klasifikační kritéria GPA podle American College of Rheumatology 1990 [24,27]: 1. Zánět v oblasti dutiny ústní a dutiny nosní (ulcerace v dutině ústní, purulentní nebo hemoragická rinitida), 2. Abnormální nález na rtg hrudníku s nálezem nodulů, infiltrátů či dutin; 3. Abnormální močový sediment (mikroskopická hematurie s/bez přítomnosti leukocyturie), 4. Granulomatózní zánět stěn cév či perivaskulární oblasti biopticky prokázaný. Přítomnost dvou a více kritérií potvrzuje diagnózu s 88 % senzitivitou a 92 % specificitou. Dle novějších kritérií má klinický průběh dvě typické části. Iniciální fáze je charakterizována granulomatózní, ulcerativní a vůči antibiotikům rezistentní rinitidou, sinusitidou nebo mastoitidou. Ve druhé fázi je přítomna plicní manifestace a nekrotizující glomerulonefritida [29,32].
Klinický obraz:
Asi u 30–50 % pacientů jsou udávány počáteční nespecifické projevy systémových chorob jako je subfebrilie, únava, slabost, malátnost, úbytek hmotnosti. Častými projevy GPA jsou epistaxe, chronická rýma a chronická sinusitida. Postižení horních i dolních cest dýchacích je uváděno v 90 % případů a může se projevit jako nespecifický akutní nebo chronický zánět. Záněty mají nekrotizující charakter se sklonem k ulceracím, mohou vést k destrukci nosních chrupavek se vznikem tzv. sedlovitého nosu. Recidivující oboustranné záněty středouší a převodní porucha sluchu ve středním věku jsou v 90 % projevem GPA, možné je i postižení laryngu [30,49]. Kašel, bolesti na hrudníku se mohou projevit i hemoptýzou [12,29]. Až u 75 % pacientů dochází k postižení ledvin v průběhu onemocnění. Začátky mohou být asymptomatické, později s projevy mikroskopické hematurie až rychle progredující glomerulonefritidy s akutně se rozvíjejícím obrazem renální insuficience [10,11,27,32]. Obvyklé jsou artralgie, myalgie, artritida postihuje především velké klouby bez následných deformací. Při postižení kůže jsou patrné makulopapulózní exantém a podkožní uzlíky až vředy. Kožní projevy GPA jsou uváděny v 50 %. Perikarditida, dilatační kardiomyopatie, arytmie, změny na chlopních, postižení intramurálních nebo epikardiálních koronárních arterií jsou vzácné. Diabetes insipidus je projevem granulomatózního postižení pituitární žlázy. Vzácnější jsou neurologické projevy GPA od mononeuritis multiplex, polyneuropatie, paréz hlavových nervů, až po cévní mozkové příhody, vzácně s tvorbou granulomů a abscesů [10,11,16,19,32,43].
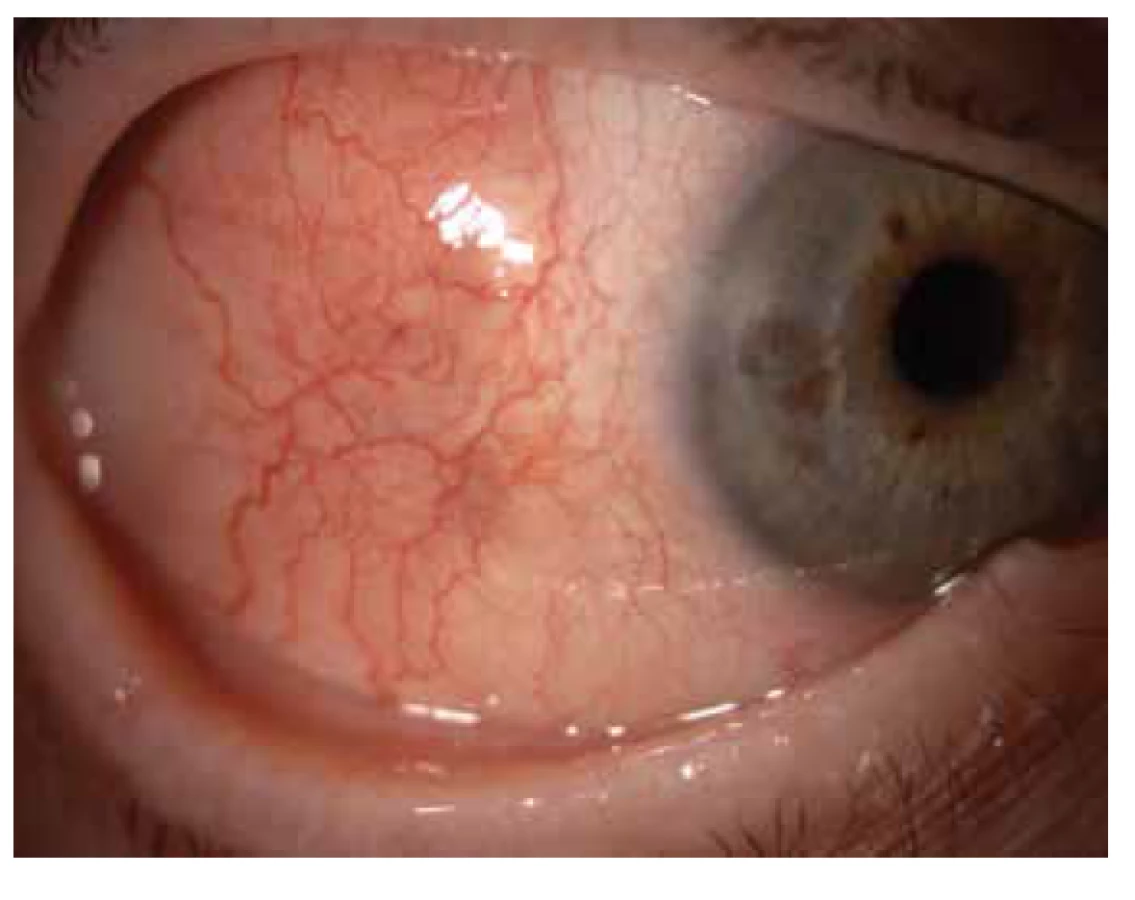
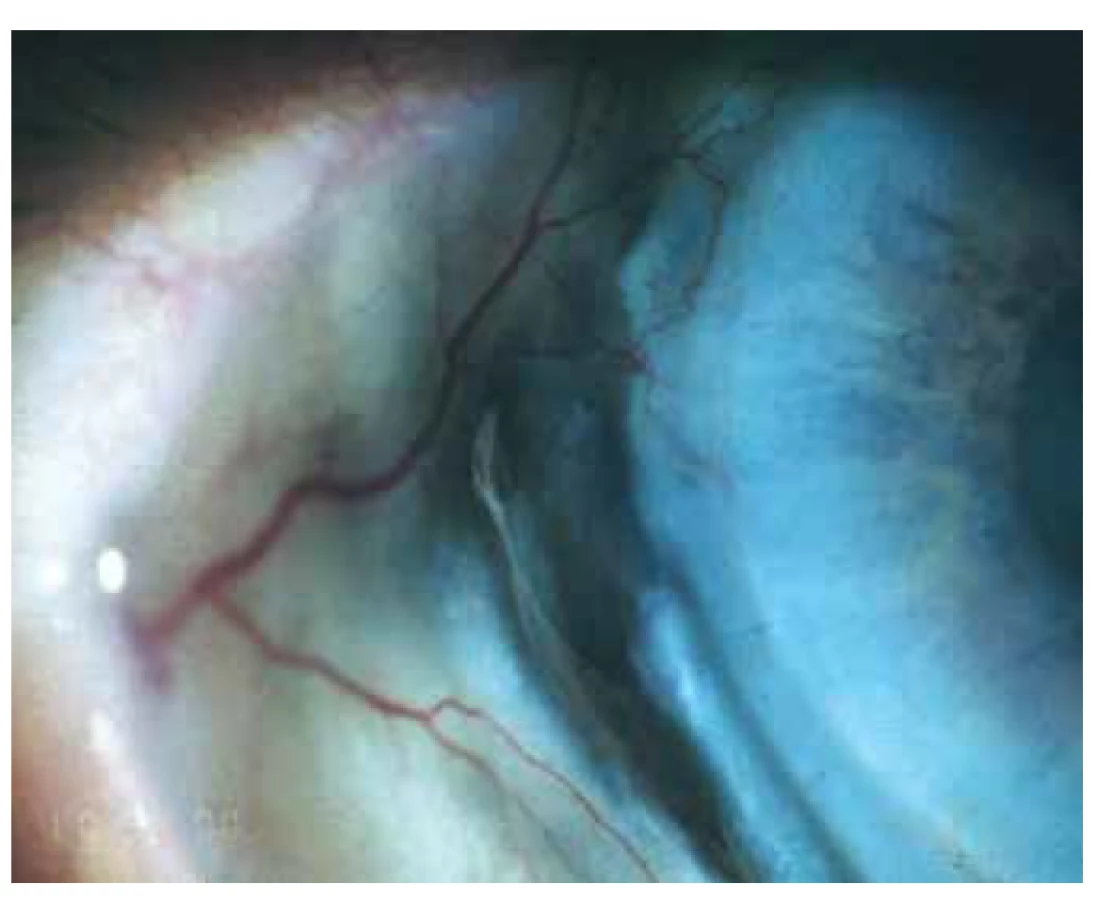
