
Stability testing of alginate-chitosan films
Authors:
Miloslava Rabišková; Kateřina Dvořáčková; Lenka Kofroňová
Authors‘ workplace:
Department of Pharmaceutics, Faculty of Pharmacy, University of Veterinary and Pharmaceutical Sciences Brno
Published in:
Čes. slov. Farm., 2012; 61, 26-33
Category:
Original Articles
Overview
Pellets containing rutin prepared by the extrusion/spheronization method were coated with sodium alginate-chitosan film. Important quality parameters in the pellets before coating were determined, and after coating the dissolution profiles of the drug were evaluated in dissolution media of the pH corresponding to the conditions in the gastrointestinal tract. Samples of coated pellets were located in the boxes for stability testing under different conditions, i.e. 25 °C and 60% of relative humidity (RH); 30 °C and 65% RH and 40 °C and 75% RH. After 1, 3, 6, 9 and 12 months (or 1, 3 and 6 months), the dissolution test was repeated and compared with the original profiles using similarity factors. All similarity factor values above 50 indicate excellent stability of alginate-chitosan films.
Keywords:
alginate • chitosan • films • colon drug delivery • stability
Introduction
Many natural polysaccharides, such as chondroitin sulphate, pectin, alginates, dextran, etc., and also some carriers derived from them, for instance chitosan, have been investigated in recent years as promising pharmaceutical excipients for their potential to provide colon-specific drug delivery or to form pH sensitive gels. Alginate and chitosan are finding widespread applications not only in pharmaceutical but also in food industry.
Alginate is extensively used as a thickener, emulsifier and stabilizer. Commercially available alginates are extracted from several species of brown algae, e.g. Laminaria hyperborea, Ascophyllum nodosum, and Macrocystis pyrifera1). Alginate exists as a mixed salt of various cations (Mg2+, Sr2+, Ba2+, Na+) found in seawater2).
Alginate is a water-soluble linear polysaccharide composed of alternating blocks of 1–4 linked α-L-guluronic and ß-D-mannuronic acid residues (Fig. 1). Particular shapes of the monomers, their modes of linkage in the polymer, the composition and extent of the sequences, and the molecular weight determine the physical properties of alginates.

The most important property of alginates is their ability to form gels by reaction with divalent cations such as Ca2+, Sr2+, Zn2+ or Ba2+. Monovalent cations and Mg2+ ions do not induce gelation3). The gelation and cross-linking of the polymers are mainly achieved by the exchange of sodium ions from guluronic acids with divalent cations, and the stacking of these guluronic groups to form the characteristic egg-box structure. The divalent cations bind to the α-L-guluronic acid blocks in a highly cooperative manner and the size of the cooperative unit is more than 20 monomers. Each alginate chain dimerizes to form junctions with many other chains and as a result gel networks are formed4). Depending on the amount of divalent cation (e.g. calcium) present in the system, either temporary highly viscous solutions at low levels, or permanent associations of the chains at higher levels can be formed. Chemical structure, molecular size and the cation have significant influence on gel properties, such as porosity, swelling behavior, gel strength, biocompatibility, biodegradability and stability5).
Oral administration of alginate was reported to be safe and non-toxic. Its mucoadhesive properties may be utilized as a potential carrier for drug delivery to mucosal tissues such as the gastrointestinal (GI) tract. Due to the adherence of alginate particles to the mucosal tissues, their transit time is delayed and the included drug is localized on the mucosal surfaces. This improves drug bioavailability and effectiveness. Drug release from alginate based dosage forms at low pH is significantly reduced. In the gastric fluid, hydrated sodium alginate is converted into insoluble alginic acid. Once passed into the higher pH of the intestinal tract, alginic acid is transformed to a soluble viscous layer. This pH-dependent behavior of alginate can be utilized in delayed release formulations. However, the rapid dissolution of alginate matrices in the higher pH ranges may result in burst drug release6). The above-mentioned properties make alginate a promising excipient for oral drug delivery.
Chitosan as such is rare in nature, except in certain fungi7). Usually it is obtained by N-deacetylation from chitin. The most commonly obtained form of chitosan is the α-chitosan from crustacean chitin obtained from crab - and shrimp shell wastes8). Production of chitosan from these sources is inexpensive and easy.
Chitosan is a linear semi synthetic polysaccharide composed of D-glucosamine and N-acetyl-D--glucosamine linked with ß-1→4 glycosidic bonds8) (Fig. 2). It is a cationic polymer which has been found to be highly biocompatible and biodegradable as it is metabolized by certain human enzymes5). Many commercially available chitosans exhibit fairly good mucoadhesive properties in vitro9). In addition to the adhesion by hydration, many other mechanisms, such as hydrogen bonding and ionic interactions, might also be involved. Ionic interactions are created between positively charged amino groups in chitosan and the negatively charged mucus gel layer10). The interactions are strong at acidic and slightly acidic pH levels, at which the charge density of chitosan is high11). The increase in the molecular weight of chitosan results in stronger adhesion9). He et al.12) showed that amounts of chitosan microspheres adhering to the intestine were greatest when the density of cross-linking of chitosan was least, i.e. when the number of free amino groups in chitosan was greatest. This finding also suggests that the adhesive properties of chitosan should become more marked as the degree of deacetylation increases, and cross-linking reduces the mucoadhesive effects of chitosan5). In recent years, chitosan has attracted a lot of attention as a potential absorption enhancer across mucosal epithelia due to the opening of epithelial tight junctions13). Chitosan is able to enhance the paracellular route of absorption, which is important for the transport of hydrophilic compounds such as therapeutic peptides and antisense oligonucleotides across the membrane. The mechanism underlying this permeation enhancing effect seems to be based on the positive charges of the polymer, which interact with the cell membrane resulting in a structural reorganization of tight junction-associated proteins14). The ability of chitosan to work as an absorption enhancer was proven on Caco-2 cells, which serve as a model of intestinal epithelium15, 16), as well as in in vitro experiments on nasal, buccal, vaginal and urinary bladder mucosa of different animals17–21). Chitosan exhibits a pH-sensitive behavior as a weak poly-base due to the large quantities of amino groups on its chain. It dissolves easily at low pH while it is insoluble at higher pH ranges. The mechanism of pH-sensitive swelling involves the protonation of amino groups of chitosan under low pH conditions leading to chain repulsion, diffusion of proton and counter ions together with water inside the gel and dissociation of secondary interactions22). This property causes a limitation to deliver drugs into the intestine. Therefore, many modifications were investigated to improve the stability of chitosan in the stomach, e.g. thiolated chitosan23), trimethylated chitosan24), carboxymethyl chitosan25).

Alginate-chitosan complexes can be of important use in oral drug delivery systems. Alginate has the property of shrinking in low pH and getting dissolved in higher pH, whereas chitosan dissolves in low pH and is insoluble in higher pH ranges. In view of these limitations encountered in pure alginate and chitosan, the concept of alginate-chitosan polyelectrolyte complexes gained acceptance26). Upon mixing, the carboxyl residues of alginate and the amino groups of chitosan interact to form a polyelectrolyte complex. Properties of this complex are affected by the composition of the alginate used and the molecular weight and deacetylation degree of chitosan. Complexation of chitosan with alginate decreases the leakage of the included drugs. Thus, the easy solubility of chitosan in low pH is prevented by the alginate network since alginate is insoluble in low pH conditions. The possible dissolution of alginate at higher pH is prevented by chitosan which is stable at higher pH values5). As both of these polymers are degraded by the enzymes of the colonic microbiota, their complex as a carrier could be an interesting possibility to deliver the drugs into the colon.
In our experiment, alginate/chitosan coated pellets containing rutin, chitosan and microcrystalline cellulose intended for the local therapy of inflammatory bowel disease are presented. The extrusion/spheronization process, a well established and reproducible pelletization method, was used to prepare pellets which were subsequently coated with sodium alginate/chitosan coating to prevent chitosan pellets from the dissolution under acidic conditions in the upper GI tract. Characteristics of uncoated pellets and coated pellets including their dissolution profiles in vitro were determined and samples of coated pellets were checked for their stability.
Experimental section
Materials
Rutin as the active ingredient and sodium alginate as the coating excipient were purchased from Sigma-Aldrich (Steiheim, Germany). Microcrystalline cellulose (MCC) Avicel® PH 101 as the spheronization enhancer was supplied by FMC (Cork, Ireland), chitosan of deacetylation degree 82%, m.w. 880 kDa and viscosity 20000 cPs by Sigma-Aldrich (Praha, Czech Republic), and acetic acid 99% by Penta (Chrudim, Czech Republic). Phosphate buffers of pH 3.0, 6.8 and 6.0, and acetic buffer of pH 4.0 were prepared according to pharmacopoeia recommendations. The active ingredient and all excipients were of pharmaceutical grade.
Pellet preparation
Dry powder mixture (100 g) containing of 30% of rutin, 45% of chitosan and 25% of MCC, was homogenized in a mixer (Tefal Kaleo, Rumilly, France) for 5 minutes and then wetted with 111 g of 0.25% acetic acid solution to dissolve chitosan. One screw extruder (Pharmex 35T, Wyss & Probst, Ettlingen, Germany) fitted with one axial located die (die thickness of 1 mm, extrusion perforations of 0.8 mm in diameter) was used to form the extrudate. The wetted mass was fed through a hopper on a rotating screw. The extruder operated at a constant speed of 110 rpm. The prepared extrudate was subsequently spheronized in a radial plate spheronizer (Pharmex 35T, Wyss & Probst, Ettlingen, Germany) with a 23-cm diameter serrated plate at a rotating speed of 640 rpm for 15 minutes. Formed pellets were transferred into a ventilated oven (Horo, Ostfildern, Germany) and dried at 40 °C for 3 hours. Ten pellet batches were prepared to verify the reproducibility of the process.