Oční manifestace u GPA je v literatuře uváděna u 50 % (16–78 %) případů. Oční projevy GPA mohou být součástí systémové choroby, v 29 % prvotními příznaky dosud nepoznané choroby, ale i jediným orgánovým projevem limitované formy GPA až ve 30 % [17]. Závažné oční projevy GPA se podílejí na snížení vízu až slepotě v 8–37 % [51,53]. Klinické příznaky zánětu se projeví různou intenzitou bolesti, zarudnutím či edémem víček, diplopií, protruzí bulbů, epiforou, červeným okem, eferentním pupilárním defektem, snížením zrakové ostrosti či výpadky zorného pole (Obrázek 1). Subjektivní potíže a klinické nálezy jsou závislé na intenzitě postižení kterékoli části oka, nejčastějšími ale i nejzávažnějšími projevy GPA jsou sklerokeratitida a zánětlivá infiltrace očnice [43,46,51,54]. Postižení orbity je udáváno v 15–60 % [24,34]. Granulomatózní zánět měkkých tkání orbit může probíhat primárně, častěji je spojen se zánětem paranazálních dutin [19,43]. Ve 14–30 % je uváděno bilaterální postižení orbit (Obrázek 2), bolest je doprovodným symptomem pouze u 1/4 pacientů [34]. Pseudotumor byl popsán u dětí jako první projev GPA [27,49]. Infiltrace měkkých tkání v orbitě se projevují prosáknutím a otokem víček, protruzí, omezením pohyblivosti oka, diplopií a poruchami vízu. Retrobulbární granulomatózní masy utlačují optický nerv i cévní svazek a mohou vést k atrofii optického nervu s ireverzibilním snížením vízu. Extenzivní masy mohou způsobit destrukci kostěných struktur orbity [34,59]. Vaskulitida zadních ciliárních cév či centrální sítnicové artérie může být příčinou úbytku zraku v důsledku ischemické neuropatie (Obrázek 3) [19,43]. Následkem fibrózy tkání v důsledku chronického pseudotumoru orbity dochází asi u 1/3 pacientů s GPA k prstencovité fibróze tkáně optiku za bulbem, fibrózní přestavbě postižených okohybných svalů s následnou poruchou hybnosti, retrakcí, chronickou bolestí a enoftalmem refrakterními na imunosupresivní léčbu [10,43,48,53,55]. Výrazná fibróza v místě lamina cribrosa a okolí optického nervu spojená s atrofií s útlakem nervových vláken byla dokumentovaná i histopatologicky [10]. Neuritidy zrakového nervu nejsou častým projevem GPA a mohou zanechat trvalé změny s výrazným snížením vízu. Záněty okohybných svalů (Obrázek 4) – orbitální myozitidy – se projevují náhlou bolestí s poruchou hybnosti oka, mohou postihnout kterýkoli okohybný sval, přímé svaly jsou častěji postiženy oboustranně [34,48]. Dakryoadenitida může být součástí zánětlivé infiltrace orbity a subjektivní potíže – edém víčka, bolest při pohybu oka – jsou zastřeny. Postižení víček není časté a projevy jsou velmi různorodé. Granulomatózní zánět může destruovat víčka (Obrázek 5), následná deformace je příčinou ektropia či entropia spojeného s trichiázou [47]. Obstrukce ductus nasolacrimalis je většinou spojena se záněty sinonazálních dutin a postihuje přibližně 7 % pacientů s GPA, obvykle je jejím pozdním projevem [5,12,40,34]. Postižení předního segmentu oka je při GPA časté. Konjunktivitidy s