Coating of pellets
Pellets (760 g; size of 0.5–1.0 mm) were charged into the process chamber of a Wurster type fluid bed unit (Aeromatic MP-1, Aeromatic-Fielder, Bubendorf, Switzerland) and heated up to 45 °C for 5 minutes. The coating solution contained 2.6% of sodium alginate, 0.14% of chitosan, 8.95% of solution of acetic acid (10%), and 88.31% of water and was prepared by mixing of chitosan solution in acetic acid and sodium alginate solution in water. The obtained solution was sprayed onto the pellets using a 1.0 mm diameter spray nozzle and a peristaltic pump with 170 kPa of atomization pressure. The spray rate was kept at 20 g/min and the inlet air temperature at 55 °C. The amount of final coat represented 18% of the total pellets mass. Pellets were dried at the temperature of 45 °C for 10 min. Dry coating contained 95% of sodium alginate and 5% of chitosan.
Pellet characterization
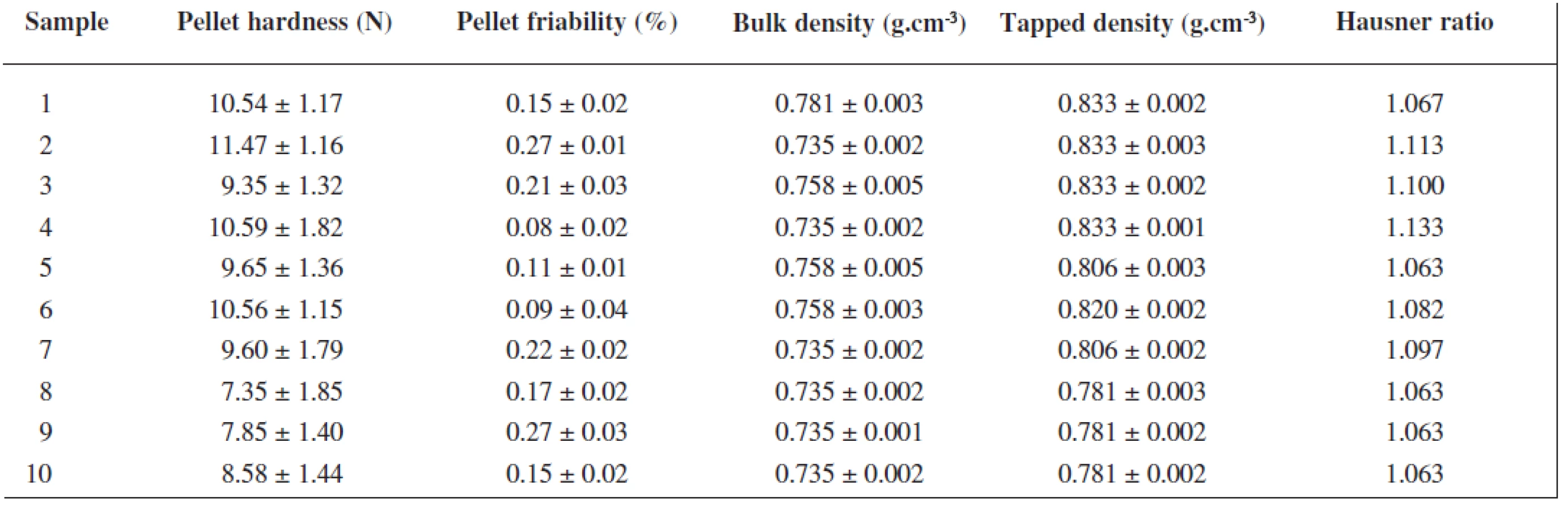
Pellet size, size distribution and their flow properties were evaluated, and then rutin content, pellet sphericity, their intraparticular porosity, hardness and friability were determined in the uncoated pellet sample fraction of 0.8–1.0 mm. Particle size and size distribution were measured by sieve analysis using a set of stainless steel sieves with apertures ranging in between of 125–2000 μm (Retsch, AS 200, Haan, Germany). The results were expressed as a percentage of the weight retained on each sieve. Pellet flow properties as Hausner ratio values were calculated from bulk and tapped densities according to the pharmacopoeia recommendations (n = 3). Pellet intraparticular porosity was determined from the difference of pycnometric density of pellets and that of initial powder mixture according to Ph. Eur. recommendations (helium pycnometr, Pycnomatic-ATC, Porotec GmbH, Germany). For pellet sphericity, the image analysis (Leco IA, Leco Instruments, St. Joseph, USA) in 200 particle sample was performed to characterize pellet shape and evaluate their sphericity using an optical microscope (DN 45, Lambda, Prague, Czech Republic) connected with a CCD camera (Alphaphot, Nikon, Tokyo, Japan)27). The hardness of ten randomly selected pellets was tested in a Tablet Hardness & Compression tester (Engineering System, Nottingham, United Kingdom) equipped with a C5 cell for pellet evaluation. The average hardness value and ± standard deviation were calculated. For friability testing, 10 g of pellets were precisely weighed, placed into a stainless steel drum of the friabilator (Erweka TAR 10, Ensenstam, Germany) together with 200 pieces of 4 mm glass beads and rotated for 10 minutes at 20 rpm. The dust was thereafter removed and pellets were reweighed. The friability was expressed as the percentage of the weight loss after agitation. Rutin content in the pellets was determined spectrophotometrically at a wavelength of 360 nm (Specord® 205, Analytik Jena, Jena, Germany). Firstly, uncoated pellets were pulverized in a porcelain mortar with a pestle. Ten mg of powdered pellets were precisely weighed, transferred to a 100 ml volumetric flask and dissolved in phosphate buffer of pH 6.8. The actual drug content was calculated using a calibration curve. The measurements of pellet friability and drug content were repeated three times and the results were expressed as an arithmetic mean ± standard deviation.
In vitro drug release
The dissolution test was examined using a basket dissolution method at a rotation speed of 100 rpm at 37.0 ± 0.5 °C (a Sotax AT 7 Smart on-line, Donau Lab, Basel, Switzerland). Dissolution method of changing pH values and thus better corresponding to the conditions in the GI tract was provided. For the dosage forms showing very slow drug release or those intended for the drug release in the colon, it is necessary to consider lower pH value when comparing with small intestine caused by acidic fermentation products of bacterial microbiota around pH 5.528). Even lower pH value of 4.7 in this part of the GI tract was reported in patients suffering from inflammatory bowel disease (IBD) by Raimundo et al.29) and ranging between 2.3–3.4 in the study of Fallingborg et al.30).
To mimick the real conditions in GI tract of patients with inflamatory bowel disease, dissolution test with changing pH values of the buffers for reported times30–33) was used to evaluate our coated pellets:
0–2 h: samples of coated pellets corresponding to the dose of 15 mg of rutin in a basket were placed into 500 mL of phosphate buffer pH 3.0 (human stomach value in fed state and also rat stomach value with respect to following test in vivo on rats)34);
2–5 h: 2.1 g of Na3PO4 . 12 H2O were added to this dissolution medium, increasing thus pH value to 6.8 simulating small intestine;
5.0–22 h: considering the pH value of 4–6 in patients with IBD, these values were selected in the remaining time up to 22 h, i.e. either phosphate buffer pH 6.0 or acetic buffer pH 4.0. The buffers needed to be exchanged after 5 h of the testing. Samples were withdrawn every 30 min in 0–5.5 h and then every hour.
Released drug amount was measured spectrophotometrically at the wavelength of 360 nm (Lambda 25, Perkin Elmer Instruments, Shelton, USA). All experiments were performed in triplicate and results are expressed as the mean ± SD of the active substance in %, dissolved at the given sampling time.
Stability testing
Samples of pellets coated with alginate/chitosan were placed into stability boxes (Binder, Tuttlingen, Germany) under 25 °C and 60% (25/60) of relative humidity (RH); 30 °C and 65% RH (30/65); 40 °C and 75% RH (40/75). Stability tests were provided for 6 months (40/75) and for 12 months (25/60; 30/65), respectively. At the times of 0, 1, 3, 6, 9 and 12 months (or 0, 1, 3 and 6 months), samples were withdrawn and their dissolution profiles were determined using pH changing dissolution method described above, and compared.
Statistical analysis
The results were expressed as mean values ± S.D. For analysis of statistical significance, the Student’s t-test was applied for all pairwise comparison. In all cases, P < 0.05 was considered to be significant.