petechiemi, granulomy, ulceracemi, syndromem suchého oka s různě těžkými projevy na spojivce či rohovce (Obrázek 6) jsou uváděny asi v 16 % [12]. Spojivka je snadno dostupným vzorkem k histologickému vyšetření. Postižení spojivky bývá často spojeno se zánětem subglotis [49]. Skleritida je častou manifestací systémových chorob a GPA je po revmatoidní artritidě uváděna jako druhá nejčastější související systémová choroba [51]. Skleritida je ve 12 % uváděna jako první příznak upozorňující na GPA. Klinicky se manifestuje výraznou bolestivostí a zarudnutím. Skleritida je většinou oboustranná, projevující se častěji jako přední než zadní zánět skléry, častěji nodulární (Obrázek 7) než difuzní a u mužů až v 67 % nekrotizující formou skleritidy GPA (Obrázek 8) [17,21]. Plocha nekrotizující skleritidy je avaskulární, ischemická, se sklerálním ztenčením a často i prosvítající choroideou. U těžkých nedostatečně léčených forem může přecházet až na oční struktury devastující perforující formu zánětu (Obrázek 9), s následnou sekundární infekcí či ftízou oka [5,8,47,56]. Spontánní perforace jsou vzácné, ale mohou být výsledkem i minimálního traumatu. Na nekrotizující skleritidu vyvolanou oční operací (katarakta, glaukom, pars plana vitrektomie) upozornili angličtí autoři [41]. Až u 35 % pacientů se skleritidou jsou patrné i perilimbální drobné infiltrace rohovky (Obrázek 10), které upozorňují na těžkou formu systémové vaskulitidy [21,35]. Periferní ulcerózní keratitida (Obrázek 11) je velmi častým projevem GPA. V začátcích se projevuje perilimbálními infiltracemi rohovky a progreduje po obvodu se šířící ulcerací. Okluzivně nekrotizující vaskulitida arteria ciliaris anterior vede k periferní ulcerozní keratitidě často oboustranné a díky společnému cévnímu zásobení spojené i se skleritidou. Ztenčení až perforace postižené rohovky (Obrázek 12) jsou komplikací nedostatečně léčeného základního onemocnění [24,35]. Přední uveitida u GPA je vzácná (Obrázek 13) a je často spojená se keratoskleritidou [12,24]. Incidence projevů GPA na zadním segmentu oka je udávána v 16 %. Uveitidy intermediální či zadní granulomatózní jsou uváděny asi ve 2,5 %. Sklero-choroidální granulomy spojené s choroidálními pruhy, efuzí, exsudativní amocí či akutní zadní multifokální pigmentová epiteliopatie byly popsané jako jednotlivé očnÍ projevy u pacientů s GPA [7,18,20,33]. Vaskulitidy jsou mírné intenzity. Retinální vaskulopatie, retinální okluze s krvácením i do sklivce jsou uváděny častěji než vaskulitidy [23,28,30]. Retinopatie – vatovitá ložiska a intraretinální krvácení jsou vzácná u GPA a mohou souviset i s nefropatií a hypertenzí při této chorobě. Okluze centrální sítnicové vény byly publikovány i u mladých pacientů s GPA bez přítomnosti vaskulitidy v angiografickém obraze [30,57].