Results and discussion
Pellets Evaluation
The extrusion/spheronization method was used to prepare rutin/chitosan/MCC pellets and acetic acid was decided to act as the wetting agent and the solvent for partial dissolution of chitosan to obtain matrix pellets35). Pellets shape and size are significantly depending on the moisture content of the extrudate36), therefore the optimum amount of acetic acid was evaluated experimentally to be 111 g for 100 g of the powder mixture, thus forming a plastic mass appropriate for extrusion (Table 1). Pellet size distribution was found in a narrow interval, i.e. 0.5–1.0 mm (66.75–93.45 %), which is typical for extrusion/spheronization method. It showed two main fractions, i.e. 0.5 – 0.8 mm (21.69–62.30 %) and 0.8–1.0 mm (31.15–47.61 %), giving the mean diameter of 0.74–0.96 mm (Table 2). The pellet fraction (0.80–1.00 mm) was used for further evaluation. Pellets shape showed sphericity values 0.8124–0.8460 (Table 2) which are considered as suitable37). Rutin content in pellets was found to be 28.12–28.50%, i.e. 93.74–95.0% of its theoretical value. Pellet intraparticular porosity was very low (0.94–3.99%), indicating good compactness of the used substances. Mechanical properties of pellets, i.e. pellet friability (0.08–0.27%) and their hardness (7.35–11.47 N) (Table 3), indicated good pellet quality sufficient to withstand further processing. These pellet characteristics together with a spherical shape and smooth surface are important in the following coating process and directly influence the coating quality. Flow properties of pellets were very good with Hausner ratio values of 1.063–1.133 corresponding to excellent flow (Table 3). For the coated pellets, rutin represented 25% of their total weight. A sample of coated pellets is shown in Figure 3. From the cross-section of a coated pellet, the thickness of the coat was approximately 28 μm. The glinting parts are chitosan particles undissolved in acetic acid. Thus the pellet matrix was composed of rutin, microcrystalline cellulose and partially dissolved chitosan particles (Fig. 3).



Dissolution Tests
After pellets characterization, in vitro dissolution profiles of coated pellets were determined. The dissolution test involved buffers with changing pH values according to the in vivo conditions and recommended in the literature, i.e. 2 h in pH 3.0 following 3 h in pH 6.8. Then the dissolution medium was changed to obtain a lower pH value found in some IBD patients for the remaining time, i.e. 4.0 and 6.0, respectively. The obtained results are depicted in Figure 4.
At the time 0, only 12% of rutin after 2 h, and 15 % of rutin in the following 3 h were released, respectively. This finding corresponds to the requested demands, i.e. low drug dissolution under upper GI tract and small intestine conditions. These results also prove a good protective role of alginate/chitosan coating. When pH was changed to 4.0, rutin release increased to about 70% (66.13%) at the 6th h of dissolution testing (Fig. 4a), while in pH 6.0, rutin release was slow with the maximum of 55.56% at the end of the testing (Fig. 4b). This different behavior could be explained as follows. Sodium alginate is practically insoluble in aqueous acidic solutions with pH less than 3, but slowly soluble in water, forming a viscous colloidal solution38). The pH change to 6.8 could start slow sodium alginate dissolution. At the end of the 5th h of the testing, the coating was probably dissolved and only the chitosan/MCC matrix, at this pH insoluble, did not allow fast drug release39). Afterwards the change to pH 4.0 caused chitosan dissolution and enhanced rutin release. Conversely, when phosphate buffer of pH 6.0 was applied, chitosan/MCC matrix did not dissolve causing thus prolonged rutin release within all the time of dissolution testing. These results were expected as for real simulation of colon conditions the enzymes produced by the colonic microbiota decomposing chitosan (ß-glusidase) and probably also alginate (alginate lyase40)) would be needed. Thus when reaching the colon, the rest of the coating if any, and the chitosan matrix would be decomposed by the enzymes and the drug released.


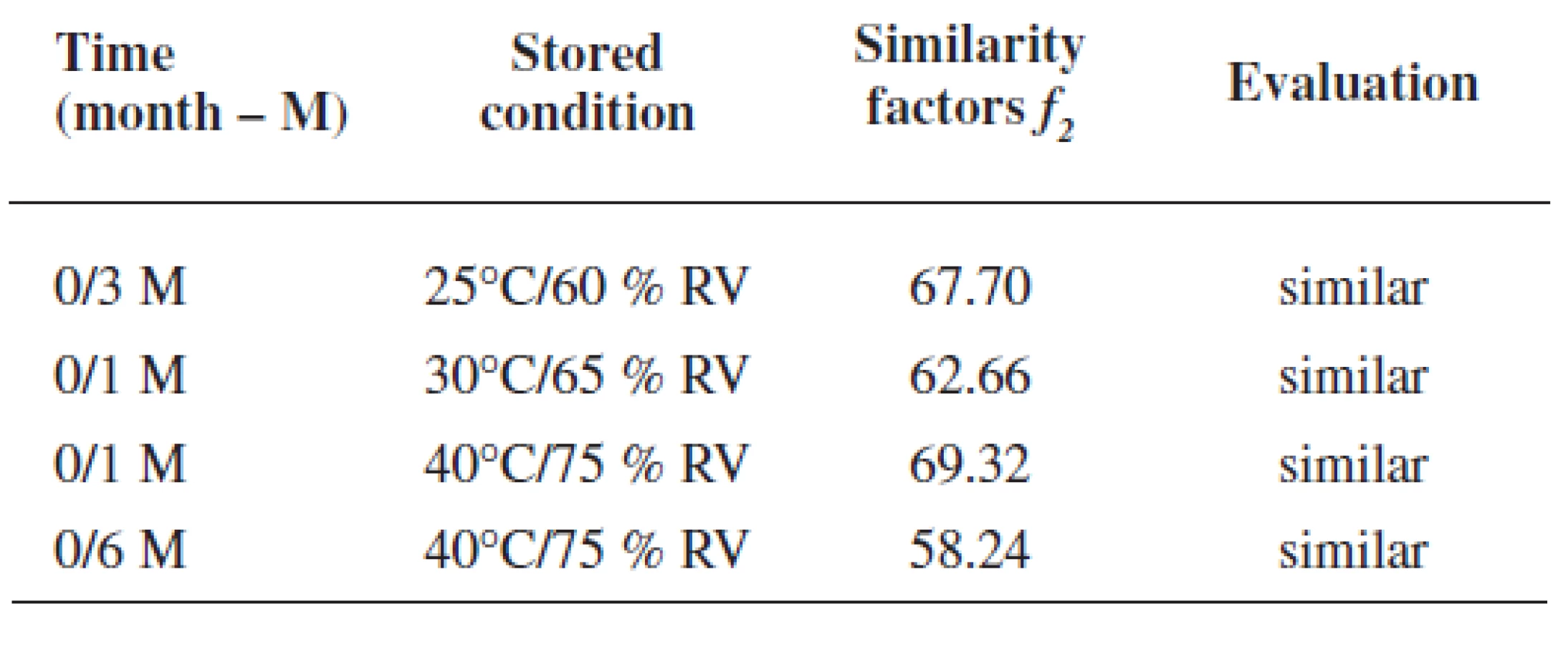
Rutin dissolution profiles from coated pellets at the time 0 were compared with dissolution profiles in stability tests under different conditions. Figure 4 presents rutin dissolution profiles under the conditions of 25 °C and 60% RH within 12 months. As it can be seen from Figure 4a, only slightly lower values were observed in the first 5 hrs and on the contrary, a higher rutin amount was measured after this time of dissolution testing. In Figure 4b, also small insignificant differences in dissolution profiles were found, i.e. less than 7%. Similar differences in rutin release were determined also under accelerated stability conditions as shown in Figure 5 for 30 °C and 65% RH and in Fig. 6 for 40 °C and 75% RH, respectively. The maximum difference was found in the stability regimen 40/75 in Figure 6b using the final buffer pH 6.0: while slightly slower rutin release was found after 1 month of the testing with the difference -4.64 %, faster drug release was determined at the end of the experiment, i.e. after 6 months, with the difference value of +8.21 %, respectively, which together gives the difference of 12.85% after 14 hrs of the dissolution testing. Similarity factors could be calculated only for dissolution profiles using the final buffer pH 6.0 (dissolution profiles depicted in Fig. 4b-6b). For this purpose the most different dissolution profile from the original one (time 0) was chosen from each regimen, i.e. 25/60; 30/65 and 40/75, respectively, and the values are presented in Table 4. All f2 values were higher than 50% proving thus the similarity of dissolution profiles.




In spite of small differences found in dissolution profiles and indicating excellent stability of the pellets, one can admit the tendency in profiles change within all stability regimens (Fig. 4a–6a), i.e. slowing down of the rutin release within the first five hours and mild acceleration in the release since the 5.5 h of the testing. This tendency is in harmony with the demanded dissolution profile, i.e. no or low amount of the drug released under conditions simulating the upper GI tract and fast release of the drug under the conditions mimicking the colon.
Conclusion
Rutin pellets based on chitosan were successfully prepared and showed very good characteristics. To withstand the upper GIT conditions and deliver the drug to the colon, pellets were subsequently coated with sodium alginate/chitosan. Coated pellets presented promising rutin dissolution profiles and excellent stability under different testing conditions and times. This is a reason to evaluate their therapeutic effect in vivo in a rat model in future.
Acknowledgement: The support of the project No. 44/2011/FaF IGA VFU entitled “Development of chitosan microparticles intended for drug delivery into the colon” is announced.
Conflicts of interest: none.
Received 29. September 2011 / Accepted 26. October 2011
prof. PharmDr. Miloslava Rabišková, CSc.
Department of Pharmaceutics, Faculty of Pharmacy, University of Veterinary and Pharmaceutical Sciences Brno
Palackého 1–3, 612 42 Brno
e-mail: rabiskovam@vfu.cz
Sources
1. Sutherland I. W. Alginates. In: Byrom, D. ed. Biomaterials - Novel Materials from Biological Sources, NewYork: Stockton 1991, pp. 309–331.