Diagnóza GPA je stanovena na základě korelace klinického obrazu s laboratorními nálezy, výsledky zobrazovacích metod a eventuálně tkáňové biopsie postiženého orgánu. Vyšetření specialistou oboru dle orgánového postižení s cíleným dotazem na GPA je nezbytné. Všechny příznaky granulomatózy nemusí být v době diagnózy vyjádřeny současně. Různorodost projevů někdy nespecifických pro GPA způsobuje opoždění správné diagnózy v průměru dvou let. Limitované formy GPA postihují zvláště respirační trakt a oči. Součástí diagnostické rozvahy je i důkladná diferenciální diagnostika [6,8,9,17,43]. Z laboratorních vyšetření je konstantně zvýšená hodnota sedimentace erytrocytů. Anémie je přítomna asi v 50 %. Obvykle jsou zvýšeny C-reaktivní proteiny (CRP), gamaglobuliny (především IgA) a cirkulující imunokomplexy (CIK). Může být přítomna erytrocyturie, proteinurie. V případě renálního poškození stoupá urea a kreatinin a klesá glomerulární filtrace. Významným diagnostickým kritériem jsou protilátky proti cytoplazmě neutrofilů typu – ANCA. Nález ANCA protilátek v plazma, obvykle podtypu c-ANCA proti PR3, se vyskytuje v iniciální fázi asi u 50 % pacientů, ve fázi generalizace kolem 90 %. Senzitivita i specificita vyšetření PR3-ANCA je až 90 %, kolísá s aktivitou onemocnění a je jedním z kritérií posouzení efektu léčby. Výkyvy hladiny c-ANCA však nemusí souviset s aktivitou očních projevů GPA. U izolované oční formy mohou být titry ANCA negativní [18,19,29,32]. Zobrazovací metody jsou nezbytným doplňkem diagnózy u postižení horních cest dýchacích, paranazálních dutin, orbity i neurologické formy GPA. Radiologické nálezy mohou prokázat infiltraci orbity, ztluštění či fibrózu optického nervu a okohybných svalů, erozi orbitální stěny a jsou jistě přínosem pro diagnózu GPA, ale nejsou specifické [9,43]. Proto histologické vyšetření granulomu nebo infiltrované tkáně, nejen orbity, ale kteréhokoliv postiženého orgánu, může diagnózu GPA podpořit [22,39]. Při diagnostické rozvaze GPA je nutné zvažovat i další choroby, jako jsou ANCA asociované vaskulitidy, Gravesova orbitopatie, orbitocelulitida, nádorová infiltrace, sarkoidóza, polyarteriitis nodosa, systémový lupus erytematodes, revmatoidní artritida, IgG4 - related onemocnění. Oční záněty infekčního a neinfekčního původu a některé polékové reakce (maskující syndromy) je nutné vzít v úvahu při stanovení diagnózy či relapsu GPA [29,43,53,60].
Léčba GPA se řídí závažností postižených orgánů, aktivitou onemocnění a věkem pacienta. Standardní postup léčby GPA je rozdělen na indukční a udržovací. Hlavním cílem indukční léčby je navození rychlé remise, účelem druhé fáze je udržení dlouhodobé remise. Všeobecně uznávanou a doporučovanou léčbou je kombinovaná imunosuprese. Cyklofosfamid s kortikoidy jsou léky první volby. Úvodní dávky prednisonu 1 mg/kg/den a cyklofosfamidu 2 mg/kg/den většinou projevy GPA zklidní. U rychle progredující nebo fulminantní formy GPA jsou doporučovány v pulzní intravenózní formě [1,5,29,37,54]. V současné době je užití cyklofosfamidu minimalizováno pro karcinogenní riziko. Nesteroidní léčba ani monoterapie kortikoidy obvykle projevy GPA dostatečně neovlivní. Azatioprin může být alternativou cyklofosfamidu, ale je méně účinný, zvláště v akutní fázi choroby. Metotrexát, cyklosporin A, mykofenolát mofetil jsou dalšími léky, které mohou udržet remisi některých forem GPA [42]. Biologická léčba (infliximab a rituximab jsou v současné době preparáty, které mohou navodit a udržet remisi těžších forem GPA [14,25,55,61]. Rituximab podaný s kortikoidy je velmi účinný v akutní fázi GPA a může navodit dlouhodobou remisi. Dle světové literatury je léčba rituximabem srovnatelná s cyklofosfamidem a má menší vedlejší účinky. Léčba rituximabem se zdá být účinnější při projevech nekrotizující vaskulitidy refrakterní na léčbu cyklofosfamidem, než u granulomatózních projevů GPA [32]. U orbitální formy GPA je důležité rozlišovat aktivní zánětlivé onemocnění GPA od již neaktivní fibrotické formy, která nebude reagovat na imunomodulační léčbu. V literatuře je kladně hodnocena i dlouhodobá léčba infliximabem u pacienta s oboustrannou infiltrací orbit [52], ale žádné významné pozitivní výsledky s léčbou etanerceptem [58]. V naší republice jsou oba preparáty off-label, ale u pacientů s těžkou formou nemoci či při intoleranci cyklofosfamidu je možné je podávat na specializovaných pracovištích. V léčbě je nutné pokračovat podle klinického obrazu a laboratorních parametrů dlouhodobě. Remise GPA znamená úplné vymizení