2. Andriamanantoaninaa H., Rinaudo M. Characterization of the alginates from five madagascan brown algae. Carbohydr Polym 2010; 82, 555–60.
3. Draget I. K., Tailor C. Chemical, physical and biological properties of alginates and their biomedical implications. Food Hydrocolloids 2011; 25, 251-256.
4. Dupuy B., Arien A., Minnot A. P. FT-IR of membranes made with alginate/polylysine complexes-variations with the mannuronic or guluronic content of the polysaccharides. Artif Cells Blood Substit Immobil Biotechnol 1994; 22, 71–82.
5. George M., Abraham T. E. Polyionic hydrocolloids for the intestinal delivery of protein drugs: Alginate and chitosan – a review. J. Control. Release 2006; 114, 1–14.
6. Chen S. C., Wu Y. C., Mi F. L., Lin Y. H., Yu L. C., Sung H. W. A novel pH sensitive hydrogel composed of N,O-carboxymethyl chitosan and alginate cross-linked by genipin for protein drug delivery. J. Control. Release 2004; 96, 285–300.
7. Nwe N., Chandrkrachang S., Stevens W., Maw T., Tan T., Khor E., Wong S. Production of fungal chitosan by solid state and submerged fermentation, Carbohydr Polym 2002; 49, 235–237.
8. Rhazi M., Desbriéres J., Tolamaite A., Alagui A., Vottero P. Investigation of different natural sources of chitin: influence of the source and deacetylation process on the physicochemical characteristics of chitosan. Polym Int 2000; 49, 337–344.
9. Muzzarelli R. A. A. Human enzymatic activities related to the therapeutic administration of chitin derivatives. Cell Mol Life Sci 1997; 53, 131–140.
10. Lehr C. M., Bouwstra J. A., Schacht E. H., Junginger H. E. In vitro evaluation of mucoadhesive properties of chitosan and some other natural polymers. Int J Pharm 1992; 78, 43–48.
11. Deacon M. P., McGurk S., Roberts C. J., Williams P. M., Tendler S. J., Davies M. C., Davis S. S., Harding S. E. Atomic force microscopy of gastric mucin and chitosan mucoadhesive systems. Biochem J 2000; 348, 557–563.
12. He P., Davis S. S., Illum L. In vitro evaluation of the mucoadhesive properties of chitosan microspheres. Int J Pharm 1998; 166, 75–88.
13. Kotze A. F., Luessen H. L., Thanou M., Verhoef J. C., de Boer A. G., Junginger H. E., Lehr C. M. Chitosan and chitosan derivatives as absorption enhancers for peptide drugs across mucosal epithelia. In: Mathiowitz, E., Chickering, D. E., Lehr, C. M. eds. Bioadhesive Drug Delivery Systems, New York: Marcel Dekker Inc. 1999, pp. 341–385.
14. Schipper N. G. M., Olsson S., Hoogstraate J. A., deBoer A. G., Varum K. M., Artursson P. Chitosans as absorption enhancers for poorly absorbable drugs: 2. Mechanism of absorption enhancement. Pharm Res 1997; 14, 923–929.
15. Artursson P., Lindmark T., Davis S. S., Illum L. Effect of chitosan on the permeability of monolayers of intestinal epithelial cells (Caco-2). Pharm Res 1994; 11, 1358–1361.
16. Smith J., Wood E., Dornish M. Effect of chitosan on epithelial cell tight junctions. Pharm Res 2004; 21, 43–49.
17. Natsume H., Iwata S., Ohtake K., Miyamoto M., Yamaguchi M., Hosoya K., Kobayashi D., Sugibayashi K., Morimoto Y. Screening of cationic compounds as an absorption enhancers for nasal drug delivery. Int J Pharm 1999; 185, 1–12.
18. Senel S., Hincal A. A. Drug permeation enhancement via buccal route: Possibilities and limitations. J Control Release 2001; 72, 133–144.
19. Grabnar I., Bogataj M., Mrhar A. Influence of chitosan and polycarbophil on permeation of a model hydrophilic drug into the urinary bladder wall. Int J Pharm 2003; 256, 167–173.
20. Sinswat P., Tengamnuay P. Enhancing effect of chitosan on nasal absorption of salmon calcitonin in rats: comparison with hydroxypropyl - and dimethyl-ß-cyclodextrins. Int J Pharm 2003; 257, 15–22.
21. Sandri G., Rossi S., Ferrari F., Bonferoni M.C., Muzzarelli C., Caramella C. Assessment of chitosan derivatives as buccal and vaginal penetration enhancers. Eur J Pharm Sci 2004; 21, 351–359.
22. Yao K. D., Peng T., Feng H. B., He Y. Y. Swelling kinetics and release characteristic of crosslinked chitosan-polyether polymer network (semi - IPN) hydrogels. J Polym Sci A, Polym Chem 1994; 32, 1213–1223.
23. Kast C. E., Schnürch A. B. Thiolated polymers-thiomers: development and in vitro evaluation of chitosan-thioglycolic acid conjugates. Biomaterials 2001; 22, 2345–2352.
24. Thanou M., Kotzé A. F., Scharringhausen T., Lueßen H. L., de Boer A. G., Verhoef J. C., Junginger H. E. Effect of degree of quaternization of N-trimethyl chitosan chloride for enhanced transport of hydrophilic compounds cross intestinal Caco-2 cell monolayers. J Control Release 2000; 64, 15–25.
25. Cheng G. X., Liu J., Zhao R. Z., Yao K. D., Sun P. C., Men A. J., Wang W. H., Wei L. Studies on dynamic behavior of water in crosslinked chitosan hydrogel. J Appl Polym Sci 1998; 67, 983–988.
26. Mi F. L., Sung H. W., Shyu S. S. Drug release from chitosan-alginate complex beads reinforced by a naturally occurring crosslinking agent. Carbohydr Polym 2002; 48, 61–72.
27. Scala-Bertola J., Gajdziok J., Rabišková M., Bonneaux F., Lecompte T., Sapin A., Maincent P. for oral administration of low-molecular-weight heparin. Drug Dev. Ind. Pharm. 2009; 35, 1503-1510.
28. Gander B., Ventouras K., Gurny R., Doelker E. In vitro dissolution medium with supramicellar surfactant concentration and its relevance for in vivo absorption. Int. J. Pharm. 1985; 27, 117–124.
29. Raimundo A. H., Evans D. F., Rogers J., Silk, D. B. A. Gastrointestinal pH profiles in ulcerative colitis. Gastroenterology 1992; 102, A681.
30. Fallingborg J., Christensen L. A., Jacobsen B. A., Rasmussen S. N. Very low intraluminal colonic pH in patients with active ulcerative colitis. Dig Dis Sci 1993; 38, 1989–1993.
31. Washington N., Washington C., Wilson C. G. Drug delivery to the large intestin and rectum. In: Physiological Pharmaceutics: Barriers to Drug Absorption, 2nd ed., New York: Taylor and Francis 2001, 143–180.
32. Rajabi-Siahboomi A. R., Bowtell R. W., Mansfield P., Henderson A., Davis M. C., Melia C. D. Structure and behaviour in hydrophilic matrix sustained release dosage forms: 2. NMR-imaging studies of dimensional changes in the gel layer and core of HPMC tablets undergoing hydration. J Control Release 1994; 31, 121–128.
33. Ashby L. J., Beezer A. E., Buckton G. In vitro dissolution testing of oral controlled release preparations in the presence of artificial food stuffs. I. exploration of alternative methodology: Microcalorimetry. Int J Pharm 1989; 51, 245-251.
34. Eastman I. M., Miller E. G. Gastrointestinal pH in rats as determined by the glass electrode. J Biol Chem 1935; 255–262.
35. Dvořáčková K., Škrabáková G., Rabišková M. Influence of formulation technology on theophylline release from chitosan-based pellets. Čes slov Farm 2009; 58, 216–224.
36. Peréz J. P., Rabišková M. Influence of the drying technique on theophylline pellets prepared by extrusion-spheronization. Int J Pharm 2002; 242, 349–351.
37. Deasy P. B., Law M. F. L. Use of extrusion-spheronization to develop an improved oral doage form of indomethacin. Int J Pharm 1997; 148, 201–209.
38. Rowe R. S., Sheskey P. J., Owen S. C. Handbook of Pharmaceutical Excipients. 5th ed. Washington: Pharmaceutical Press 2006, pp. 146–147.
39. Dvořáčková K., Bautzová T., Rabišková M. Dissolution study in the evaluation of oral preparations with controlled release of drugs. Chem Listy 2011; 105, 50–54.
40. Jedrzejas M. J. Structural and functional comparison of polysaccharide-degrading enzymes. Critical Reviews in Biochemistry and Molecular Biology 2000; 35, 221–251.
Labels
Pharmacy Clinical pharmacologyArticle was published in
Czech and Slovak Pharmacy

2012 Issue 1-2
Most read in this issue
- The possibilities of innovation of extemporaneous preparation in pharmacies in the Czech Republic
- Compatibility of phosphates with calcium salts in parenteral nutrition
- Metabolomics in research of phytotherapeutics
- Intended pharmacotherapeutical approaches of Alzheimer’s disease therapy