nejen klinických projevů choroby, ale i návrat laboratorních vyšetření k obvyklým hodnotám. GPA s trvale zvýšenými hodnotami d-ANCA a CRP má obvykle horší prognózu a častěji dochází k relapsům. K recidivě GPA dochází většinou při snížení dávek léků nebo po ukončení jejich aplikace. Po úpravě léčby nemusí recidiva choroby dosáhnout předchozího stupně. Léčbu vede specialista daného oboru a mezioborová konzultace klinických projevů a léčby je nezbytná. Lokální léčbu očních projevů GPA vede oftalmolog. Lokální protizánětlivá terapie může zmírnit zánětlivé projevy předního segmentu oka, ale bez celkové léčby je málo účinná. Lubrikans vystačí při léčbě syndromu suchého oka, ale zánět předního segmentu je nutné intenzivně léčit kortikoidy ve formě kapek či mastí. Dle klinických projevů nekrotické skleritidy či infiltrace rohovky doporučujeme antibiotické preparáty, někdy v kombinaci s kortikoidem nebo cyklosporinem A ve formě kapek [5,35,51]. Komplikace zánětu, jako je výrazné ztenčení či perforace rohovky nebo skléry, může být řešena cyanoakrylátovým lepidlem, překrytím spojivkou, amniem, sklerálním štěpem či keratoplastikou postižené – ztenčené či perforované – tkáně [13,35,45]. Dobrý pooperační efekt dakryocystorhinostomie u stenózy ductus nasolacrimalis může být komplikován pooperační nekrózou jizvy nebo kožní fistulí [40]. Orbitální projevy GPA často progredují i při imunosupresivní terapii, dekomprese orbity může být účinná v některých případech [49]. Orbitální infekce, fistula, čí absces byly popsané jako vážné komplikace orbitálních projevů [11,13,34]. Katarakta a sekundární glaukom jsou komplikací očních projevů GPA a dlouhodobé kortikoidní léčby, operaci je doporučeno provést v remisi GPA s navýšením celkové medikace alespoň po dobu 1 měsíce [13,24]. Depotní kortikoidy aplikované periokulárně potencují nekrotické pochody skléry, proto nejsou u GPA doporučovány. Pacienti s GPA jsou léčeni imunosupresivními léčivy dlouhodobě a jsou jako imunonekompetentní jedinci rizikoví k očním infekcím jako cytomegalovirové retinitidě, herpes zoster keratitidě [3,6,14] nebo orbitálním abscesům [11]. Biologika paradoxně mohou navodit oční změny podobné nitroočnímu zánětu – například sněhové koule ve sklivci, makulopatii, vaskulopatii podobnou systémovému lupus erytematodes či sarkoidóze [60]. Některá lokální léčiva snižující nitrooční tlak, jako brimonidin, nejsou doporučována při akutních projevech zánětu předního segmentu oka.
Prognóza GPA je závislá na rozsahu onemocnění – četnosti a tíži orgánového postižení. GPA je charakterizována systémovou vaskulitidou, ale existují značné rozdíly mezi limitovanou a generalizovanou formou choroby. Prognóza GPA bez léčby je infaustní, pacient umírá do dvou let, většinou pro selhání ledvin [56]. Imunosupresivní léčba výrazně zlepšila prognózu tohoto život a zrak ohrožujícího onemocnění, až 80 % pacientů přežívá 10 let [34,38,39,43]. Pacienti s limitovanou formou GPA jsou v počátku onemocnění mladší než pacienti s generalizovanou formou a častěji jsou to ženy. Izolovaná forma přechází často v chronický zánět horních cest dýchacích se známkami destrukce, jako je např. sedlovitý nos. Tito pacienti mají lepší prognózu s dobrou odpovědí na imunosupresivní léčbu [40]. Kompletní remise GPA je uváděna asi u 70 % pacientů, ale u poloviny z nich dojde k relapsu při snížení udržovací léčby nebo po jejím ukončení, někdy o i mnoho let později. Obnovení nebo navýšení léčby zklidní klinické projevy GPA, ale remise nemusí být plná [24,29,32]. Léčba by měla být vždy výsledkem mezioborové spolupráce. Prognóza očních projevů GPA a výsledný vízus jsou závislé na lokalizaci zánětu a tíži postižení očních tkání. Za nejčastější a nejzávažnější zrak ohrožující projevy GPA jsou považovány postižení orbity a sklerokeratitida. Postižení sítnice či terče zrakového nervu zanechávají často ireverzibilní poškození i v remisi celkové choroby. Riziko snížení vízu je vyšší u pacientů s obtížněji navozenou remisí, s vyšším počtem relapsů a refrakterním typem GPA [17,24]. Recidivy očních projevů GPA mohou být první příznakem relapsu zánětů i jiných orgánů postižených touto chorobou, oftalmolog ve spolupráci se specialisty daných oborů celkovou léčbu upravuje dle klinických projevů GPA a tolerance léčiva pacientem. Pro určení správné a včasné diagnózy a vedení cílené léčby GPA je nezbytná dobrá multidisciplinární spolupráce s nefrologem, imunologem, revmatologem, otorhinolaryngologem, dermatologem, gastroenterologem, radiologem, histopatologem [46].
Souhrn:
GPA je vzácné, život a zrak ohrožující onemocnění. Včasná diagnóza a správná cílená léčba může navodit dlouhodobou remisi choroby, předejít komplikacím souvisejícím s touto chorobou a zlepšit kvalitu života pacientů. Oční projevy GPA mohou být prvním příznakem dosud neprojevené či nediagnostikované celkové choroby a umožňují tak oftalmologovi podílet se na diagnostice této nemoci. Skleritida je nejčastějším očním projevem GPA a zánětlivá infiltrace tkání orbity je nejzávažnějším zrak ohrožujícím projevem této systémové choroby.
Děkujeme za laskavou spolupráci při diagnostice a léčbě pacientů s GPA sledovaných na Oční klinice 1. LF UK a VFN v Praze panu prim. MUDr. J. Žabkovi, CSc., B. Braun-Avitum Praha a kolektivu lékařů Nefrologické kliniky 1. LF UK a VFN v Praze.
Části práce byly předneseny na sympoziu XVII. Jihočeské Timrovy dny v Hluboké nad Vltavou v květnu 2017 a na Klinickém semináři Oční kliniky 1. LF UK a VFN a v Praze v březnu 2018.
Autoři práce prohlašují, že vznik a téma odborného sdělení není ve střetu zájmu a není podpořeno farmaceutickou firmou.
Do redakce doručeno dne: 4. 6. 2018
Do tisku přijato dne: 31. 10. 2018
doc. MUDr. Eva Říhová, CSc.
Oční klinika 1. LF UK a VFN v Praze
U Nemocnice 2
128 08 Praha 2
Sources
1. Alloway, JA., Cupps, TR.: High–dose methylprednisolone for retro-orbital Wegener‘s granulomatosis. J Rheumatol. 20(4); 1993 : 752-754.
2. Banerjee, SH., Grayson, PC.: Vasculitis Around the World: Epidemiologic insights into causality and a need for global partnership. J Rheumatol. 44(2); 2017 : 136-139.
3. Bertelmann, E., Liekfeld, A., Pleyer, U. et al.: Cytomegalovirus retinitis in Wegener‘s granulomatosis: case report and review of the literature. Acta Ophthalmol Scand. 83(2); 2005 : 258-261.
4. Blaise, P., Robe-Collignon, N., Andris, C. et al.: Wegener granulomatosis and posterior ischemic optic neuropathy. Eur J Intern Med. 18(4); 2007 : 326-327.
5. Charles, SJ., Meyer, PA., Watson, PG.: Diagnosis and management of systemic Wegener‘s granulomatosis presenting with anterior ocular inflammatory disease. Br J Ophthalmol. 75(4); 1991 : 201-207.
6. Charlier, C., Henegar, C., Launay, O. et al.: Risk factors for major infections in Wegener granulomatosis: analysis of 113 patients. Ann Rheum Dis. 68(5); 2009 : 658-663.
7. Chiquet, C., Lumbroso, L., Denis, P. et al.: Acute posterior multifocal placoid pigment epitheliopathy associated with Wegener‘s granulomatosis. Retina 19(4); 1999 : 309-313.
8. Cocho, L., Gonzales-Gonzales, LA., Molina-Prat, N. et al.: Scleritis in patients with granulomatosis with polyangiitis (Wegener). Br J Ophthalm. 100(8); 2016 : 1062-1065.
9. Courcoutsakis, NA., Langford, CA., Sneller, MC. et al.: Orbital involvement in Wegener‘s granulomatosis: MR findings in 12 patients. J Comput Assist Tomogr. 21(3); 1997 : 452-458.
10. Cutler, WM., Blatt, IM.: The ocular manifestations of lethal midline granuloma (Wegener‘s granulomatosis). Am J Ophthalmol. 42(1); 1956 : 21-35.
11. de Silva, DJ., Cole, C., Luthert, P. et al.: Masked orbital abscess in Wegener‘s granulomatosis. Eye 21(2); 2007 : 246-248.
12. Fortney, AC., Chodosh, J.: Conjunctival ulceration in recurrent Wegener granulomatosis. Cornea 21(6); 2002 : 623-624.
13. Foster, CS.: Peripheral ulcerative keratitis in Melki SA., Fava LA.: Cornea and refractery atlas of clinical wisdom, chapter 17, 2011, SLACK corp, print, USA.
14. Freeman, WR., Stern, WH., Gross, JG. et al.: Pathologic observations made by retinal biopsy. Retina. 10(3); 1990 : 195-204.
15. Freidlin, J., Wong, I.G., Acharya, N.: Rituximab treatment for peripheral ulcerative keratitis associated with Wegener‘s granulomatosis. Br J Ophthalmol. 91(10); 2007 : 1414-1416.
16. Greco, A., Marinelli, C., Fusconi, M. et al.: Clinic manifestations in granulomatosis with polyangiitis. Int J Immunopath and Pharmacol. 29(2); 2016 : 151-159.
17. Harper, SL., Letko, E., Samson, CM. et al.: Wegener‘s granulomatosis: the relationship between ocular and systemic disease. J Rheumatol. 28(5); 2001 : 1025-1032.
18. Hejcmanová, D., Koblížek, V., Ruta, J.: Wegenerova granulomatóza - kazuistické sdělení, Cesk Slov Oftalmol, 63(1); 2007 : 55-62.
19. Hoffman, GS., Kerr, GS., Leavitt., RY. et al.: Wegener‘s granulomatosis: an analysis of 158 patients. Ann Intern Med. 116(6); 1992 : 488-498.
20. Huong du, LT., Tran, TH., Piette, JC.: Granulomatous uveitis revealing Wegener‘s granulomatosis. J Rheumatol. 33(6); 2006 : 1209-1210.
21. Jabs, DA., Mudun A., Dunn J.P. et al.: Episcleritis and scleritis: clinical features and treatment results. Am J Ophthalmol. 130(4); 2000 : 469–476.
22. Kalina, PH., Lie, JT., Campbell. RJ. et al.: Diagnostic value and limitations or orbital biopsy in Wegener‘s granulomatosis. Ophthalmology 99(1); 1992 : 120-124.
23. Kamei, M., Yasuhara, T., Tei, M. et al.: Vitreous hemorrhage from a ciliary granuloma associated with Wegener granulomatosis. Am J Ophthalmol. 132(6); 2001 : 924-926.
24. Kubaisi, B., Abu Samra, K., Foster, CS.: Granulomatosis with polyangiitis: an apdated rewiev of ocular disease manifestation. Intractable Rare Dis Res. 5(2); 2016 : 61-69.
25. Lasave, AF., You, C., Ma, L. et al.: Long-term outcomes of rituximab therapy in patients with noninfectious posterior uveitis refractory to conventional immunosuppressive therapy. Retina 38(2); 2018 : 395-402.
26. Leavitt, RY., Fauci, AS., Bloch, DA. et al.: The American College of Rheumatology 1990 criteria for the diagnosis of Wegener‘s granulomatosis. Arthritis Rheum. 33(8); 1990 : 1101-1107.
27. Levi, M., Kodsi, SR., Rubin, SE. et al.: Ocular involvement as the initial manifestation of Wegener‘s granulomatosis in children. J AAPOS. 12(1); 2008 : 94-96.
28. Lida, T., Spaide, RF., Kantor, J.: Retinal and choroidal arterial occlusion in Wegener‘s granulomatosis. Am J Ophthalmol. 133(1); 2002 : 151-152.
29. Lošťáková, V., Kolek, V., Vašáková, M. et al.: Granulomatóza s polyangiitidou (Doporučený postup při diagnostice, terapii a sledování vývoje onemocnění) [KAP. 6.4] Sekce intersticiálních plicních procesů ČPFS – dostupné na www.pneumologie.cz
30. Lukáš, J., Hroboň, M., Rambousek, P. et al.: Lokalizovaná forma Wegenerovy granulomatozy s postižením ucha a oka. Otolaryng Foniat. 52(1); 2003 : 29-32.
31. Lutalo, MK., DCruz, DP.: Diagnosis and classification of granulomatosis with polyangiitis. J Autoimun. 48-49; 2014 : 94-98.
32. Martínková, K., Valkovský, I., Handlos P.: Granulomatóza s polyangiitidou. Interni Med. 16(5); 2014 : 199-201.
33. Masuda, T., Izumi, Y., Takeshita, H. et al.: Granulomatosis with polyangiitis presenting as a choroidal tumor. Case Rep Rheumatol. 2015; 2015 : 271823. doi: 10.1155/2015/271823.
34. McNab, AA.: The 2017 Doyne Lecture: the orbit as a window to systemic disease. Eye (Lond). 32(2); 2018 : 248-261.
35. Messme, EM., Foster, CS.: Vasculitic peripheral ulcerative keratitis. Surv Ophthalmol. 43(5); 1999 : 379-396.
36. Metee, A., Kimyon, S., Saygili, O. et al.: Bilateral acute angle-closure glaucoma as a first presentation of granulomatosis with polyangiitis (Wegener’s) Arq Bras Oftalmol. 79(5); 2016 : 336-338.
37. Meyer, PA., Watson, PG., Franks, W. et al.: Pulsed immunosuppressive therapy in the treatment of immunologically induced corneal and scleral disease. Eye (Lond) 1(4); 1987 : 487-495.
38. Mohammad, AJ., Jacobsson LT., Westman, KW. et al.: Incidence and survival rates in Wegener‘s granulomatosis, microscopic polyangiitis, Churg-Strauss syndrome and polyarteritis nodosa. Rheumatology (Oxford) 48(12); 2009 : 1560-1565.
39. Muller, K., Lin, JH.: Orbital granulomatisis with polyangiitis: clinical and pathological findings. Arch Pathol Lab Med. 138(8); 2014 : 1110-1114.
40. Eloy, P., Leruth, E., Bertrand, B. et al.: Successful endonasal dacryocystorhinostomy in a patient with Wegener‘s granulomatosis. Clin Ophthalmol. 3; 2009 : 651-656.
41. O‘Donoghue, E., Lightman, S., Tuft, S. et al.: Surgically induced necrotising sclerokeratitis (SINS) -precipitating factors and response to treatment. Br J Ophthalmol. 76(1); 1992 : 17-21.
42. Pagnoux, C., Mahr, A., Hamidou, MA. et al.: Azathioprine or methotrexat maintenance for ANCA-associated vasculitis. N Engl J Med. 359(26); 2008 : 2790-2803.
43. Pakalniskis, MG., Berg, AD., Policeni, BA.: The Many Faces of granulomatosis with polyangiitis: A review of the head and neck imaging manifestations. Am J Roentgenol. 205(6); 2015 : 619-629.
44. Phillip, R., Luqmani. R.: Mortality in systemic vasculitis: a systematic review. Clin Exp Rheumatol. 26(5); 2008 : 94-104.
45. Pakrou, N., Selva, D., Leibovitch, I.: Wegener‘s granulomatosis: ophthalmic manifestations and management. Semin Arthritis Rheum. 35(5); 2006 : 284-292.
46. Reinhold-Keller, E., Beuge, N., Latza, U. et An indesciplinary approach to the care of patients with Wegenerś granulomatosis. Long-Term Outcome in 155 Patients. Arthritis Rheum. 43(5); 2000 : 1021-1032.
47. Říhová, E., Havlíková, M., Michalová, K. et al.: Diagnóza a léčba Wegenerovy granulomatózy na podkladě očních změn. Cesk Slov Oftalmol. 53(4); 1997 : 223-228.
48. Salam, A., Meligonis, G., Malhotra, R.: Superior oblique myositis as an early feature of orbital Wegener‘s granulomatosis. Orbit 27(3); 2008 : 203-206.
49. Santiago, YM., Fay, A.: Wegener‘s granulomatosis of the orbit: a review of clinical features and updates in diagnosis and treatment. Semin Ophthalmol. 26(4-5); 2011 : 349-355.
50. Seo, P., Specks, U., Keogh, KA.: Efficacy of rituximab in limited Wegener‘s granulomatosis with refractory granulomatous manifestations. J Rheumatol. 35(10); 2008 : 2017-2023.
51. Soukiasian, SH.: Wegeners granulomatosis in Foster et Vitale: Diagnosis and treatment of uveitis. Saunders company, Philadelphia, 2011 : 661-672.
52. Svozílková, P., Říhová, E., Brichová, M. et al.: Infliximab v léčbě Wegenerovy granulomatózy. Cesk Slov Oftalmol, 62(4); 2006 : 280-286.
53. Talar-Williams, C., Sneller, M.C., Langford, C.A. et al.: Orbital socket contracture: a complication of inflammatory orbital disease in patients with Wegener‘s granulomatosis. Br J Ophthalmol. 89(4); 2005 : 493-497.
54. Tarabishy, AB., Schulte, M., Papliodis, GN. et al.: Wegeners granulomatosis: Clinical manifestations, differential diagnosis and management of ocular and systemic disease. Surv Ophthalmol. 55(5); 2010 : 429-443.
55. Taylor SR., Salama, AD., Joshi, L et al.: Rituximab is effective in the treatment of refractory ophthalmic Wegener‘s granulomatosis. Arthritis Rheum. 60(5); 2009 : 1540-47.
56. Vogiatzis, KV.: Bilateral blindness due to necrotizing scleritis in a case of Wegener‘s granulomatosis. Ann Ophthalmol. 15(2); 1983 : 185-188.
57. Wang, M., Khurana, RN., Sadda, SR.: Central retinal vein occlusion in Wegener‘s granulomatosis without retinal vasculitis. Br J Ophthalmol. 90(11); 2006 : 1435-1436.
58. WGET Research Group: Design of the Wegener‘s granulomatosis etanercept trial (WGET). Control Clin Trials. 23(4); 2002 : 450-468.
59. Woo, TL., Francis, IC., Wilcsek, GA. et al.: Australasian orbital and adnexal Wegener‘s granulomatosis. Ophthalmology 108(9); 2001 : 1535-43.
60. You, C., Sahawneh, HF., Ma, L., Kubaisi, B. et al.: A review and update on orphan drugs for the treatment of noninfectious uveitis. Clin Ophthalmol. 11; 2017 : 257-265.
61. You, C., Ma, L., Lasave, AF. et al.: Rituximab induction and maintenance treatment in patients with scleritis and granulomatosis with polyangiitis (Wegener‘s). Ocul Immunol Inflamm. 19; 2017 : 1-8.
Labels
OphthalmologyArticle was published in
Czech and Slovak Ophthalmology

2018 Issue 5
Most read in this issue
- OČNÍ PROJEVY GRANULOMATÓZY S POLYANGIITIDOU
- MERKELŮV KARCINOM VÍČEK (KLINICKO - HISTOLOGICKÁ STUDIE)
- PACHYCHOROIDNÍ CHOROBA MAKULY – KAZUISTIKA
- PILOTNÍ VÝSLEDKY IMPLANTACE NOVÉ HYDROFOBNÍ NITROOČNÍ ČOČKY ZEISS LUCIA 611P